

Abstract
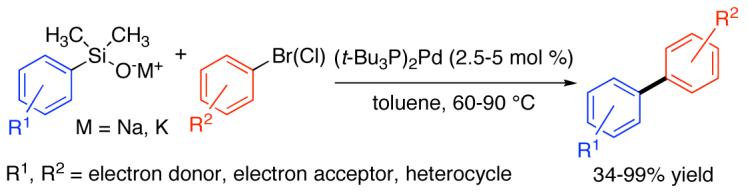
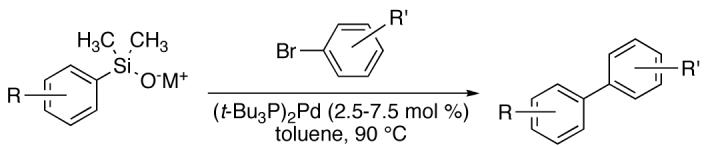
The alkali-metal salts (potassium and sodium) of a large number of aryl- and heteroarylsilanols undergo efficient cross coupling with a wide range of aromatic bromides and chlorides under mild conditions to form polysubstituted biaryls. The critical feature for the success of these coupling reactions and their considerable scope is the use of bis(tri-tert-butylphosphine)palladium. Under the optimized conditions, electron-rich, electron-poor, and sterically hindered arylsilanolates afford cross-coupling products in good yields. Many functional groups are compatible with the coupling conditions such as esters, ketones, acetals, ethers, silyl ethers, and dimethylamino groups. Two particularly challenging substrates, (2-benzofuranyl)dimethylsilanolate and (2,6-dichlorophenyl)dimethylsilanolate prepared as their sodium salts showed excellent activity in the coupling reactions, in the former case also with aromatic chlorides. General methods for the efficient synthesis of a wide range of aromatic silanols are also described.
Introduction
Biaryls represent a common structural motif in both natural products and biologically active compounds (Chart 1). For example, vancomycin is a glycopeptide antibiotic that contains a configurationally defined biaryl subunit.1 Additionally, a number of pharmaceutical compounds containing a biaryl moiety have been developed including Losartan and Irbesartan.2 The prevalence of the biaryl subunit is further evident in the development of highly conjugated aromatic polymers that possess interesting optical and electronic properties.3 Lastly, the biaryl scaffold is an integral part of the ligand design for many catalytic processes. Chiral ligands such as BINAP, BINOL, MOP, SEGPHOS, SYNPHOS and their derivatives have been utilized in many asymmetric transformations.4
Chart 1.
Given the importance of the biaryl motif, a myriad of methods have been developed for the synthesis of biaryl compounds.5 In 1901, Ullmann discovered the homocoupling reaction of aryl halides in the presence of copper powder at elevated temperatures (> 200 °C).6 Since this seminal report, extensive reaction development has produced milder conditions for copper-, nickel-, or palladium-catalyzed couplings.7 Although this method is useful for the synthesis of symmetrical biaryls, high yielding preparations of unsymmetrical biaryls often require pairing an electron-rich aryl halide with an electron-deficient aryl halide.
Among the modern methods of carbon-carbon bond formation, the transition-metal catalyzed cross-coupling reaction has ascended to a strategy level reaction in both industrial and academic sectors.8 For example, a survey of the reactions scaled in the GMP facilities at Pfizer-Groton shows that the use of cross-coupling reactions has increased by 10.2% between 1985 and 2002.9 Of particular importance is the palladium-catalyzed, cross-coupling reaction for the synthesis of biaryls. Various organometallic donors have been studied for this purpose, including organomagnesium (Kumada), organozinc (Negishi), organotin (Stille), organoboron (Suzuki) and organosilicon (Hiyama) reagents.8 Although Grignard reagents can couple effectively, their high nucleophilicity and basicity is not compatible with many functional groups. Organozinc reagents are more tolerant of sensitive functionality; however, the instability of arylzinc reagents has hampered large-scale development.10 In contrast, organostannanes are mild, stable and readily prepared reagents, but a major drawback is the inherent toxicity of the tin reagents and byproducts.11 Consequently, recent investigations have focused on using organoboron-based reagents and many synthetically challenging biaryls have been prepared by judicious choice of catalysts and ligands.12 Despite the broad application of this cross-coupling strategy, these methods are not without drawbacks. For example, some boronic acids are difficult to handle, difficult to purify, unstable to extended storage and suffer protiodeborylation under cross-coupling conditions. Variants of boron reagents such as borate esters and trifluoroborates13 have been developed to address some of these problems.14 In recent years arylsilicon nucleophiles have emerged as useful nucleophiles for cross-coupling reactions. The specific advantages of organosilicon-based donors have been described in detail in a number of review articles and will be summarized only briefly below.15
Background
Aryl(dihalo)silanes represent the first organosilicon reagents used for biaryl synthesis.16 Because of the sensitivity of halosilanes to moisture, acid or base, the more robust trialkoxysilanes have been advocated as more practical reagents for cross coupling with aryl iodides and bromides.17 However, the reaction with aryl chlorides provides the biaryl products in moderate yields (47-71%).18 To address this limitation, stable triallylarylsilanes were introduced for the cross coupling of a range of aryl chlorides.19 A major disadvantage of all the aforementioned arylsilicon donors is the need for fluoride activators in superstoichiometric amounts to facilitate transmetalation. The widespread use of silicon-based protecting groups in complex molecule synthesis along with the cost and aggressive nature of soluble fluoride sources precludes their use as activators. Accordingly, alternative methods of activation have been developed for arylsilicon reagents. In 2005, Hiyama and coworkers reported the use of aryl[2-(hydroxymethyl)phenyl]dimethylsilanes which bear a pendant nucleophilic tether.20 In the presence of potassium carbonate and copper iodide, these reagents deliver aryl groups to aromatic halides. The iterative cross coupling of these organosilicon reagents was elegantly demonstrated for the synthesis of oligoarenes.21
As part of our longstanding interest in the use of the organosilanes in cross-coupling reactions,15c-f,h,i we have investigated use of these compounds in the synthesis of biaryls. One of the initial illustrations of this class of reagents stemmed from using arylsiletanes as aryl transfer substrates.22 The palladium-catalyzed cross-coupling of aryl(halo)silacyclobutanes could be achieved via a fluoride-promoted reaction with aryl iodides to provide biaryls in good yields. Mechanistic studies (with alkenylsilacyclobutanes) demonstrated that treatment of silacyclobutanes with tetrabutylammonium fluoride (TBAF) led to cleavage of the siletanes and formation of organosilanols.23
The introduction of (organo)dimethylsilanols as coupling partners has allowed the development of non-fluoride promoted cross-coupling reactions. An early report from Hiyama and coworkers described the use of silver(I) oxide as a stoichiometric promoter in conjunction with (Ph3P)4Pd to prepare biaryls from silanol precursors.24 Although good yields were obtained, large amounts of a silver activator and long reactions times are needed.
Subsequent investigations from these laboratories identified Cs2CO3 as an effective promoter for the cross-coupling of (4-methoxyphenyl)dimethylsilanol with a range of aryl iodides and bromides.25 These studies also addressed the problem of homocoupling from the aryl halide through the judicious choice of ligands. Unfortunately, attempts to expand the scope of the arylsilanol afforded lower yields. More forcing conditions (110 °C) and other bases (CsOH) were required because of the lower rate of transmetalation of other arylsilanols and their tendency to exist as their thermodynamically more stable disiloxanes.
In the course of mechanistic studies on the cross-coupling reactions of arylsilanols, it was discovered that arylsilanolate salts are stable, isolable compounds that are resistant to dimerization to the corresponding disiloxane.26 The ease of preparation of these salts made them attractive targets for investigating their utility as organosilicon nucleophiles.27 As part of this study, the use of phosphine oxides as stabilizing ligands was also discovered. A significant rate enhancement was observed using both bisphosphine oxides derived from common bidentate phosphines as well as triphenylphosphine oxide.27 These results were transformed into a useful and effective set of reaction conditions that could be employed for the cross coupling of potassium (4-methoxyphenyl)dimethylsilanolate with a range of aryl bromides.27 However, these conditions were not uniformly applicable. Because no strongly coordinating ligands are present, significant amounts of homocoupling products from the aryl bromides are formed and the overall yields of the desired biaryls correspondingly decreases. Moreover, the extension of this method to other arylsilanolates was unsuccessful as unproductive dimerization to the disiloxane was observed.
The variable levels of success achieved with palladium-catalyzed cross coupling of aryldimethylsilanols and its derivatives meant that a more robust and general set of reaction conditions for the preparation of biaryls is still required. In light of these limitations, the following objectives were formulated: (1) prepare a wide range of arylsilanols and their silanolate salts, (2) determine the stability of these reagents, (3) evaluate the competence of these alkali-metal silanolates for the palladium-catalyzed cross-coupling reactions and (4) develop conditions that allow the facile cross coupling of a range of aryl halides. We report herein the successful realization of these goals in the development of a practical fluoride-free, cross coupling of a wide range of arylsilanolates with a wide range of aryl halides that provides unsymmetrical biaryls. Moreover, this reaction has been implemented in the cross coupling of heterocyclic silanolates that have not been disclosed previously.
Results
1. Preparation of Silanols and Silanolates28
To set the stage for our studies, a large number of arylsilanols and their derived silanolates were needed to determine their chemical stability, ease of handling and suitability for cross-coupling reactions with aryl electrophiles. In general, the arylsilanols used herein were prepared by three different protocols: (1) direct installation of the dimethylsilanol unit through halogen-lithium exchange and trapping with a silicon electrophile,29 (2) catalytic, oxidative hydrolysis of dimethylsilanes30 and (3) palladium-catalyzed silylation of aryl halides.31 The selection of the most appropriate method was based on a number of considerations including the availability of a suitable starting material and compatibility of any functional groups in the molecule. Each approach is described in detail below.
1.1 Preparation of Arylsilanols via Halogen-Lithium Exchange
Direct installation of the dimethylsilanol moiety was accomplished by treatment of an aryl iodide (e.g. 4-iodoanisole or 1-iodonaphthalene) with n-butyllithium at low temperature followed by trapping the organolithium intermediate with hexamethylcyclotrisiloxane (D3) to afford (4-methoxyphenyl)dimethylsilanol 1 and (1-naphthyl)dimethylsilanol 2 in 88% and 84% yields, respectively (Table 1, entries 1-2). Although aryl iodides readily participate in halogen-metal exchange, the respective aryl bromides are more attractive substrates because of their greater availability and lower cost.
Table 1.
Preparation of Arylsilanols and the Corresponding Alkali-Metal Arylsilanolates.
 | ||||||
|---|---|---|---|---|---|---|
| entry | starting reagent | conditionsa | arylsilanol yieldb, % |
silanol | conditionsc | arylsilanolate yieldb, % |
| 1 | 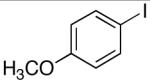 |
A | 88 | 1 | D | 91 |
| 2 | 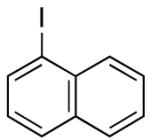 |
A | 84 | 2 | D | 99 |
| 3 |  |
A | 93 | 3 | D | 94 |
| 4 |  |
A | 78 | 4 | D | 98 |
| 5 |  |
A | 72 | 2 | D | 95 |
| 6 |  |
A | 95 | 6 | D | 90 |
| 7 |  |
B | 79 | 7 | D | 90 |
| 8 |  |
B | 94 | 8 | D | 97 |
| 9 |  |
A | 87 | 9 | D | 95 |
| 10 | A | 87 | 10 | D | 95 | |
| 11 | A | 79 | 11 | D | 99 | |
| 12 |  |
A | 54 | 12 | D | 99 |
| 13 | 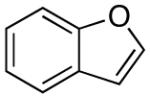 |
B | 93 | 13 | E | 98 |
| 14 | 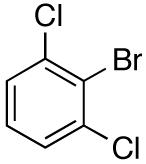 |
B | 95 | 14 | F | 79 |
| 15 | 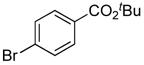 |
C | 82 | 15 | D | 91 |
Methods for silanol preparations: A. 1. n-BuLi (1.0 equiv) or t-BuLi (2.0 equiv), Et2O, -78°C 2. D3 (0.33 equiv), Et2O, -78 °C to rt. B. 1. n-BuLi (1.0 equiv) or t-BuLi (2.0 equiv), Et2O, -78 °C 2. Me2SiCl(H), -78 °C to rt 3.[RuCl2(p-cymene)]2 or [Ir(1,5-cyclooctadiene)Cl]2 (1-2 mol %), H2O (2-5 equiv), CH3CN, rt. or aq. TBAOH (1.5 equiv, 1 M), CH3CN, 0 °C C. 1. (EtO(CH3)2Si)2 (1.5 equiv), [allylPdCl]2 (5 mol %), BPTBP (10 mol %), NMP, 50 °C 2. pH = 5 OAc buffer, HS(CH2)2NH2.HCl, CH3CN
Yields correspond to isolated, purified products
Methods for silanolate preparation: D. KH (1.2 equiv), THF or C6H6, rt. E. NaHMDS, THF, rt. F. NaH (1.2 equiv), THF, rt.
Thus, the scope of the direct process was further illustrated with a diverse array of aryl bromides. In these cases tert-butyllithium was used to generate the organolithium intermediate. Electron-rich aryl bromides such as tert-butyldimethylsilyl (TBS) protected 4-bromophenol, 3- and 2-bromoanisole, and 4-bromostilbene all furnished the aryldimethylsilanols in good to excellent yields of analytically pure products (72-95%) (Table 1, entries 3-6). Similarly, electron-deficient aryl bromides containing trifluoromethyl, chloro, and fluoro substituents showed comparable reactivity and overall yields (79-87%) (Table 1, entries 9-11).
The direct metalation of dibenzothiophene using n-BuLi in refluxing ether gave the 4-dibenzothienyllithium intermediate that underwent further reaction with D3 to afford analytically pure (4-dibenzothienyl)dimethylsilanol 12 in 54% yield (Table 1, entry 12). These procedures were easily scalable, e.g. the preparation of 12 was routinely performed on a 20 mmol scale (5 g of product).
1.2 Catalytic Oxidative Hydrolysis of Arylsilanes
For less reactive or less stable aryllithium intermediates (such as those from 4-bromo-N,N-dimethylaniline, benzofuran, and 2,6-dichlorobromobenzene) the more reactive electrophile dimethylchlorosilane was employed to provide the arylsilanes in high yields. Selective oxidation of these silyl hydrides was accomplished with [RuCl2(p-cymene)]2 (1-5 mol %) in MeCN/H2O to yield the desired silanols in 79-95% yield (Table 1, entries 7-8, 13-14). Although, successful oxidative addition could be effected under these conditions with (2,6-dichlorophenyl)dimethylsilane, the reaction was quite sluggish (> 8 h). The more active catalyst, [Ir(COD)Cl]2 led to the formation of the desired silanol in 45 min (in the presence of 5 equiv of H2O). These processes are reproducible, e.g., (2-benzofuranyl)dimethylsilane can be prepared consistently on an 85 mmol scale, while the oxidative hydrolysis of this silane is routinely performed on > 20 mmol scale.
Another silanol that initially proved challenging to prepare was (4-(1,3-dioxolan-2-yl)phenyl)dimethylsilanol 7. The the lower reactivity of the aryllithium intermediate (from the corresponding bromide) with D3 led to significant amounts of disiloxane formation. Preparation of the (4-(1,3-dioxolan-2-yl)phenyl)dimethylsilane as described above was straightforward, but attempts to effect the oxidative hydrolysis using either Ru or Ir catalysis was problematic. Significant amounts of 4-(hydroxydimethylsilyl)benzaldehyde (4–10%) were observed and separation from the desired silanol was not easily achieved. Gratifyingly, simply treating the (4-(1,3-dioxolan-2-yl)phenyl)dimethylsilane with 1.5 equiv of tetrabutylammonium hydroxide32 led to quantitative formation of the desired silanol in 79% yield (Table 1, entry 7).
1.3 Palladium-Catalyzed Silylation of Aryl Halides
The installation of the dimethylsilanol unit into substrates possessing functional groups that are incompatible with organolithium reagents (esters, ketones, nitriles, etc.) employed previously developed catalytic reaction with 1,2-diethoxy-1,1,2,2-tetramethyldisilane.31 For example, tert-butyl 4-(hydroxydimethylsilyl)benzoate was prepared from tert-butyl 4-bromobenzoate, allylpalladium chloride dimer ([allylPdCl]2), 2-(di-tert-butylphosphino)biphenyl (BPTBP),33 and di-isopropylethylamine and the disilane in NMP. The silyl ether was hydrolyzed to the silanol 15 with 1.0 M acetic acid in 82% yield, over two steps (Table 1, entry 15). The preparation of 15 was routinely carried out on a 10 mmol scale.
1.4 Preparation of Arylsilanolates from the Parent Silanols
Alkali metal arylsilanolate salts can be prepared from arylsilanols (pKa ∼ 9-11)28 by any of three different protocols: (1) irreversible deprotonation of the arylsilanol in situ with a strong Brønsted base, (2) irreversible deprotonation of the arylsilanol with a strong base and isolation of the silanolate salt,34 and (3) silanolate exchange with disiloxanes.35 Isolation of the arylsilanolate has a number of advantages that deserve mention, the most important of which is operational simplicity. The salt can be charged directly into the reaction mixtures and no additional base is needed. Moreover, isolation of the arylsilanolate avoids the formation of disiloxane from the parent silanol under the reaction conditions. Accordingly, metal hydrides and disilazanes were chosen as the bases to promote rapid and quantitative deprotonation that allowed the isolation of the arylsilanolate salts. For the most part, potassium silanolate salts were selected because they are more nucleophilic than sodium silanolates thus avoiding the complicating factor of a rate limiting displacement of the palladium halide in the catalytic cycle.36
The potassium arylsilanolates were prepared by the dropwise addition of a solution of the arylsilanol (in THF or benzene) to either KH or KHMDS in the same solvent. After 15 min, the reaction mixture was Schlenk filtered to remove any particulates. The solution was concentrated in vacuo to afford solids that were triturated with hexanes and dried to afford the arylsilanolate salts as stable powders in most cases (Table 1, entries 1-12, 15).37 Overall, excellent yields of the potassium salts were obtained but the less than quantitative recovery was occasionally seen because of partial solubility of these compounds in hexanes.
Unfortunately, the preparation of both potassium (2-benzofuranyl)dimethylsilanolate and (2,6-dichlorophenyl)dimethylsilanolate by the addition of the corresponding silanol to KH or KHMDS in THF led to rapid desilylation to benzofuran and 1,3-dichlorobenzene, respectively. This problem has been encountered previously in the preparation of potassium (2-indolyl)dimethylsilanolates.38 The instability of the potassium salts of heterocyclic compounds was circumvented by preparing the sodium silanolate Na+13 and Na+14- using either NaH or NaHMDS. Generally, NaHMDS is the preferred base because it provides higher yields and no filtration is required prior to concentration (Table 1, entry 13). These sodium salts were stable solids as well.
2. Optimization of Cross-Coupling Reaction Conditions
2.1. Substrate Choice
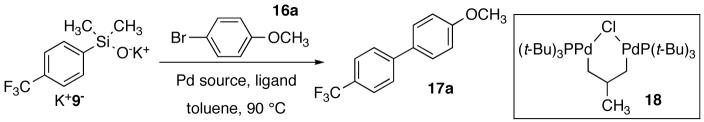
The selection of an appropriate substrate pair for the initial optimization was crucial in view of the limited substrate scope seen in previous studies.25 Whereas highly reactive silanolates from electron-rich silanols did react smoothly, less activated silanolates required more vigorous reaction conditions and gave poorer yields. Thus, to ensure a successful demonstration of broad scope, we selected very challenging substrates for optimization studies with the expectation that conditions that work well for the poorest substrates, should be applicable to better ones in general. Accordingly, potassium (4-trifluoromethylphenyl)dimethylsilanolate K+9- and 4-bromoanisole 16a were selected because of: (1) the poor nucleophilicity of K+9-, (2) the slower oxidative addition by Pd(0) catalysts to 16a, and (3) the increased propensity for 16a to undergo homocoupling. Orienting experiments with K+9- (1.5 equiv) and 16a under conditions developed for couplings of arylsilanolates previously (Pd(dba)2 and Ph3 P(O) in toluene at 90 °C)27 provided low conversion and product yield (Table 2, entry 1). Other Pd(II) and Pd(0) sources successfully catalyzed the cross-coupling reaction albeit in low yields (Table 2, entries 2 and 4). Moreover, the use of an N-heterocyclic carbene-based palladium catalyst gave a poor yield of 17a (entry 3).
Table 2.
Optimization of the Ligand/Palladium Combination for the Cross Coupling of K+9- with 16a.a
 | |||||
|---|---|---|---|---|---|
| entrya | Pd source, 5 mol% Pd |
ligand, 1/1 Pd/L |
time, h |
conversion, %b |
product yield, %c |
| 1 | Pd(dba)2 | Ph3P(O) | 7 | 24 | 7 |
| 2 | (C3H5)CpPd | dppp | 20 | 100 | 30 |
| 3 | PdCl(C3H5)(Ii-Pr) | - | 20 | 43 | 10 |
| 4 | (Ph3P)4Pd | - | 20 | 68 | 37 |
| 5 | [allylPdCl]2 | - | 7 | 37 | 9 |
| 6 | [allylPdCl]2 | dppp(O)2 | 7 | 95 | 45 |
| 7 | [allylPdCl]2 | dppp(O) | 7 | 54 | 30 |
| 8 | [allylPdCl]2 | dppp | 3 | 69 | 51 |
| 9 | [allylPdCl]2 | Ph3As | 7 | 22 | 12 |
| 10 | [allylPdCl]2 | SPhos | 7 | 71 | 46 |
| 11 | [allylPdCl]2 | t-Bu3P | 5.5 | 99 (100)d | 79 (89)d |
| 12 | [allylPdCl]2 | t-Bu3P•HBF4 | 20 | 88 | 68 |
| 13 | 18 | - | 7 | 90 | 73 |
| 14 | (t-Bu3P)2Pd (19) | - | 5 | 100 (100)e | 88 (92)e |
Reactions conditions: 1.5 equiv of arylsilanolate K+9- and 1.0 equiv of 16a
Conversion was based on consumption of aryl bromide as determined by GC analysis using an internal standard.
Yield determined by GC analysis using an internal standard.
Yield in parentheses based on 2:1 ratio of ligand/Pd.
Conversion and yield in parentheses refers to the use of (t-Bu3P)2Pd purchasedfrom Aldrich Chemical Co.
2.2. Palladium Source and Ligands
Clearly, a thorough optimization of reaction conditions was needed which began with studies on the influence of ligands. Because [allylPdCl]2 is cleanly and quantitatively reduced to ligandless Pd(0) in the presence of arylsilanolates, this complex was chosen for the ligand studies.39 When [allylPdCl]2was employed in the absence of ligands for the cross-coupling of K+9- (1.5 equiv) and 16a only a 9% yield of 17a was obtained (Table 2, entry 5). Employing [allylPdCl]2 and dppp(O)2 afforded a substantial increase in conversion and yield (entry 6) compared to the ligandless conditions. Under the reasonable assumption that a more coordinating ligand was required to prolong the catalyst lifetime, both dppp(O) and dppp were tested but proved to be ineffective (Table 2, entries 7-8). Furthermore, the use of ligands that have been successful for the cross coupling of aryl-25 and alkenylsilanols40 including Ph3As and SPhos41 provided poor yields of 17a (entries 9 and 10 respectively). Gratifyingly, the use of t-Bu3P in combination with [allylPdCl]2 provided complete conversion of 16a and a high yield of biaryl 17a (entry 11). Although an acceptable yield of 17a was achieved, additional studies were needed to clarify the optimal catalyst loading and ligand stoichiometry.
If indeed the active catalytic species is t-Bu3P-Pd(0), other catalyst combinations that could produce this complex should also be effective. However, the more air and moisture stable t-Bu3P·HBF4 salt led to a slightly diminished overall yield (entry 12). The use of the palladacycle catalyst 18 (previously described for the cross-coupling of indolyl-2-silanols)34b provided a comparable conversion and product yield at elevated temperatures (Table 1, entry 13). Interestingly, increasing the ligand/Pd loading from 1:1 to 2:1 using t-Bu3P and [allylPdCl]2 provided an additional increase in yield of 17a to 89% (Table 2, entry 11). Finally, the preformed “Fu’s catalyst” ((t-Bu3P)2Pd (19)) with the desired ligand/Pd stoichiometry,42 led to complete consumption of both aryl halide and arylsilanolate to afford 17a in 88% yield (entry 14). At this point, only minor, substrate specific, modifications (solvent, temperature and stoichiometry) were needed to create a general set of reaction conditions.
2.3. Solvent, Temperature, and Stoichiometry
Solvents representing a wide range of polarities were tested (Table 3). Toluene (ε=2.4) was an effective solvent for the cross coupling of K+9-(1.5 equiv), providing the biaryl in 92% yield within 5 h (entry 1). Benzotrifluoride, a slightly more polar aromatic solvent (ε=9.2), afforded 17a in 90% yield in 3 h (entry 2). Ethereal solvents, such as dioxane (entry 5), led to productive cross coupling albeit in slightly lower yields. The use of highly polar solvents, such as NMP or DMF, led to a rapid, unproductive consumption of aryl bromide in only 15 min (entries 3-4). No biaryl product was obtained and multiple products were observed including reduction of the aryl bromide to anisole. Ultimately, toluene was chosen as the solvent because of its widespread use and lower cost compared to benzotrifluoride.
Table 3.
Optimization of Solvent for the Cross Coupling of K+9- with 4-Bromoanisole.a
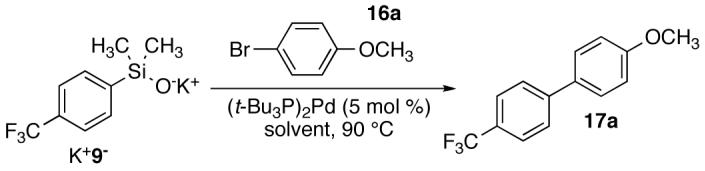 | |||||
|---|---|---|---|---|---|
| entrya | solvent | dielectric constant |
time, h |
conversion, %b |
yield, %b |
| 1 | toluene | 2.4 | 5 | 100 | 92 |
| 2 | C6H5CF3 | 9.2 | 3 | 98 | 90 |
| 3 | NMP | 32.2 | 0.25 | 100 | 0 |
| 4 | DMF | 36.7 | 0.25 | 100 | 0 |
| 5 | dioxane | 2.2 | 5 | 88 | 74 |
Reactions employed 1.5 equiv of K+9-.
Conversion and yield were determined by GC analysis using an internal standard
Next, the reaction temperature was optimized using toluene as the solvent (Table 4). Initial studies began at 90 °C following earlier experiences with aryldimethylsilanols. At this temperature, the cross coupling of K+9>- (1.5 equiv) afforded 17a in excellent yield and short reaction time (Table 4, entry 3). A 10 or 20 °C decrease in the temperature significantly.
Table 4.
Optimization of Temperature for Cross Coupling of K+9-.a
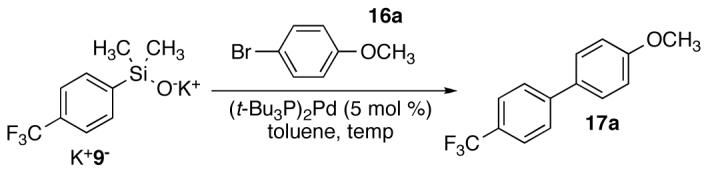 | ||||
|---|---|---|---|---|
| entry | temperature, °C | time, h | conversion, %b | yield, %b |
| 1 | 70 | 20 | 66 | 61 |
| 2 | 80 | 20 | 72 | 66 |
| 3 | 90 | 5 | 100 | 92 |
| 4 | 100 | 3 | 98 | 86 |
| 5 | 110 | 1 | 100 | 86 |
Reaction employed 1.5 equiv of K+9-.
Conversion and yield were determined by GC analysis using an internal standard.
decreased both conversion (72 and 66 %) and yield to only 66 or 61 % in 20 h, respectively (entries 1-2). Although increased temperatures led to more rapid consumption of 16a, no increase in yield was obtained (entries 4 and 5).
The final dimension of optimization concerned the catalyst loading, reaction concentration and silanolate stoichiometry (Table 5). Decreasing the catalyst loading to 0.01 equiv relative to the aryl halide dramatically decreased the reaction rate (20 h) and product yield 46% (entry 1). Using 0.025 equiv of the catalyst gave a comparable yield of 17a but the reaction stalled at 89% conversion (entry 2). Performing the reaction at a higher concentration (0.5 M with respect to 16a) resulted in a heterogeneous mixture at room temperature (entry 3). However, upon heating to 90 °C, the mixture became homogeneous and within 3 h, a 92% yield of 17a was recorded (entry 3, GC analysis). An increase in the catalyst loading at the higher concentration (0.5 M) did not provide an increase in product yield. Attempts at decreasing the amount of K+9- (from 1.5 equiv to 1.1 or 1.3 equiv) for the cross coupling afforded lower yields (entries 4-5). The optimized cross-coupling conditions (1.5 equiv of K+9-, per aryl bromide, (t-Bu3P)2Pd (2.5 mol %) in toluene at 90 °C) provide a simple and practical protocol that will be used in the substrate survey that follows.43
Table 5.
Optimization of the Catalyst Loading for the Preparation of 17a.a
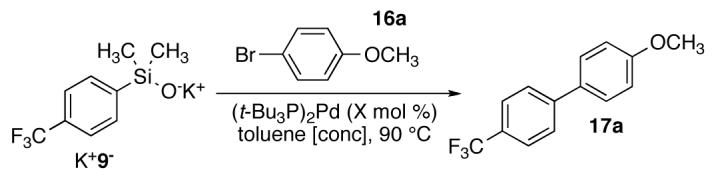 | |||||
|---|---|---|---|---|---|
| entry | cat. loading, equivb |
concentration, Mb |
time, h |
conversion, %c |
yield, %c |
| 1 | 0.01 | 0.25 | 20 | 55 | 46 |
| 2 | 0.025 | 0.25 | 11 | 89 | 77 |
| 3 | 0.025 | 0.50 | 3 | 98 | 92 |
| 4d | 0.025 | 0.50 | 5 | 79 | 74 |
| 5e | 0.025 | 0.50 | 5 | 91 | 82 |
| 6 | 0.05 | 0.25 | 5 | 100 | 92 |
| 7 | 0.05 | 0.50 | 3 | 100 | 91 |
Reactions employed 1.5 equiv of K+9-.
Calculated based upon 1.0 equiv. of aryl bromide.
Conversion and yield were determined by GC analysis using an internal standard.
Reaction employed 1.1 equiv of K+9-.
Reaction employed 1.3 equiv of K+9-.
3. Preparative Cross-Coupling Reactions of Aryl Silanolates
3.1 Cross Coupling of Electron-Rich Alkali-Metal Silanolates with Aryl Halides
For the initial survey of cross-coupling reactions with aryl bromides under the newly developed conditions, K+1- was chosen as the silanolate coupling partner (Table 6). This arylsilanolate was used successfully in previous studies and should react in a similar fashion under the reaction conditions developed above. Indeed, both electron-rich (entries 1-2) and electron-deficient (entry 3) aryl bromides reacted smoothly with K+1- to afford biaryls in good yields and reasonable reaction times (3-4 h). 2-Bromotoluene was also reactive (entry 2) and provided the biaryl product efficiently within that reaction time. Heterocyclic bromides were of particular interest because these substrates previously provided only modest success in the coupling with arylsilanolates.25,44 Gratifyingly, 3-bromoquinoline 16e afforded the coupling product in high yield (entry 4).
Table 6.
Cross Coupling of Methoxy-Substituted Potassium Arylsilanolates with Aryl Halides using (t-Bu3P)2Pd.a
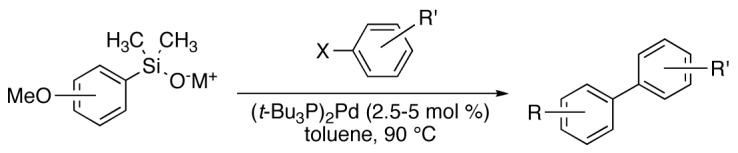 | ||||||||
|---|---|---|---|---|---|---|---|---|
| entry | R | no. | aryl halide | no. | catalyst loading. mol % |
time, h |
product | yield, %b |
| 1 | 4-OMe | K+1- |  |
16b | 2.5 | 3 | 20b | 88 |
| 2 | 4-OMe | K+1- |  |
16c | 2.5 | 3 | 20c | 88 |
| 3 | 4-OMe | K+1- |  |
16d | 5.0 | 4 | 20d | 78 |
| 4 | 4-OMe | K+1- |  |
16e | 5.0 | 5 | 20e | 85 |
| 5 | 4-OMe | K+1- |  |
16f’ | 5.0 | 5 | 20f | 88 |
| 6 | 4-OMe | K+1- |  |
16b’ | 5.0 | 7 | 20b | 81 |
| 7 | 4-OMe | K+1- |  |
16c’ | 5.0 | 7 | 20c | 84 |
| 8 | 3-OMe | K+4- |  |
16g | 5.0 | 3.5 | 21g | 82c |
| 9 | 3-OMe | K+4- |  |
16c | 5.0 | 3 | 21c | 78 |
| 10 | 3-OMe | K+4- |  |
16h | 2.5 | 5.5 | 21h | 80 |
| 11 | 2-OMe | K+5- |  |
16a | 5 | 8 | 22a | 83 |
| 12 | 2-OMe | K+5- |  |
16h | 5 | 8 | 22h | 71 |
| 13 | 2-OMe | K+5- | 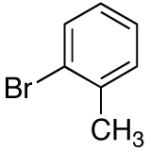 |
16c | 5 | 6 | 22c | 59 |
Reactions employed 1.5 equiv of arylsilanolate
Yields are for isolated, chromatographically homogeneous products.
Yields are for analytically pure products
As has now become the state of the art, aryl chlorides are recognized as more useful substrates in view of their greater availability and lower cost, making them attractive candidates for these studies.45 Although aryl chlorides are less reactive toward oxidative addition by palladium because of the stronger C-Cl bond,46 the use of strong electron-donating phosphine ligands such as t-Bu3 P, has overcome this challenge.42,45 Thus, the examination of aryl chlorides under identical conditions was undertaken. When arylsilanolate K+1- was combined with a representative set of aryl chlorides, the biaryls were obtained in yields equivalent to those obtained from aryl bromides (Table 6, entries 5-7). Whereas, chlorobenzene reacted in 5 h to provide an 88% yield of 20f (entry 5), aryl chlorides bearing a methyl group at either the 2- or the 4-position required slightly longer reaction times (7 h, entries 6-7).
Two other substituted arylsilanolates, K+4- and K+5-,which bear a methoxy group at either the 3- or the 2-position, led to good yields with all of the aryl bromides tested (Table 6, entries 8-13). Interestingly, K+4- reacted with 4-bromochlorobenzene to provide a 4-chlorobiaryl (entry 10). Although the reaction times for (2-methoxyphenyl)dimethylsilanolate K+5- were longer (8 h), the biaryl products were formed efficiently (including the 2,2’-diortho-substituted biaryl from coupling with 2-bromotoluene (entry 13)).47
Next, other electron-rich arylsilanolates bearing different substituents as well as electron-neutral silanolates were examined (Table 7). Thus, potassium (N,N-dimethylaminophenyl)-dimethylsilanolate K+8- reacted with a number of aryl electrophiles in short reaction times. No immediate difference was encountered as electron-rich and electron-deficient aryl bromides smoothly reacted to give the desired biaryl products in good yields (entries 1-3).
Table 7.
Cross Coupling of Electron-Rich and Electron-Neutral Potassium Arylsilanolates with Aryl Bromides Using (t-Bu3P)2Pd.a
 | ||||||||
|---|---|---|---|---|---|---|---|---|
| entry | R | no. | aryl halide | no. | catalyst loading. mol % |
time h, |
product | yield, % |
| 1 | 4-NMe2 | K+8- | 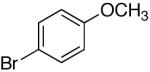 |
16a | 2.5 | 3 | 23a | 88 b |
| 2 | 4-NMe2 | K+8- | 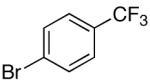 |
16i | 2.5 | 3 | 23i | 80 b |
| 3 | 4-NMe2 | K+8- |  |
16e | 2.5 | 3 | 23e | 73 c |
| 4 | 4-OTBS | K+3- |  |
16a | 5 | 3.5 | 24a | 62 c |
| 5 | 4-OTBS | K+3- |  |
16c | 5 | 3.5 | 24c | 34 c |
| 6 | d | K+2- |  |
16a | 2.5 | 4 | 25a | 82 b |
| 7 | d | K+2- | 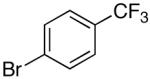 |
16i | 5 | 5 | 25i | 81 b |
| 8 | d | K+2- | 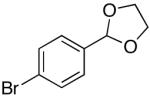 |
16d | 5 | 4 | 25d | 71 c |
| 9 | (trans) 4-(2- phenyl)ethenyl |
K+6- | 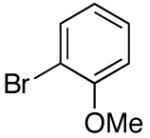 |
16j | 2.5 | 10 | 26j | 51 c |
Reactions employed 1.5 equiv of arylsilanolate
Yields are for isolated, chromatographically homogeneous products.
Yields are for analytically pure products.
Potassium (1-naphthyl)dimethylsilanolate
The cross coupling of the electron-rich arylsilanolate derived from tert-butyldimethylsilyl (TBS) protected phenol (K+3-) was investigated with varying results. Although a direct reaction involving phenol-based nucleophiles was unsuccessful because of the similarity of the pKa’s of the silanol and phenol, simply protecting the phenol as a silyl ether was effective. The cross coupling of K+3- and 16a afforded a modest yield of 24a. However, reactions involving sterically-hindered and electron-deficient aryl bromides such as tert-butyl 4-bromobenzoate (16l) were plagued with significant amount of deprotection to phenol-based products.
The cross coupling of electronically neutral silanolates K+2- proceeded uneventfully. Silanolate K+2- was tested with a number of aryl bromides including 4-(1,3-dioxolane)bromobenzene (entry 8) providing further evidence for the mildness of these conditions. Interestingly, the cross coupling of 2-bromoanisole with an alkenyl-substituted arylsilanolate K+6- required 10 h to reach completion affording 51% of 26j. Other aryl bromides smoothly coupled with K+6-, but difficulty in purification prevented including these substrates in this report. Overall, a variety of substituents on the electrophile were compatible and only minor variations were observed between substrates. With these promising results in hand, we were encouraged to investigate the more challenging electron-deficient aryl silanolates.
3.2 Cross Coupling of Electron-Deficient Alkali-Metal Silanolates with Aryl Halides
Previous reports from these laboratories revealed that electron-deficient silanolates performed poorly in cross-coupling reactions, most likely because of a slow transmetalation of the silanolate.25 This failure was a major motivation for the current study and the reason that K+9- was selected for the optimization. The demonstration that K+9- performed well underthe modified reaction conditions stimulated the investigation of other electron-deficient arylsilanolates to determine their competency as nucleophilic partners for the preparation of biaryls. Under the optimized conditions, K+9- combined with a number of aryl bromides (Table 8) encompassing electron-rich (entries 1-3), electron-poor (entry 4), sterically-hindered (entry 3) and heterocyclic substrates (entry 5) in moderate to excellent yields (68-92%). Moreover, aryl chlorides bearing reactive functionalities such as phenols with silicon-based protecting groups, esters, and ketones, underwent smooth cross coupling with K+9- (entries 6-8).
Table 8.
Cross Coupling of Fluorine Containing Potassium Arylsilanolates with Aryl Halides Using (t-Bu3P)2Pd.a
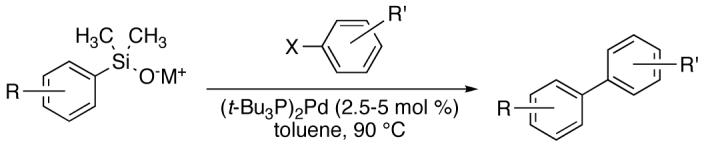 | ||||||||
|---|---|---|---|---|---|---|---|---|
| entry | R | no. | aryl halide | no. | catalyst loading. mol % |
time, h |
product | yield, %b |
| 1 | 4-CF3 | K+9- |  |
16a | 2.5 | 3 | 17a | 92 |
| 2 | 4-CF3 | K+9- |  |
16k | 5.0 | 5 | 17k | 90 |
| 3 | 4-CF3 | K+9- |  |
16j | 5.0 | 5 | 17j | 78 |
| 4 | 4-CF3 | K+9- |  |
16l | 5.0 | 5 | 17l | 68 |
| 5 | 4-CF3 | K+9- | 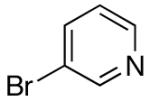 |
16m | 5.0 | 5 | 17m | 73 |
| 6 | 4-CF3 | K+9- | 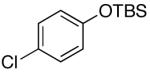 |
16n’ | 5.0 | 5 | 17n | 58 c |
| 7 | 4-CF3 | K+9- | 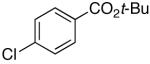 |
16l’ | 2.5 | 5 | 17l | 62 c |
| 8 | 4-CF3 | K+9- | 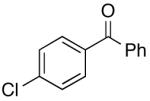 |
16o | 5.0 | 5 | 17o | 65 |
| 9 | 4-F | K+11- |  |
16a | 2.5 | 3 | 27a | 86 |
| 10 | 4-F | K+11- |  |
16p | 2.5 | 4 | 27p | 91 c |
| 11 | 4-F | K+11- |  |
16c | 2.5 | 4 | 27c | 81 |
| 12 | 4-F | K+11- |  |
16l | 2.5 | 3 | 27l | 70 c |
| 13 | 4-F | K+11- | 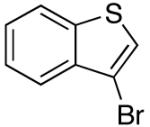 |
16q | 2.5 | 5 | 27q | 65 |
| 14 | 4-F | K+11- | 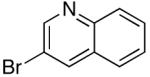 |
16e | 2.5 | 5 | 27e | 84 |
Reactions employed 1.5 equiv of arylsilanolate
Yields are for isolated, chromatographically homogeneous products.
Yields are for analytically pure products
Additionally, (4-fluorophenyl)dimethylsilanolate, K+11-, also afforded good yields of the desired products for a range of electrophiles (Table 8, entries 9-14). Both electron-rich and electron-deficient aryl bromides afforded biaryls in good yields (entries 9-12). Heteroaryl bromides were also competent electrophiles in coupling with K+11- and provided similar yields compared to those obtained from reactions with electron-rich aryl silanolates (entries 13 and 14).
Because both K+9- and K+11- were effective nucleophiles under the optimized conditions, the use of other electron-deficient arylsilanolates bearing more sensitive functional groups was of interest (Table 9). Both tert-butyl ester K+15- (entries 1-3) and acetal-derived K+7- arylsilanolates (entries 4-5) reacted smoothly and in good yields without any observable saponification or deprotection. In each case, 4-bromochlorobenzene 16h was used and preferential cross-coupling reaction occurred at the more reactive bromine bearing carbon, leaving the aryl chloride available for further manipulations. Moreover, the failed cross coupling of the silyloxy-substituted silanolate (K+3-) and tert-butyl 4-bromobenzoate (16l) could be remedied by reversing the roles of the substrates. Thus, combining K+15- with 16n and 2.5 mol % of (t-Bu3P)2Pd provided 28n in moderate yield in 6 h (entry 1). Even potassium 4-(chloro)dimethylsilanolate K+10- was competent under the cross-coupling conditions. Biaryls containing an aryl chloride were formed in good yields and no products arising from multiple cross-coupling events were detected (entries 7-11). However, employing 16l as the electrophile led to a low yield of the cross-coupling product. The yield could be improved slightly by making recourse to the more bulky tert-heptyl ester 16s which provided the biaryl in 56% yield (entry 10).
Table 9.
Cross Coupling of Electron-Deficient Potassium Arylsilanolates with Aryl Bromides Using (t-Bu3P)2Pd.a
 | ||||||||
|---|---|---|---|---|---|---|---|---|
| entry | R | no. | aryl halide | no. | catalyst loading. mol % |
time, h |
product | yield, % |
| 1 | 4-CO2t-Bu | K+15- | 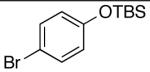 |
16n | 2.5 | 6 | 28n | 57d |
| 2 | 4-CO2t-Bu | K+15- |  |
16j | 2.5 | 7 | 28j | 69 d |
| 3 | 4-CO2t-Bu | K+15- |  |
16h | 2.5 | 6 | 28h | 79 d |
| 4 | 4-[2-(1,3- dioxolane)] |
K+7- |  |
16a | 5.0 | 3 | 29a | 77 c |
| 5 | 4-[2-(1,3- dioxolane)] |
K+7- |  |
16h | 5.0 | 3 | 29h | 77 d |
| 6 | 4-Cl | K+10- |  |
16n | 2.5 | 3.5 | 30n | 57 d |
| 7 | 4-Cl | K+10- |  |
16r | 5 | 3.5 | 30r | 60 d |
| 8 | 4-Cl | K+10- |  |
16c | 5 | 3.5 | 30c | 68 c |
| 9 | 4-Cl | K+10- |  |
16l | 2.5 | 3.5 | 30l | 42 d |
| 10 | 4-Cl | K+10- |  |
16s | 2.5 | 3.5 | 30s | 56 d |
| 11 | 4-Cl | K+10- |  |
16q | 5.0 | 3.5 | 30q | 61 c |
| 12 | 2,6-Clb | Na+14- |  |
16g | 7.5 | 3 | 31g | 68 e |
| 13 | 2,6-Clb | Na+14- |  |
16k | 7.5 | 3 | 31k | 76 d |
| 14 | 2,6-Clb | Na+14- |  |
16h | 7.5 | 3 | 31h | 70 e |
Reactions employed 1.5 equiv of arylsilanolate
Reaction employed 2.0 equiv of arylsilanolate in THF
Yields are for isolated, chromatographically homogeneous products.
Yields are for analytically pure products.
Products were determined to be >99% pure by 1H NMR spectroscopy.
Another interesting class of compounds that has gained attention in the medicinal chemistry sector are multi-substituted chlorinated arenes. These compounds have been investigated for VEGFr2 and the Src family (Src and YES) kinase inhibition-based studies and required the use of 2,6-dichlorophenyl nucleophiles.48 Although the arylboronic acid is the most commonly used donor, these couplings lead to varying efficiencies and overall yields. To improve the effectiveness of this class of nucleophiles, (2,6-dichlorophenyl)dimethylsilanolate Na+14- was prepared and investigated. Orienting experiments with 4-tert-butylbromobenzene 16g under the optimized reaction conditions led to modest conversions (ca. 80%) as the reactions stalled (palladium black observed from catalyst death). Conversions were only moderately improved by increasing both the amount of Na+14- and (t-Bu3P)2Pd respectively as unacceptable amounts of protiodesilylation were also observed. To improve the nucleophilicity of the sodium silanolate, 1,4-dioxane was used as the solvent. Surprisingly, the reaction rate diminished dramatically but 14 was detected by GC analysis after 2 h and only trace amounts of protiodesilylation were observed. Unpredictably in THF at reflux (66 °C), no desired product was observed. However when the reaction was performed in THF in a Teflon-sealed vessel at 90 °C using 2.0 equiv of Na+14- and 7.5 mol% of (t-Bu3P)2Pd the complete consumption of 16g was observed. This process was successfully employed for a number of aryl bromides to provide the desired biaryl in 68-76% yields (Table 9, entries 12-14).
3.3 Cross Coupling of Heterocyclic Silanolates
In view of the enormous interest in heterocycles in the pharmaceutical industry and to extend the scope of silanolates developed in these laboratories,38 the cross coupling of heterocyclic silanolates adopting the standard reaction condition for the synthesis of biaryls was investigated. Orienting studies were conducted with the readily prepared potassium (dibenzothienyl)dimethylsilanolate K+12-. The scope of the reaction was illustrated using a representative set of aryl bromides (Table 10). In general, the cross-coupling products from electron-rich and electron-poor bromides were obtained in excellent yields (73-96%) in 3 h.
Table 10.
Cross Coupling of Potassium (Dibenzothienyl)dimethylsilanolate K+12- with Aryl Bromides Using (t-Bu3P)2Pd.a
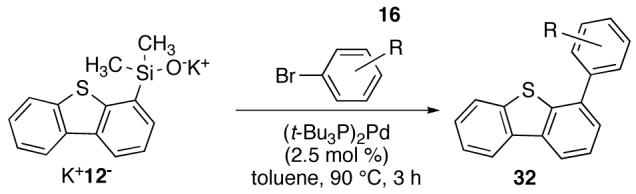 | ||||
|---|---|---|---|---|
| entry | R | no. | product | yield, %b |
| 1 | 4-OCH3 | 16a | 32a | 96 (84) c |
| 2 | 4-CF3 | 16i | 32i | 93c |
| 3 | 2-OCH3 | 16j | 32j | 89 (81) c |
| 4 | 3-bromothiophene | 16q | 32q | 73 (65) c |
Reactions employed 1.5 equiv of arylsilanolate
Yields are for isolated, chromatographically homogeneous products.
Yields in parentheses are for analytically pure products
The successes described above for a wide range of silanolate nucleophiles, encouraged the extension of these conditions to other substrates that have been elusive to date. Sodium (2-benzofuranyl)dimethylsilanolate Na+13-, which failed to provide cross-coupled products using previously reported conditions, was reinvestigated.38 Using Na+13- and (t-Bu3P)2Pd as the catalyst, a significant amount of benzofuran (from protiodesilylation) was observed in the coupling with aryl bromides at 90° C. However, simply lowering the temperature to 60° C provided good yields of the cross-coupling products with significantly less protiodesilylation (Table 11). Both electron-rich (entries 1-3) and sterically hindered electrophiles (entry 4) afforded the biaryl products in good to excellent yields (71-99%). Notably, the TBS protected 4-bromophenol reacted with Na+13- providing 33n in nearly quantitative yield (entry 2). Finally, N-Boc-5-bromoindole coupled in high yield thereby highlighting the mildness of these reaction conditions (entry 5).
Table 11.
Cross Coupling of Sodium (2-Benzofuranyl)dimethylsilanolate Na+13- with Aryl Bromides Using (t-Bu3P)2Pd.a
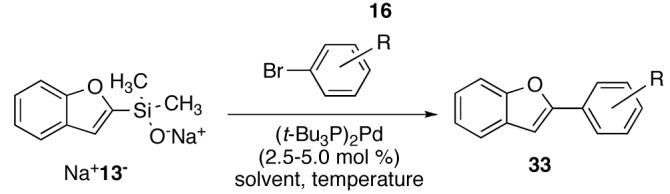 | |||||||
|---|---|---|---|---|---|---|---|
| entry | R | no. | solvent | temp., °C |
time, h |
product | yield, %b |
| 1c | 4-OCH3 | 16a | toluene | 60 | 5 | 33a | 82 |
| 2 | 4-OTBS | 16n | toluene | 60 | 5 | 33n | 99d |
| 3 | 3-CH2OTBS | 16r | toluene | 60 | 5 | 33r | 71e |
| 4 | 2-CH3 | 16c | toluene | 60 | 8 | 33c | 95 |
| 5 | f | 16t | toluene | 60 | 5 | 33t | 91d |
| 6 | 4-NO2 | 16u | THF | 60 | 8 | 33u | 58e,g |
| 7 | 4-CN | 16v | dioxane | 70 | 3.5 | 33v | 58e,g |
Reactions employed 1.5 equiv of arylsilanolate
Yields are for isolated, chromatographically homogeneous products
2.5 mol% (t-Bu3P)2Pd was used
Yields are for analytically pure products
The yield is low because of sacrificial purification from polysiloxane side products..
N-Boc-5-bromoindole
See footnote 49
Preliminary experiments with electron-poor aryl bromides and Na+13- did not provide the biaryl products although both silanolate and aryl bromide were gradually consumed. In conjunction with the results for the cross coupling of Na+14-, ethereal solvents appear to provide a beneficial effect with sodium silanolates. Therefore, simply changing the solvent to THF successfully provided 2-(4’-nitrophenyl)benzofuran 33u in moderate yield (Table 11, entry 6).49 For the cross-coupling involving 16v, a significant amount of protiodesilylation of the silanolate was observed in both THF and DME. However, performing the cross coupling in dioxane at 70 °C provided 33v in 58% yield (entry 7).50
With the success of the cross coupling in the aryl bromide series, the focus shifted towards aryl chloride substrates. Surprisingly, employing the optimized conditions for the cross coupling of Na+13- with 4-chloroanisole 16a’ yielded no desired product. Although the majority of the aryl chloride remained, a significant amount of benzofuran from protiodesilylation of Na+13- was observed. Even executing the reaction at elevated temperatures (90 °C) did not provide the desired cross-coupling product in any appreciable amount. At this stage, further optimization was warranted to identify appropriate conditions for this class of substrates.
The success of the cross coupling of alkenyldimethylsilanolates with a broad range of aryl chlorides, inspired the application of these reaction conditions.40 To our delight, employing SPhos41 and APC in THF smoothly promoted the cross coupling of a variety of aryl chlorides with Na+13-(Table 12). Both the silanolate and the aryl chloride were completely consumed within 5 h to afford good yields (64-85%) of the biaryl products (entries 1-4). Although the coupling of 3-chloropyridine did not reach completion in 6.5 h (entry 5), the desired product was still obtained in 67% yield.
Table 12.
Cross Coupling of Sodium (2-Benzofuranyl)dimethylsilanolate Na+13- with Aryl Chloridesa
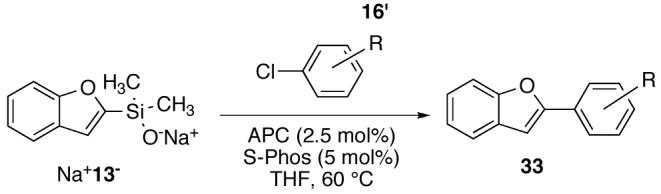 | |||||
|---|---|---|---|---|---|
| entry | R | no. | time, h |
product | yield, %b |
| 1 | 4-OCH3 | 16a’ | 3.5 | 33a | 77 |
| 2 | 2,6-(CH3)2 | 16w’ | 5 | 33w | 85 |
| 3 | 4-CF3 | 16i’ | 3 | 33i | 82 |
| 4 | 4-CN | 16v’ | 3.5 | 33v | 64d |
| 5 | c | 16m’ | 6.5 | 33m | 67 |
Reactions employed 1.5 equiv of arylsilanolate relative to aryl chloride (1.0 mmol)
Yields are for isolated, chromatographically homogeneous products.
3-Chloropyridine
See footnote 49.
4. Discussion
The results above represent a significant expansion of substrate scope for the aryl-aryl cross-coupling process. Two critical advances are responsible for these improvements: (1) the introduction and use of stable arylsilanolate salts, and (2) the discovery that (t-Bu3P)2Pd efficiently catalyzes the coupling while substantially suppressing the homocoupling, reduction and desilylation that plagued the earlier protocols. The ease of preparation of a wide variety of arylsilanolate salts and the simplicity of the coupling procedure makes these reagents viable alternatives to the more common organostannanes and -boranes used for similar transformations. The broad generality of the method has been demonstrated through an extensive survey of both nucleophilic and electrophilic partners bearing a variety of electron demand and substitution patterns.
4.1 Preparation of Arylsilanols
The methods employed for the synthesis of the arylsilanol component employed two different kinds of silicon partners, electrophiles (D3, Me2SiClH) and a nucleophile ((EtO)Me2Si)2. The preparation of arylsilanols via halogen-lithium exchange and capture with D3 proceeded uneventfully to yield the products in good to excellent yields in most cases. For substrates that gave either unstable or unreactive organolithium compounds, the use of the more electrophilic dimethylchlorosilane was needed. In general, the resulting silanes were readily oxidized to the silanols with water under catalysis by a Ru or Ir complex. However, one unexpectedly challenging case warrants further comment. Initial attempts to prepare (4-[1,3-dioxolane]phenyl)dimethylsilanol 7 by trapping the organolithium species with D3 led to low yields and dimerization to the disiloxane. Nevertheless, trapping the aryllithium with dimethylchlorosilane furnished the arylsilane in 99% yield. Unfortunately, further difficulties were encountered in the oxidative hydrolysis using either [RuCl2(cymene)]2 or [IrCl(COD)]2 in MeCN/H2O in which significant amounts of acetal cleavage (3-10%) were observed. Although the oxidative hydrolysis was successful in basic medium (0.1 M NaHCO3 solution), deprotection of the acetal was still observed. The problem was easily solved by performing the oxidative hydrolysis with tetrabutylammonium hydroxide in aqueous acetonitrile.32,51 The ability to maintain a basic, metal-free medium was crucial to afford the desired silanol in excellent yield.
4.2 Preparation of Silanolate Salts
The potassium salts of the arylsilanols were used almost exclusively in this study because they allowed for rapid combination with the palladium(II) halide intermediates to form the requisite palladium(II) silanolates without promoting undesirable base-induced reactions.52 More importantly, with only one exception, the reagents are isolable, free-flowing solids that are indefinitely stable and require no additional activation. The majority of arylsilanolates were prepared by simple deprotonation of the silanols with KH in THF, except silanolate K+3- which was prepared in benzene because of its instability in THF. Although the cause of the decomposition is unknown, the higher coordinating ability of THF most likely leads to greater charge separation and the ability to cleave the TBS group.
Two other silanolates were prepared as their sodium salts for similar reasons. (2-Benzofuryl)dimethylsilanolate (Na+13-) was prepared by deprotonation of 13 with NaH or NaHMDS in THF. Attempts to prepare the corresponding potassium silanolate led to rapid protiodesilylation to afford benzofuran. More surprising was the inability to prepare the potassium silanolate from silanol 14. Treatment of 14 with either KH or KHMDS in a number of solvents provided a dark, viscous oil which contained no identifiable products. Here again, making recourse to the sodium silanolate led to the isolation of Na+14- as a stable, colorless solid. The stability and ease handling of the arylsilanolate salts were significant because most arylsilanols are liquids and are prone to dimerization to form disiloxanes in the presence of trace amounts of acid or base. However, the silanolate salts are indefinitely stable at room temperature in a moisture free environment (similar to sodium methoxide) and their physical properties facilitate handling, transportation, and storage.
4.3. Mechanistic Insights Into the Cross Coupling of Arylsilanolates
To facilitate the discussion of the influence of reaction parameters on the optimization and outcome of the coupling process, a brief summary of the current mechanistic hypothesis is warranted. On the basis of previous52 and ongoing26 mechanistic investigations, the current formulation of the catalytic cycle is illustrated in Figure 1. This scheme has been adapted to incorporate the catalyst activation step relevant to the discussion below. The general picture for coupling of metal silanolate salts involves the displacement of the X group on arylPdLnX (i) to form a discrete arylPd(L)-O-SiMe2(aryl) (ii) species. These compounds have been implicated by kinetic studies52 as well as from independent synthesis and isolation.26 Transmetalation from ii to arylPd(L)aryl (iii) is generally believed to be turnover limiting. The reductive elimination from iii to form the biaryl product and regenerate the catalyst completes the catalytic cycle.
Figure 1.
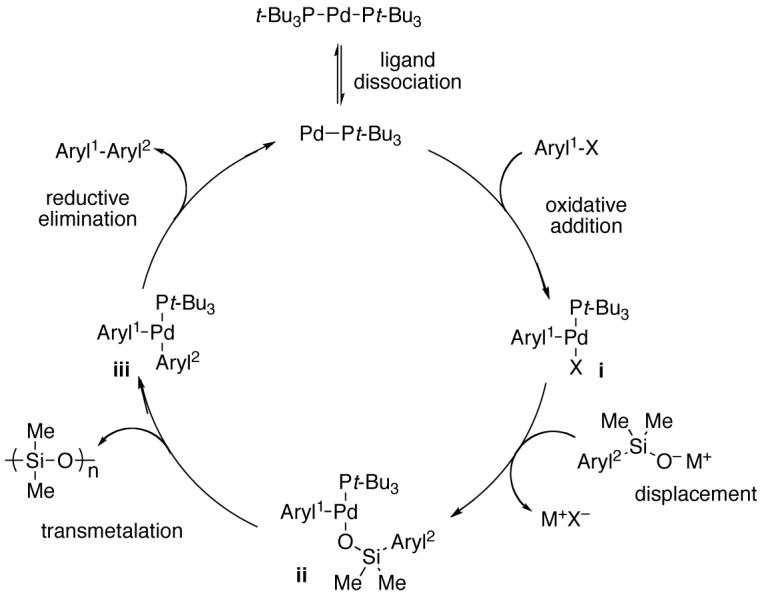
General catalytic cycle for cross coupling of silanolate salts.
Although in-depth mechanistic studies are still incomplete, the results obtained in this investigation are consistent with this picture. Among the most important observation is the effectiveness of the current catalyst/ligand combination compared with other reports involving arylsilanols. Previous conditions employed APC in combination with bidentate phosphine ligands to promote the cross-coupling reaction as well as to prevent unwanted homocoupling processes. However, we have recently discovered the crucial requirement of an open coordination site for promoting transmetalation from ii.26 Thus, the use of a bidentate phosphine effectively decreases the opportunity for transmetalation to occur and slows the overall catalytic process. Fortunately, the use of a bulky ligand such tri(tert-butyl)phosphine prevents a second phosphine from coordinating to the palladium center thus providing an open coordination site throughout the course of the reaction. Additionally, the amount of homocoupling is also negligible with the current catalyst system. The origin of this result is likely the increased steric bulk around the palladium center that decreases the opportunity for either a second oxidative insertion of the aryl halide or simply a bimolecular recombination from occurring.
Although these studies do not provide direct evidence for the turnover-limiting step for the catalytic cycle, circumstantial evidence supports the postulate that with potassium salts, transmetalation is turnover limiting. Experimental evidence for this notion comes from lower temperature required for the coupling of sodium (2-benzofuranyl)dimethylsilanolate (Na+13-) compared to other sodium arylsilanolates (60 °C compared to 90 °C). Whereas productive cross coupling for the arylsilanolates is observed at lower temperatures, these reactions quickly stall because of the slower rate of transmetalation. This difference can be attributed to the key transmetalation event that forms the Pd-sp2 carbon bond prior to reductive elimination. Previous mechanistic studies performed in these laboratories have demonstrated that displacement of an arylPd(X)Ln with silanolate salts to form ii (without ligands) occurs readily at lower temperatures.26b,52 Even though the exact molecular events in transmetalation are currently unknown, the aromaticity of the arene ring must be disrupted when palladium atom bonds to the ipso carbon of the silanolate. Thus, fused heteroarylsilanolates, particularly the π-excessive types, are expected to undergo more rapid transmetalation because of the lesser energetic cost in interrupting aromaticity53 and greater electron density at the ipso carbon.
4.4. Optimization of Reaction Conditions
The reaction conditions developed herein have allowed the successful cross coupling of a broad range of arylsilanolate reagents containing various functional groups. Previous reports from these laboratories described the use of arylsilanols in combination with external activators such as fluoride or Brønsted bases.22,25 Unfortunately, the substrate scope using these protocols was limited to electron-rich arylsilanols. The introduction of preformed arylsilanolate salts described herein represents significant operational and preparative advantages. The arylsilanolate nucleophile can be charged directly into the reaction mixture with an aryl electrophile in the presence of a palladium catalyst.
Performing optimization studies with K+9- proved critical to the development of a general set of conditions for the cross coupling of aryl silanolates. The diminished reactivity of this electron-deficient silanolate should affect two key steps in the catalytic cycle. The rate of both the displacement of the palladium halide and the transmetalation of the palladium silanolate would be affected by the lower electron density of this benzene ring.
The identification of (t-Bu3P)2Pd as the catalyst was crucial to the success of this program. “Fu’s catalyst” allowed for rapid oxidative addition with aryl bromides, and rapid transmetalation thus avoiding unwanted side processes. The use of a palladium(0) source avoided the necessity for the reduction of a palladium(II) precatalyst (e.g. APC) and proved advantageous. Recent studies from these laboratories have demonstrated that the reduction of APC requires a stoichiometric amount of the arylsilanolate (with respect to palladium(II)) to produce palladium(0).39 Alternatively, phosphines can serve as the reducing agent for the palladium(II) precatalyst by formation of the corresponding phosphine oxides.54 This is significantly more wasteful, especially for expensive and sensitive phosphines that would thus have to be used in superstoichiometric amounts (with respect to palladium(II)). Moreover, the phosphine oxides are much less Lewis basic than the strongly electron-rich phosphines and are ineffective at maintaining catalyst life and promoting facile oxidative additions to aryl halides.
4.5. Scope of Arylsilanolate Structure
The most significant advance described in this work is the expansion of scope of the arylsilanolate beyond the 4-anisylsilanolate, K+1-, reported previously.25 For the silicon-based, cross-coupling reaction to be competitive with other methods for the synthesis of biaryls, a much wider range of both coupling partners was needed. Fortunately, the combination of preformed silanolate salts and (t-Bu3P)2Pd enabled that expansion of scope. The discussion below follows the presentation of the different classes of substrates in the Results section.
4.5.1. Electron-rich Arylsilanolates
Although the cross coupling of K+1- was already well documented, the limited scope of these studies prompted a full investigation of the functional group compatibility under the newly developed conditions.25 As expected, K+1- reacted smoothly with a number of aryl bromides in good yields (78-88%). In addition, K+4- and K+5- in which the position of the electron-donating methoxy group was moved to the meta and ortho positions also underwent cross coupling in comparable yields. The reaction times with K+5- were slightly longer than the other two substrates and the coupling with the sterically demanding 2-bromotoluene was successful albeit in slightly diminished yield.
The ability to employ other electron-rich silanolates containing sensitive functional groups illustrates the mildness and generality of the method. For example, the successful couplings of the presence of the TBS protected phenol in K+3-, and the dimethylamino group in K+8- further demonstrated the mildness of this protocol. Obviously, the use of fluoride sources to promote the coupling precludes the use of silicon protective groups. The coupling of the stilbene-derived silanolate K+6- illustrated that an alkene is also compatible. The double bond did not adversely affect the desired cross coupling and the products were obtained with similar yields compared to other electron-rich arylsilanolates. Additionally, no products resulting from a Heck-type process were observed.
4.5.2. Electron-deficient Arylsilanolates
The cross coupling of electron-deficient arylsilanols represented one of the greatest challenges to this enterprise. Only one example of this class of arylsilanols had been reported previously (9) and the results were far from optimal (low yields with CsOH as the activator at 110 °C in toluene). The forcing conditions limited the scope of aryl electrophiles that could participate. Presumably, the problem is associated with the poor equilibrium concentration of active silanolate because of the ready and favorable formation of the disiloxane. In addition, the electron-deficient nature of the silanolate leads to slower displacement and transmetalation (see Section 4.3). However, these problems were effectively overcome by the use of the preformed potassium silanolate K+9-. Silanolate K+9- was resistant to disiloxane formation and underwent efficient cross coupling with a wide range of electrophiles in moderate to good yields. Similarly, 4-fluorophenylsilanolate K+11- afforded biaryls in very good yields from aryl bromides representing a wide range of electronic and steric character. In addition, 4-chlorophenylsilanolate K+10- also underwent cross coupling with range of aryl bromides albeit in lower yield compared to K+11-. The reason for the attenuated yields is not clear, but no products arising form cross coupling at the chlorine-bearing carbon were detected.
Arylsilanolate K+14- is not only electron deficient, but it also contains the reactive carboalkoxy functional group. The ester survived the reaction conditions to provide biaryls in good yields. The decreased nucleophilicity of this arene was reflected in the increased reaction times (6-7 h) required to reach completion and lack of cleavage of the tert-butyl ester compared to other silanolates.55 In addition, the decreased nucleophilicity of K+14- allowed for the successful formation of 28n by combination with 16n. Attempts to prepare 28n from the alternative pair wise combination of K+3- with 16l led to increased amounts of phenol-based products resulting from deprotection of 16l.
Although silanolate K+7- containing a dioxolane protective group is not rigorously an electron deficient arene, it nonetheless could be prepared easily and underwent smooth cross coupling with a number of aryl bromides in excellent yields. The parent aldehyde was not compatible with these reaction conditions.
Silanolate Na+14- also proved to be an extremely interesting example both preparatively and mechanistically. Attempts to directly translate the optimized conditions to this substrate were ineffective. Although the product was detected, the reaction quickly stalled with the formation of palladium black. Moreover, significant amounts of protiodesilylation were observed by GC/MS leading to an overall decrease in the concentration of active silanolate. These two observations lead to the hypothesis that the sodium silanolate along with the steric environment were dramatically slowing down the displacement step. To improve the nucleophilicity, ethereal solvents were surveyed and were found to improve the lifetime of the sodium salt with only trace amount of protiodesilylation observed. Presumably, by increasing the charge separation of the two ions, the nucleophilicity of Na+14- was improved and allowed for the reaction to proceed as the concentration of the silanolate decreased. In addition, the more rapid displacement prevented protiodesilylation from occurring.
4.5.3. Heterocyclic Silanolates
The importance of biaryls containing one or more heterocyclic nucleus in medicinal chemistry and material science cannot be overstated. As part of this ongoing program we have already reported the ability to synthesize and successfully couple π-excessive heterocyclic silanolates without significant protiodesilylation.38 Two heterocyclic silanolates K+12- and Na+13- were investigated here.56 The cross coupling of K+12- proceeded smoothly with a variety of aryl bromides in excellent yields without any modification of the standard conditions. This result is not surprising because the dibenzothiophene has no serious liabilities.
On the other hand, the cross coupling of 2-benzofurylsilanolate Na+13- failed under standard conditions and a significant amount of protiodesilylation was detected. As was also the case in the cross coupling of 2-indolylsilanolate, carrying out the reactions at lower temperatures led to the smooth cross coupling of Na+13- while maintaining the stability of the silanolate throughout the course of the reaction.38 Attempts to cross couple Na+13- with electron-deficient aryl bromides failed using (t-Bu3P)2Pd in toluene, but simply replacing toluene with ethereal solvents successfully promoted the desired cross-coupling. The use of ethereal solvents must serve to improve the nucleophilicity of sodium silanolate by increasing the charge separation. Presumably, the consequence of this transformation improves the rate of displacement of the palladium halide, thereby creating a more efficient catalytic cycle.
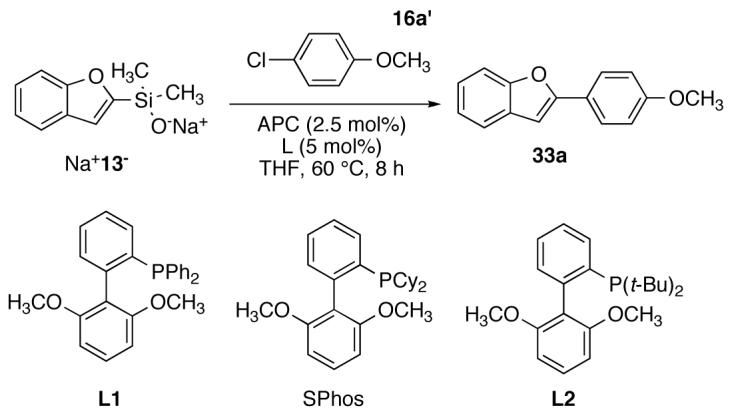
This hypothesis became more apparent in attempts to cross couple Na+13- with aryl chlorides. Under conditions that were successful with aryl bromides, both 16a’ and 16c’ failed to provide the desired products. However, conditions recently developed for the cross coupling of aryl chlorides with alkenylsilanolates (APC, SPhos, THF, 60 °C) were successful.40 Good yields of the biaryl products were achieved in short reaction times. These results were surprising because the mechanism of cross-coupling reaction for bromides and chlorides is believed to be similar. Both bromides and chlorides are known to undergo the oxidative addition by oxidative addition with (t-Bu3P)2Pd57. Additionally, once the displacement has occurred, the pathways converge for the different halide families and transmetalation followed by a facile reductive elimination must proceed for both. This analysis leaves the displacement as the only problematic step in the catalytic cycle for the aryl chlorides. Although no direct evidence has been secured for the differences under these conditions, these results can be explained by a combination of factors that influence the rate of displacement on monoligated, T-shaped arylpalladium halide complexes. First, the displacement of a bromide should be more rapid compared to displacement of a chloride on the basis of the affinities of the halides for Pd(II).58 Second, the sodium silanolate is a poorer nucleophile compared to the potassium silanolates that coupled successfully with chlorides using (t-Bu3P)2Pd.25b,40 Finally, that the lesser steric bulk around the phosphorus atom in SPhos compared to (t-Bu3P)2Pd also contributes to the more facile attack of the silanolate on the palladium center. To understand the effect of these ligands, various derivatives of SPhos were employed in the cross-coupling reaction of 4-chloroanisole in THF at 60°C (Scheme 1). The use of biphenyl ligand L1 provided no trace of biaryl product although the silanolate was unproductively consumed to benzofuran. Interestingly, the more bulky tert-butyl substituted ligand L2 promoted the cross coupling, albeit more slowly compared to SPhos which bears two cyclohexyl substituents. The small change in the steric environment around the palladium center is critical to the success of this reaction but a concrete conclusion cannot be provided at this time.59
Scheme 1.
4.5.4. Nucleophilicity and Basicity of Aryl Silanolates
Previous studies in these laboratories demonstrated that the counter ion of an in situ generated silanolate has a dramatic effect on the rate of reaction.25b Activators such as Na2CO3 and K2CO3 did not promote the cross-coupling of 1 with ethyl 4-iodobenzoate. In contrast, 8% and 45% conversion of the iodide was observed using Rb2CO3 and Cs2CO3 in 24 h, respectively Furthermore, the dependence of the alkali metal was also seen in the alkenyl-aryl cross-coupling reactions in which the potassium salt of (E)-heptenylsilanolate reacted significantly faster than did the sodium salt.40 Thus, the expected correlation between the size of the alkali metal and the nucleophilicity of the silanolate is seen.60 Larger alkali metals typically form more ionic metal-oxygen bonds and consequently, these alkali metal silanolates are more nucleophilic than the silanolates of smaller alkali metals.
With this observation in mind, Na+3- and Na+10- were prepared and the reactivity of these nucleophiles was evaluated in (t-Bu3P)2Pd-catalyzed cross-coupling reactions. Electrophiles bearing either carboxylic esters or silicon protecting groups were surveyed. These substrates were chosen because lower yields of the biaryl products 24a, 24c, 30n, 30r and 30l were obtained using the corresponding potassium salts K+3- and K+10-. It was hypothesized that decreasing the nucleophilicity of the aryldimethylsilanolate could potentially minimize the undesired side reactions such as cleavage of the TBS protected phenol and tert-butyl ester. Additionally, the formation of the phenol from 30l via SNAr pathyway may be avoided. The cross-coupling of Na+3- and Na+10- with 16a, and 16n provided the desired biaryls 24a and 30n by GC analysis. However, prolonged heating was required (>29 h and >19 h) compared to the reactions of the corresponding potassium silanolates (3.5 h and 3.5 h). The difference in reaction time implies that displacement becomes competetive as the turnover limiting step in the catalytic cycle. Significant amounts of disiloxane and unconsumed bromo ester 16l were observed in the reaction with Na+10-. From these findings, the use of the sodium arylsilanolates (Na+3- and Na+10-) did not offer any preparative advantage over the potassium analogues.
4.6. Scope of Electrophile Structure
4.6.1. Electron-rich Electrophiles
A wide range of substituted, electron-rich aryl bromides were surveyed in the cross coupling with arylsilanolates. The position of the functional group on the electrophile had little influence on the overall process. For example, the cross-coupling of K+1- with 4- and 2-bromotoluene afforded the biaryl compounds 20b and 20c in the same yield and reaction time. This result demonstrates the insensitivity the catalyst feels due to steric effects. However, this conclusion must be tempered by the results from the reaction of K+9- with 2- and 4-bromoanisole, which afforded the biaryl products 17a and 17j in 92% and 78% yield respectively. From these results it is clear that the heteroatom plays a role in the overall effectiveness of this process. Coordination of the Lewis basic aryl oxygen to an open coordination site at the palladium center lowers the reaction rate. Subsequently, partial inhibition leads to lower yields as compared with the methyl substituted systems. However, unlike other systems reported previously, the coordination retards, but does not completely inhibit the reaction.27
4.6.2 Electron-deficient Electrophiles
Generally, the cross coupling of arylsilanolates with electron-poor electrophiles afforded good to excellent yields of the biaryl products. Reactions of electron-rich silanolates K+2- and K+8- with electron-poor bromides yielded the biaryl products in good yields (71-88%). Similarly, cross coupling of electron-poor silanolates K+9- and K+11- with tert-butyl 4-bromobenzoate gave the biaryl products in almost identical yields (68% and 70%) albeit with slightly higher catalyst loading for the less nucleophilic silanolate K+9-. The mildness of this protocol is illustrated by the competency of esters, ketones, and acetals to be present without any appreciable reaction occurring at these sensitive functional groups.
4.6.3 Heterocyclic Bromides
Previously, the cross coupling of arylsilanols with heteroaryl bromides was only moderately successful. However, the reaction conditions described herein successfully promote the cross coupling without any decrease in reaction rate or yield. Reactions of 3-bromoquinoline, 3-bromopyridine, and 3-bromobenzothiophene with a variety of arylsilanolates afforded biaryl compounds in good yields (61-85%) with no significant differences in rate. The success of these reactions is attributed to facile catalyst turnover without interference of the heteroatom binding to the palladium center that would result in stalling.
4.6.4. Aryl Chlorides
The cross coupling of aryl chlorides is more challenging because oxidative addition of C-Cl bonds is more difficult compared to C-Br bonds. Furthermore, the halide displacement from Aryl-Pd(X)Ln is further attenuated by the greater affinity of chloride for palladium(II).58 This obstacle has been overcome and arylsilanolates smoothly cross couple with aryl chlorides, as illustrated by the successful reaction of K+1- with 2- and 4-chlorotoluene to afford 20b and 20c. Similarly, reaction of electron-deficient silanolate K+9- with various aryl chlorides (e.g. tert-butyldimethylsilyl (TBS) protected 4-chlorophenol, tert-butyl 4-chlorobenzoate, and 4-chlorobenzophenone) afforded the respective biaryl adducts in moderate yields. The cross coupling of 4-chlorobenzophenone is a further illustration of the mildness of the reaction as the ketone function survived the reaction conditions.
4.7. Other Cross-Coupling Reactions Using Arylsilanolates
During the course of these studies, a number of pair wise combinations were found to reach high conversions, but the products could not be obtained in high yields because of difficulties in isolation. Namely, those substrates that lacked polar functional groups were not easily separated from the polysiloxane byproducts. For example, K+2- smoothly coupled with 3-bromopyridine, reaching 100% conversion within 5 h. However, the low polarity of 25l complicated the removal of the polysiloxanes and a clean product could not be obtained. Another disappointing coupling resulted from the combination of Na+14- with 2-bromotoluene. The desired product was identified by GC/MS analysis but the isolated yield was lower than expected (<50%).
A second class of substrates that were difficult to isolate derived from those couplings involving K+6-. Cross coupling of K+6- with both 16a and 16i reached 100% conversion as determined by GC analysis. However, the products could not be separated easily from byproducts that contained the stilbene moiety. Even recrystallization was did not provide pure products as compounds containing the stilbene unit would co-crystallize. Presumably, the purification of the product could be simplified using less K+6-.
Conclusion
A broadly applicable protocol for the cross coupling of alkali-metal, aryl- and heteroarylsilanolates with aromatic bromides and chlorides has been developed. Under catalysis by (t-Bu3P)2Pd, a wide range of electron-rich, electron-poor, and sterically hindered aryldimethylsilanolates underwent smooth coupling with a wide range of aryl halides. The advantages of using the preformed silanolate salts include their ease of synthesis from inexpensive precursors, stability to storage, resistance to disiloxane formation, and self-activating properties. The broad substrate scope, functional group compatibility, and anhydrous conditions for cross coupling bode well for the adoption of this method particularly in cases where the traditional boron or tin-based reagents are problematic. The development and application of silanolates derived from both π-deficient and π-excessive heteroaromatic compounds are currently under investigation.
Experimental Section
General Experimental
See Supporting Information
Preparation of 3-(4-N,N-Dimethylaminophenyl)quinoline (23e) from 3-Bromoquinoline Using K+8-
To an oven-dried, 5-mL, round-bottomed flask equipped with a magnetic stir-bar, reflux condenser, and three-way argon adapter was charged (t-Bu3P)2Pd (12.8 mg, 0.025 mmol, 2.5 mol %) as a colorless solid in a drybox. The flask was sealed away from the atmosphere and removed to a hood. Dry toluene (2 mL) was added via syringe through the three-way adapter resulting in a colorless solution upon stirring. 3-Bromoquinoline (136 μL, 1.0 mmol) and K+8- (350 mg, 1.5 mmol, 1.5 equiv) were added sequentially by removal of the adapter, adding the reagents as liquid and solid respectively and replacing the adapter as quickly as possible. The flask was placed into a preheated 90 °C oil bath and stirred at 90 °C under a static pressure of argon. After 3 h, the reaction was cooled to room temperature and poured onto water (10 mL). The aq. layer was extracted with ethyl acetate (3 x 20 mL) and the organics washed with brine (15 mL) and dried (MgSO4, 5 g). The volatiles were removed in vacuo (30 °C, 10 mmHg) to afford a viscous, brown oil. Purification by MPLC (24 g SiO2, 30 mL/min, hexane/ethyl acetate 9:1 (3 column volumes), gradient to hexane/ethyl acetate 3:2 (5 column volumes), hexane/ethyl acetate 3:2 (6 column volumes)) followed by sublimation (80 °C, 0.1 mmHg) afforded 198 mg (80%) of 23e as analytically pure yellow plates. Data for 23e: mp: 127-129 °C; 1H NMR: (500 MHz, CDCl3) 9.19 (d, 1 H, J = 2.2 Hz, HC(2)), 8.22 (d, 1 H, J = 2.2 Hz, HC(7)), 8.11 (d, 1 H, J = 8.4 Hz, HC(3)), 7.84 (d, 1 H, J = 8.4 Hz, HC(6)), 7.67 (ddd, 1 H, J1= 1.4 Hz, J2= 7.0 Hz, J3= 8.3 Hz, HC(4)), 7.64 (d, 2 H, J = 8.9 Hz, HC(2′)), 7.54 (ddd, 1 H, J1= 1.4 Hz, J2= 7.0 Hz, J3= 8.3 Hz, HC(5)), 6.86 (d, 2 H, J = 8.9 Hz, HC(3′)), 3.03 (s, 6 H, H3C(5′)); 13C NMR: (126 MHz, CDCl3) 150.4 (C(1′)), 149.9 (C(2)), 146.7 (C(8)), 133.8 (C(4′)), 131.3 (C(7)), 129.1 (C(3)), 128.6 (C(4)), 128.3 (C(1)), 128.0 (C(2′)), 127.7 (C(6)), 126.7 (C(5)), 125.4 (C(9)), 112.8 (C(3′)), 40.5 (C(5′)), 40.4 (C(5′)); IR: (KBr) 2952 (w), 2849 (w), 2798 (w), 1602 (s), 1525 (s), 1355 (m), 1292 (w), 1212 (m), 1061 (w), 950 (w), 813 (s); MS: (EI, 70eV) 248 (M+ , 100), 232 (12), 218 (1), 204 (12), 176 (4), 151 (2), 124 (10), 102 (6), 88 (4); HRMS: Calcd. for C17H16N2 (248.1314); Found: 248.1312; TLC: Rf 0.26 (hexanes/ethyl acetate, 3:2); Anal. Calcd. for C18H20O3: C, 82.22; H, 6.49; N, 11.28, Found: C, 81.88; H, 6.46; N, 11.23.
Preparation of tert-Butyl 4′-(Trifluoromethyl)biphenyl-4-carboxylate (17l) from tert-butyl 4-Chlorocarboyxlate Using K+9-
To an oven-dried, 5-mL, round-bottomed flask equipped with a magnetic stir-bar, reflux condenser, and three-way argon adapter was charged (t-Bu3P)2Pd (12.8 mg, 0.025 mmol, 2.5 mol %) as a colorless solid in a drybox. The flask was sealed away from the atmosphere and removed to a hood. Dry toluene (2 mL) was added via syringe through the three-way adapter resulting in a colorless solution upon stirring. tert-Butyl 4-chlorocarboxylate (213 mg, 1.0 mmol) and K+9- (388 mg, 1.5 mmol, 1.5 equiv) were added sequentially by removal of the adapter, adding the reagents as liquid and solid respectively and replacing the adapter as quickly as possible. The flask was placed into a preheated 90 °C oil bath and stirred at 90 °C under a static pressure of argon. After 5 h, the reaction was cooled to room temperature and poured onto water (10 mL). The aq. layer was extracted with ethyl acetate (3 x 20 mL) and the organics washed with brine (15 mL) and dried (MgSO4, 5 g). The volatiles were removed in vacuo (30 °C, 10 mmHg) to afford a viscous, brown oil. Purification by MPLC (12 g SiO2, 30 mL/min, hexanes (3 column volumes), ramp to hexane/ethyl acetate 3:1 (20 column volumes), hexane/ethyl acetate 3:1 (5 column volumes)) followed by recrystallization (EtOH) afforded 200 mg (62%) of 17l as analytically pure, colorless plates. Data for 17l: mp: 116-117 °C; 1H NMR: (500 MHz, CDCl3) 8.09 (d, 2 H, J = 8.5 Hz, HC(3)), 7.72 (app. S, 4 H, HC(2′) and HC(3′)), 7.64 (d, 2 H, J = 8.5 Hz, HC(2)), 1.63 (s, 9 H, H3C(2′′)); 13C NMR: (126 MHz, CDCl3) 165.4 C(5), 143.7 C(1), 143.5 C(1′), 131.7 C(4), 130.1 C(2′), 130.0 (q, J = 32 Hz, C(4′)), 127.6 C(2), 127.1 C(3), 125.8 (q, J = 3.9 Hz, C(3′)), 124.1 (q, J = 271 Hz, C(5′)), 81.3 C(1′′), 28.2 C(2’’); 19F NMR: (470 MHz, CDCl3) -62.9; IR: (KBr) 2983 (w), 2935 (w), 1711 (s), 1613 (w), 1395 (m), 1375 (w), 1330 (m), 1300 (m), 1255 (w), 1171 (m), 1123 (m), 1075 (m), 1001 (w), 834 (s), 777 (s), 736 (m), 702 (w); MS: (EI, 70eV) 322 (M+, 10), 303 (4), 266 (100), 249 (44), 221 (4), 201 (17), 183 (8), 152 (20), 57 (24); TLC: Rf 0.53 (hexanes/ethyl acetate, 9:1). Anal. Calcd for C18H17F3O2: C, 67.07; H, 5.32. Found: C, 67.08; H, 5.54.
Preparation of 2-(4-Methoxyphenyl)benzofuran (33a) from 4-Chloroanisole Using Na+13-
To an oven-dried, 5-mL, round-bottomed flask containing a magnetic stir bar equipped with a reflux condenser and a 3-way adaptor fitted with a septum, APC (9.1 mg, 0.025, mmol, 2.5 mol %) and 2-dicyclohexylphosphino-2′,6′-dimethoxybiphenyl, SPhos (20.5 mg, 0.05 mmol, 5 mol %) were combined as solids. The flask was sequentially evacuated and purged with argon twice. THF (1.0 mL) was added via a syringe resulting in a faint yellow solution upon stirring. 4-Chloroanisole (125 μL, 1.0 mmol) was added followed by Na+13- (322 mg, 1.5 mmol, 1.5 equiv). The reaction vessel was placed into a preheated 60 °C oil bath and the mixture was stirred at 60 °C under a static pressure of argon. After 3.5 h, the mixture was cooled to rt, filtered through SiO2 (11 g, 20 mm diameter) and the column was eluted with Et2O (100 mL). The filtrate was concentrated and dissolved in a minimal amount of CH2Cl2 to give a brown solution. Filtration through a plug of SiO2 (20 mm × 20 mm) eluting with CH2Cl2 (50 mL) gave a yellow solution. Purification by recrystallization (EtOH) afforded 177 mg (77%) of 33a as colorless flakes. The spectroscopic data matched those from the literature and was free of major impurities.61 Data for 33a: mp 148-149 °C (EtOH); 1H NMR: (500 MHz, CDCl3) 7.81 (d, 2 H, J = 9.0 Hz), 7.57 (dd, 1 H, J1 = 7.5 Hz, J2 = 1.0 Hz), 7.52 (dd, 1 H, J1 = 7.5 Hz, J2 = 1.0 Hz), 7.27 (ddd, 1 H, J1 = J2 = 7.5 Hz, J3 = 1.5 Hz), 7.23 (ddd, 1 H, J1 = J2 = 7.5 Hz, J3 = 1.5 Hz), 6.99 (d, 2 H, J = 9.0 Hz), 6.90 (d, 1 H, J = 1.0 Hz), 3.87 (s, 3 H); 13C NMR: (126 MHz, CDCl3) 159.5, 156.0, 154.7, 129.5, 126.4, 123.7, 123.3, 122.8, 120.5, 114.2, 111.0, 99.6, 55.4; MS (EI, 70eV) 224 (M+, 100), 209 (61), 181 (32), 152 (15), 127 (3), 112 (7), 76 (3), 63 (4); Rf 0.22 (hexanes/CH2Cl2, 8:1) [silica gel, UV, CAM].
Acknowledgement
We are grateful to the National Institutes of Health for generous financial support (GM63167) and Johnson-Matthey for a gift of Pd2(dba)3. R.C.S thanks Amgen for a graduate fellowship as well as Sigma-Aldrich for an innovation award. W.T.C acknowledges the University of Illinois for a graduate fellowship. J.M.M. acknowledges the NIH for a postdoctoral fellowship.
Footnotes
Supporting Information Available: Detailed experimental procedures, optimization studies, full characterization of all products and complete reference 48. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- (1).Rao A. V. Rama, Gurjar MK, Reddy KL. Chem. Rev. 1995;95:2135–2167. [Google Scholar]
- (2).Corbet J-P, Mignani G. Chem. Rev. 2006;106:2651–2710. doi: 10.1021/cr0505268. [DOI] [PubMed] [Google Scholar]
- (3) (a).Roncali J. Chem. Rev. 1992;92:711–738. [Google Scholar]; (b) Müllen K, Wegner G, editors. Electronic Materials: The Oligomer Approach. Wiley-VCH; Weinheim, Germany: 1998. [Google Scholar]
- (4).Berthod M, Mignani G, Woodward G, Lemaire M. Chem. Rev. 2005;105:1801–1836. doi: 10.1021/cr040652w. [DOI] [PubMed] [Google Scholar]; (b) Seyden-Penne J. Chiral Auxiliaries and Ligands in Asymmetric Synthesis. Wiley; New York: 1995. [Google Scholar]
- (5) (a).Sainsbury M. Tetrahedron. 1980;36:3327–3359. [Google Scholar]; (b) Stanforth SP. Tetrahedron. 1998;54:263–303. [Google Scholar]; (c) Hassan J, Sévignon M, Gozzi C, Schulz E, Lemaire M. Chem. Rev. 2002;102:1359–1469. doi: 10.1021/cr000664r. [DOI] [PubMed] [Google Scholar]
- (6).Ullmann F, Bielecki J. Chem. Ber. 1901;34:2174–2185. [Google Scholar]
- (7).Nelson TD. Org. React. 2004;63:265–555. [Google Scholar]
- (8) (a).de Meijere A, Diederich F, editors. Metal-Catalyzed Cross-Coupling Reactions. 2nd 1 and 2. Wiley-VCH; Weinheim, Germany: 2004. [Google Scholar]; (b) Negishi E.-i., editor. Handbook of Organopalladium Chemistry for Organic Synthesis. Wiley-Interscience; New York: 2002. [Google Scholar]
- (9).Dugger RW, Ragan JA, Ripin D. H. Brown. Org. Process Res. Dev. 2005;9:253–258. [Google Scholar]
- (10).Tucker CE, Majid TN, Knochel P. J. Am. Chem. Soc. 1992;114:3983–3985. [Google Scholar]
- (11) (a).Krigman MR, Silverman AP. Neurotoxicology. 1984;5:129–139. [PubMed] [Google Scholar]; (b) Cragg ST. In: Patty’s Toxicology. Bingham E, Cohrssen B, Powell CH, editors. Wiley; Hoboken: 2001. DOI: 10.1002/0471435139.tox.093. [Google Scholar]
- (12)(a).Bedford RB, Cazin CSJ, Holder D. Coord. Chem. Rev. 2004;248:2283–2321. [Google Scholar]; (b) Littke AF, Fu GC. Angew, Chem. Int. Ed. 2002;41:4176–4211. doi: 10.1002/1521-3773(20021115)41:22<4176::AID-ANIE4176>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]; (c) Nolan SP, Navarro O. Chapter 11.01. Comprehensive Organometallic Chemistry III. 2007:1–37. [Google Scholar]
- (13).Miyaura N. Chapter 2. In: de Meijere A, Diederich F, editors. Metal-Catalyzed Cross-Coupling Reactions. 2nd Wiley-VCH; Weinheim: 2004. [Google Scholar]
- (14).Molander GA, Ellis N. Acc. Chem. Res. 2007;40:275–286. doi: 10.1021/ar050199q. [DOI] [PubMed] [Google Scholar]
- (15) (a).Hiyama T. Chapter 10. In: Diederich F, Stang PJ, editors. Metal-Catalyzed Cross-Coupling Reactions. Wiley-VCH; Weinheim: 1998. [Google Scholar]; (b) Hiyama T. J. Organomet. Chem. 2002;653:58–61. [Google Scholar]; (c) Denmark SE, Sweis RF. Chem. Pharm. Bull. 2002;50:1531–1541. doi: 10.1248/cpb.50.1531. [DOI] [PubMed] [Google Scholar]; (d) Denmark SE, Sweis RF. Acc. Chem. Res. 2002;35:835–846. doi: 10.1021/ar020001r. [DOI] [PubMed] [Google Scholar]; (e) Denmark SE, Ober MH. Aldrichimica Acta. 2003;36:75–85. [Google Scholar]; (f) Denmark SE, Sweis RF. Chapter 4. In: de Meijere A, Diederich F, editors. Metal Catalyzed Cross-Coupling Reactions. 2nd Wiley-VCH; Weinheim: 2004. [Google Scholar]; (g) Handy CJ, Manoso AS, McElroy WT, Seganish WM, DeShong P. Tetrahedron. 2005;61:12201–12225. [Google Scholar]; (h) Denmark SE, Regens CS. Acc. Chem. Res. 2008;41:1486–1499. doi: 10.1021/ar800037p. [DOI] [PMC free article] [PubMed] [Google Scholar]; (i) Denmark SE, Butler CR. Chem. Commun. 2009:20–33. doi: 10.1039/b809676g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16) (a).Hatanaka Y, Fukushima S, Hiyama T. Chem. Lett. 1989;18:1711–1714. [Google Scholar]; (b) Gouda K-I, Hagiwara E, Hatanaka Y, Hiyama T. J. Org. Chem. 1996;61:7232–7233. doi: 10.1021/jo9611172. [DOI] [PubMed] [Google Scholar]
- (17).Mowery ME, DeShong P. J. Org. Chem. 1999;64:1684–1688. doi: 10.1021/jo982463h. [DOI] [PubMed] [Google Scholar]
- (18).Mowery ME, DeShong P. Org. Lett. 1999;1:2137–2140. doi: 10.1021/ol991186d. [DOI] [PubMed] [Google Scholar]
- (19)(a).Nakao Y, Oda T, Sahoo AK, Hiyama T. J. Organomet. Chem. 2003;687:570–573. [Google Scholar]; (b) Sahoo AK, Nakao Y, Hiyama T. Chem. Lett. 2004;33:632–633. [Google Scholar]; (c) Sahoo AK, Oda T, Nakao Y, Hiyama T. Adv. Synth. Catal. 2004;346:1715–1727. [Google Scholar]
- (20).Nakao Y, Imanaka H, Sahoo AK, Yada A, Hiyama T. J. Am. Chem. Soc. 2005;127:6952–6953. doi: 10.1021/ja051281j. [DOI] [PubMed] [Google Scholar]
- (21).Nakao Y, Chen J, Tanaka M, Hiyama T. J. Am. Chem. Soc. 2007;129:11694–11695. doi: 10.1021/ja074728s. [DOI] [PubMed] [Google Scholar]
- (22).Denmark SE, Wu Z. Org. Lett. 1999;1:1495–1498. [Google Scholar]
- (23).Denmark SE, Wehrli D, Choi JY. Org. Lett. 2000;2:2491–2494. doi: 10.1021/ol006170y. [DOI] [PubMed] [Google Scholar]
- (24).Hirabayashi K, Mori A, Kawashima J, Suguro M, Nishihara Y, Hiyama T. J. Org. Chem. 2000;65:5342–5349. doi: 10.1021/jo000679p. [DOI] [PubMed] [Google Scholar]
- (25) (a).Denmark SE, Ober MH. Org. Lett. 2003;5:1357–1360. doi: 10.1021/ol034328j. [DOI] [PubMed] [Google Scholar]; (b) Denmark SE, Ober MH. Adv. Synth. Cat. 2004;346:1703–1714. [Google Scholar]
- (26) (a).Tymonko SA. Ph.D. Thesis. University of Illinois at Urbana-Champaign; 2007. Denmark SE, Tymonko SE, Smith RC, Ober MH. unpublished results from these laboratories.
- (27).Denmark SE, Smith RC, Tymonko SA. Tetrahedron. 2007;63:5730–5738. doi: 10.1016/j.tet.2007.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Lickiss PD. Adv. Inorg. Chem. 1995;42:147–262. [Google Scholar]
- (29).Frye CLS,RM, Fearon FWG, Klosowski JM, DeYoung T. J. Org. Chem. 1970;35(5):1308–1314. [Google Scholar]
- (30) (a).Lee M, Ko S, Chang S. J. Am. Chem. Soc. 2000:12011–12012. [Google Scholar]; (b) Lee Y, Seomoon D, Kim S, Han H, Chang S, Lee PH. J. Org. Chem. 2004;69(5):1741–1743. doi: 10.1021/jo035647r. [DOI] [PubMed] [Google Scholar]
- (31).Denmark SE, Kallemeyn JM. Org. Lett. 2003;5:3483–3486. doi: 10.1021/ol035288m. [DOI] [PubMed] [Google Scholar]
- (32).Denmark SE, Neuville L. Org. Lett. 2000;2:3221–3224. doi: 10.1021/ol0064112. [DOI] [PubMed] [Google Scholar]
- (33).Wolfe JP, Singer RA, Yang BH, Buchwald SL. J. Am. Chem. Soc. 1999;121:9550–9561. [Google Scholar]
- (34) (a).Denmark SE, Baird JD. Chem. Eur. J. 2006;12:4954–4963. doi: 10.1002/chem.200600034. [DOI] [PubMed] [Google Scholar]; (b) Denmark SE, Baird JD. Org. Lett. 2006;8:793–795. doi: 10.1021/ol053165r. [DOI] [PubMed] [Google Scholar]
- (35).Denmark SE, Butler CR. J. Am. Chem. Soc. 2008;130:3690–3704. doi: 10.1021/ja7100888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).The cesium and rubidium salts could be prepared also and are competent coupling partners. However the difficulty in handling these metals renders these reagents less attractive. For a discussion of the working mechanistic hypothesis see ref. 53.
- (37).>All aryldimethylsilanolates were isolated as stable powders except for potassium 3-(methoxyphenyl)dimethylsilanolate which was a viscous oil.
- (38).Denmark SE, Baird JD, Regens CS. J. Org. Chem. 2008;73:1440–1455. doi: 10.1021/jo7023784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Denmark SE, Smith RC. Synlett. 2006;18:2921–2928. [Google Scholar]
- (40).Denmark SE, Kallemeyn JM. J. Am. Chem. Soc. 2006;128:15958–15959. doi: 10.1021/ja065988x. [DOI] [PubMed] [Google Scholar]
- (41).2-Dicyclohexylphosphino-2′,6′-dimethoxybiphenyl. Barder TE, Walker SD, Martinelli JR, Buchwald SL. J. Am. Chem. Soc. 2005;127:4685–4696. doi: 10.1021/ja042491j.
- (42) (a).Dai C, Fu GC. J. Am. Chem. Soc. 2001;123:2719–2724. doi: 10.1021/ja003954y.Littke AF, Fu GC. Org. Synth. 2005;81:63–70.(t-Bu3P)2Pd can also be purchased from either Aldrich Chemical Co. or Strem Chemicals.
- (43).Small amounts of disiloxane were detected during the optimization that could account for the need for 1.5 equiv of silanolate. However the general use of 1.5 equiv of silanolate salts assured that all subsequent reactions proceeded to completion. Most of the substrates in this study were not further optimized, so it is unclear whether 1.5 equiv is needed for all combinations of substrates.
- (44).Denmark SE, Wu Z. Org. Lett. 1999;1(9):1495–1498. [Google Scholar]
- (45).For a recent review on the development of ligands for the cross-coupling of aryl chlorides see: Bedford RB, Cazin CSJ, Holder D. Coord. Chem. Rev. 2004;248:2283–2321.
- (46).Littke AF, Fu GC. Angew. Chem., Int. Ed. 2002;41:4176–4211. doi: 10.1002/1521-3773(20021115)41:22<4176::AID-ANIE4176>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- (47).In none of the coupling reactions was the product of homocoupling detected by GC-MS or NMR analysis.
- (48).Palaniki MSS, et al. J. Med. Chem. 2008;51:1546–1559. doi: 10.1021/jm7011276. [DOI] [PubMed] [Google Scholar]
- (49).The lower than expected yields were ascribed to problems in purification. Presumably, 33u and 33v can coordinate to the palladium which cannot be completely removed by aqueous workup and column chromatography. Sublimation was required to provide the palladium-free products judging by the colors of the products. For example, the yellow solid 33u became an off-white solid after sublimation even though no change in 1H NMR spectra can be identified. See Supporting Information for purification procedures.
- (50).In toluene, the cross-coupling reaction of Na+13- with 4-bromobenzonitrile 16v provided a product whose identity cannot be unambiguously confirmed at this time.
- (51).Eaborn C. Organosilicon Compounds. Butterworth; London: 1960. p. 200. [Google Scholar]
- (52).Previous mechanistic studies have demonstrated that palladium(II) silanolates are the intermediates that undergo transmetalation. See Denmark SE, Sweis RF. J. Am. Chem. Soc. 2004;126:4876–4882. doi: 10.1021/ja0372356. and reference 26.
- (53).Clar E. The Aromatic Sextet. John Wiley & Sons; London: 1972. [Google Scholar]
- (54) (a).Mason MR, Verkade JG. Organometallics. 1992;11:2212–2220. [Google Scholar]; (b) Amatore C, Jutand A, Barki’ MAM. Organometallics. 1992;11:3009–3013. [Google Scholar]; (c) Ozawa F, Kubo A, Hayashi T. Chem. Lett. 1992:2177–2180. [Google Scholar]; (d) Amatore C, Carre E, Jutand A, Barki’ MAM. Organometallics. 1995;14:1818–1826. [Google Scholar]; (e) Amatore C, Jutand A. J. Organomet. Chem. 1999;576:254–278. [Google Scholar]
- (55).Langanis ED, Chenard BL. Tetrahedron Lett. 1984;25:5831–5834. [Google Scholar]
- (56).The preparation and coupling of π-deficient heterocyclic silanolates will be the subject of a forthcoming report.
- (57).Dai C, Fu G. J. Am. Chem. Soc. 2001;123:2719–2724. doi: 10.1021/ja003954y. [DOI] [PubMed] [Google Scholar]
- (58) (a).Flemming JP, Pilon MC, Borbulevitch OY, Antipin MY, Grushin VV. Inorg. Chim. Acta. 1998:87–98. [Google Scholar]; (b) Stambuli JP, Weng Z, Incarvito CD, Hartwig JF. Angew. Chem. Int. Ed. 2007;46:7674–7677. doi: 10.1002/anie.200702809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).The primary influence of the ligand was also illustrated by carrying out the cross coupling of Na+13- and 16a’ with APC and SPhos in toluene at 90 \s=deg\C. After 5 h, complete conversion of 16a’ to 33a was observed with a negligible amount of protiodesilylation detected (GC).
- (60).Kurts AK, Sakembaeva SM, Beletskaya IP, Reutov OA. Zh. Org. Chim. 1974;10:1572–1580. [Google Scholar]
- (61).Kabalka GW, Wang L, Pagni RM. Tetrahedron. 2001;57:8107–8128. [Google Scholar]