

Abstract
This review provides an overview of the discovery, structures, and biological activities of anticancer natural products which act by inhibiting or promoting the assembly of tubulin to microtubules. The emphasis is on providing recent information on those compounds in clinical use or in advanced clinical trials. The vinca alkaloids, the combretastatins, NPI-2358, the halichondrin B analog eribulin, dolastatin 10, noscapine, hemiasterlin, and rhizoxin are discussed as tubulin polymerization inhibitors, while the taxanes and the epothilones are the major classes of tubulin polymerization promoters presented, with brief treatments of discodermolide, eleutherobin, and laulimalide. The challenges and future directions of tubulin-interactive natural products-based drug discovery programs are also discussed briefly.
Introduction
Natural products have proven to be the most reliable single source of new and effective anticancer agents. Thus Newman and Cragg have shown that 63% of anticancer drugs introduced over the last 25 years are natural products or can be traced back to a natural products source,2 and similar observations have been made by many others.3- 7 A recent review by Butler lists 79 natural products or natural product analogs that entered clinical trial as anticancer agents in the 2005-2007 timeframe.8 Natural products have not only yielded new and effective drugs, but they have also provided insight into new mechanisms of action, and cancer treatment would be immeasurably poorer without the insights and the compounds provided from Nature. The reasons for the effectiveness of natural products are at least twofold. In the first place, there is a high correlation between the properties of drugs and those of natural products.9,10 Second, natural products usually have built-in chirality, and are thus uniquely suited to bind to complex proteins and other three-dimensional biological receptors.
Among the various mechanisms of action of natural products, that of interaction with the cellular protein tubulin is one of the most important, and over 25% of the new clinical candidates listed by Butler operate by this general mechanism.8 Two major classes of anticancer drugs owe their effectiveness to this mechanism; the first class is that of the tubulin polymerization inhibitors, and the second is that of tubulin polymerization promoters. This review covers natural products or modified natural products that interact with tubulin, and that are in clinical use or are in advanced development towards clinical use.
The cellular protein tubulin is a crucial protein for cellular replication. The cell cycle involves the replication of DNA and the packaging of the resulting replicated chromosomes into two daughter cells. The separation of the daughter chromosomes in mitosis is brought about by microtubules, which are formed by the polymerization of α- and β-tubulin. The microtubules radiate in cells from centrosomes and from the poles of mitotic spindles, and are formed by attachment of GTP-tubulin to the growing end of an existing microtubule. Microtubules undergo rapid assembly and disassembly in cells, and this property enables those associated with the mitotic spindle to generate a large collection of structures and thus to produce those structures which will interact constructively with the centromeres of daughter chromosomes and generate the necessary aligned chromosomes at metaphase. The normal functioning of tubulin assembly and disassembly is thus crucial to cell division, and any interference with this process will disrupt cell division and cause cell death by apoptosis.
Although the most dramatic effect of the tubulin-interactive drugs is that of changing the extent of microtubule polymer mass, either decreasing it for the tubulin polymerization inhibitors, or increasing it for the tubulin polymerization promoters, cancer cell growth can be inhibited at concentrations significantly lower than those necessary to exert these macroscopic effects. This fact can be explained by the observation that cell growth inhibition at low concentrations is caused by the suppression of microtubule dynamics.11
The structure of the tubulin heterodimer has been solved by electron diffraction.12 Both the vinca alkaloids and the taxane drugs bind to β-tubulin, but at different locations on the protein; the vinca alkaloids bind to β-tubulin between amino acids 175 and 213,13 while paclitaxel binds both to an N-terminal unit of β-tubulin14 as well as to the region bounded by amino acids 217-231.15 Colchicine, which is not a clinically used drug for cancer but which has been studied extensively, binds between the two subunits. The epothilones also bind at the paclitaxel site.16
Inhibitors of Tubulin Polymerization
The Vinca Alkaloids
The first natural products to enter clinical use were the bisindole alkaloids vinblastine (1) and vincristine (2). These complex compounds were isolated from the Madagascar periwinkle Catharanthus roseus (L.) G. Don (previously known as Vinca rosea L.) in the late 1950s and early 1960s by two independent groups. Their discovery makes an interesting story, because one of the groups working on them, that of Robert Noble and Charles Beer at the University of Western Ontario, was actually looking for substances that could affect blood glucose levels, and the discovery of the antileukemic activity of the extract was made after the serendipitous observation of its effects on white blood cell counts. These observations led to the isolation and structure elucidation of the bis-indole alkaloid vincaleukoblastine (1); the name was later shortened to vinblastine.17 The related alkaloid leurocristine (later named vincristine, 2) was isolated by Gordon Svoboda and his colleagues at Eli Lilly.18 The alkaloids bind to β-tubulin at a different site from the taxane drugs and colchicine, and act to prevent tubulin assembly.19 The compounds consist of two subunits, an upper catharanthine ring system linked to a lower vindoline ring system by a single bond.
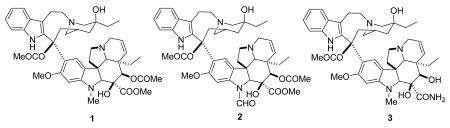
Vinblastine and vincristine have been used in clinical oncology for almost 50 years, and their use has been reviewed.20 Vincristine is used in combination chemotherapy of acute lymphoblastic leukemias and lymphomas, while vinblastine is used in combination chemotherapy to treat bladder and breast cancers. Perhaps the most significant impact of vinblastine has been as part of the curative regimen for Hodgkin's disease.
Vindesine (3) was the first analog of vinblastine to enter clinical use. It differs from vinblastine in having an amide function rather than a methyl ester on the vindoline ring, and in lacking an acetyl group on this ring system. It has a somewhat higher hematological toxicity than vincristine, but it has been incorporated into several effective combination regimens for treatment of leukemia, lymphoma, and non-small cell lung cancer (NSCLC).21,22
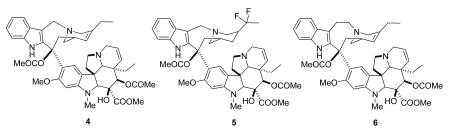
Vinorelbine (4) is a semi-synthetic derivative of vinblastine in which the bridge linking the indole ring to the piperidine nitrogen has been shortened by one carbon, and water has been eliminated from the piperidine ring. It was launched in 1989 by Pierre Fabre for the treatment of non-metastatic breast cancer and NSCLC, and it is available both in i.v. and oral formulations.23- 25
Vinflunine (Javlor®, 5) is a dihydrodifluoro derivative of vinorelbine. It can be prepared by treatment of vinorelbine (4) with HF/SbF5 in CHCl3; the proposed mechanism involves chlorination of the cation generated by protonation of the cyclohexenyl double bond and isomerization. The chloro compound then loses a hydride ion and is fluorinated to a chlorofluoro compound, which is finally converted to vinflunine.26 Vinflunine interacts with tubulin in a qualitatively similar way to vinblastine, but detailed studies indicate that it has quantitatively different properties to the classic vinca alkaloids.27 It is in Phase III trials at Pierre Fabre for the treatment of bladder cancer and NSCLC, based on the observation of clinically significant activity in Phase II studies for the treatment of bladder, non-small cell lung and breast cancers,26,28 and it is also being evaluated for second-line chemotherapy in hormone refractory prostate cancer (HRPC) and for HER2 over-expressing metastatic breast cancer (with Trastuzumab),29 and for ovarian cancer.30
Anhydrovinblastine ((Hydravin™, KRX-0403, 6) is an analog of vinblastine that differs from its parent by one molecule of water. It can also be considered a homolog of vinorelbine with an additional carbon in the indole-piperidine bridge. It entered phase I trial for the treatment of advanced solid tumors, including NSCLC, soft tissue sarcoma, and colorectal cancer. Stable disease was noted in one patient with metastatic sarcoma to the lungs and in three patients with metastatic NSCLC.31 Keryx discontinued development in 2005, but trials may still be ongoing by Prescient Neuropharma.8
Combretastatins and Analogs
The vinca alkaloids are the only inhibitors of tubulin polymerization in clinical use, but several compounds with this mechanism of action are in advanced clinical trials and will be discussed here.
Combretastatin A-4 (7, CA4) was originally isolated by Pettit et al. from the root bark of the Combretum caffrum tree, also known as the Cape Bushwillow.32 It has been shown to target the microtubule, inhibiting the polymerization of tubulin to microtubules. However, it is much more cytotoxic than its activity against tubulin would seem to warrant, and four explanations have been proposed to explain this discrepancy: (i) CA4 targets, in vivo, a subpopulation of tubulin (ii) CA4, alongside tubulin, recognizes a yet unidentified relevant target; (iii) the interaction between tubulin and CA4 is qualitatively different. (iv) CA4, for as yet unknown reasons, rapidly and preferentially disrupts in vivo cellular processes in the endothelium that require tubulin assembly.33 Combretastatin A4 phosphate (CA4P, 8), is a simple derivative of the natural product which was prepared to increase water solubility.34 CA4P has also been shown to be a vascular disrupting agent, and data from phase I studies have established that it can selectively reduce tumor blood flow at well-tolerated doses.35 Its vascular effect can be explained by its disruption of the endothelial cytoskeleton.36 CA4P is in Phase III clinical trials sponsored by Oxigene, Inc. for treatment of cervical, colorectal, NSC lung, prostate, ovarian, and thyroid cancers; reviews of the clinical results to date have appeared recently.37,38
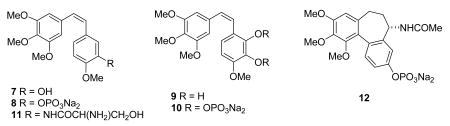
Combretastatin A-4 is accompanied in the Cape Bushwillow with numerous congeners, differing in the ring substitutions (CA series) and by reduction of the internal stilbene double bond to give the CB series.39 Combretastatin A-1 (9) is a simple hydroxyl derivative of combretastatin A-4, and it is also in Phase I clinical development as its diphosphate derivative OXi4503 (10).8 A study of its effects on mice indicated that substantial microvascular damage to liver tumors and minimal normal liver injury occurred, and it was concluded that “A combination of OXi4503 with other chemotherapeutic modalities might achieve complete tumor eradication and improve long-term survival.”40
The relative simplicity of the combretastatin skeleton has spawned the synthesis of numerous analogs. The medicinal chemistry of these compounds is beyond the scope of this review, but it is covered in detail in recent reviews.41- 43 Two of the many analogs that have been prepared are in advanced development. The serinamide derivative AVE8062A (11, AC-7700)44 is in clinical trials in Europe and the United States, and its primary therapeutic effect is to reduce blood flow to the tumor.36,45 The compound ZD-6126 (ANG453, 12) can be considered as an analog of both combretastatin and colchicine, and it is rapidly converted in vivo into N-acetylcolchinol. It too is a vascular disrupting agent.46 Its Phase II trials were halted by AstraZeneca due to problems with the method of administration, but Angiogene has solved this problem.8 The development of combretastatin A-4 and these analogs as anticancer agents has been reviewed.43 Although it is too early yet to tell which (if any) of these combretastatin analogs will enter clinical use, it is safe to predict that at least one and probably more than one drug based on the combretastatins will enter clinical use over the next few years.
NPI-2358
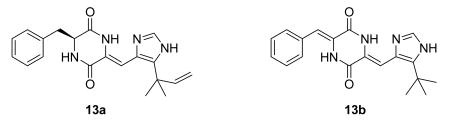
Fungi have yielded relatively few tubulin inhibitors, but one such is the diketopiperazine halimide (13a). NPI-2358 (13b) is a simple analog of halimide, which is active as a tubulin-depolymerizing agent.47 It is in Phase I trials at Nereus.8,48
Halichondrin B and Eribulin
Complex marine natural products of the halichondrin class were isolated by Uemura and Hirata from the western Pacific sponge Halichondria okadai,49 and later by Pettit et al. from an Axinella sp.50 Several members of the class showed strong cytotoxicity, with the most potent being homohalichondrin B and halichondrin B (14). These compounds were shown to bind to tubulin and to inhibit tubulin polymerization. Halichondrin B is a non-competitive inhibitor of vinblastine binding to tubulin, and has no effect on colchicine binding.51 It showed sub-nanomolar activity in the NCI 60-cell line panel,52 and excellent activity in various animal models,49 and it was thus a clear candidate for clinical development. The major obstacle to clinical development was the issue of compound supply, since it was only obtained in miniscule amounts from its marine source.
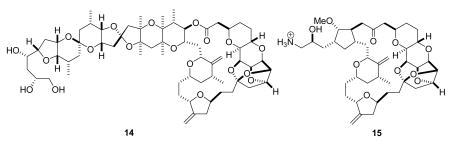
Fortunately for the future of this class of compounds, Kishi developed a total synthesis of halichondrin B.53 The synthesis was designed in a convergent fashion, and his intermediates were tested at Eisai Research Institute for activity. This led to the realization that several truncated halichondrins had significant bioactivity, and so a research program was initiated to develop a simplified and thus synthetically accessible anticancer agent based on the halichondrin B skeleton. The truncated halichondrin B analog, eribulin (15, E7389) was discovered as a result of this program, and its synthesis was effected in a highly convergent manner, although this still required over 70 steps.54,55 The synthesis of this highly cytotoxic compound on an industrial scale had its own unique challenges, but these were successfully overcome so that eribulin mesylate could enter clinical trials. Like its parent compound, halichondrin B, eribulin acts as an inhibitor of tubulin polymerization.56 Eribulin mesylate is in Phase III trials for the treatment of prostate, sarcoma, breast, NSCL, bladder, head and neck, and ovarian cancers. Its discovery and development have been reviewed,57 and its clinical potential has also been reviewed.58,59 An encouraging overall response rate of 15% was observed in a phase II trial in NSCLC patients, and the compound also showed promising results in treatment of breast cancer. One significant advantage over other tubulin-interactive agents is that eribulin appears to have a lower neurotoxicity than the other agents. It is concluded that “E7389 would probably be a very welcome addition to the available agents used to treat women with advanced breast cancer”.59
Dolastatins
Dolastatin 10 (16) was isolated from the sea hare Dolabella auricularia by Pettit as part of an extensive series of investigations; it is the most potent member of a fairly large class of related compounds.60,61 It binds to tubulin at a distinct site for peptide antimitotic agents near the exchangeable nucleotide and vinca alkaloid sites, inhibiting tubulin polymerization.62 The relative simplicity of the structure and the difficulty of sourcing the natural product made chemical synthesis the preferred approach, and dolastatin 10 has been synthesized by the Pettit group63 and others.64 Dolastatin 10 entered Phase I clinical trials in the 1990s, with the finding that 40% of patients developed moderate peripheral neuropathy.65 It then progressed to phase II trials under the auspices of the National Cancer Institute (NCI) for the treatment of several solid tumors, including liver, bile duct, gallbladder cancer, pancreatic cancer, and advanced kidney cancer, but the results of these trials were not encouraging, as summarized in two recent reviews.37,66
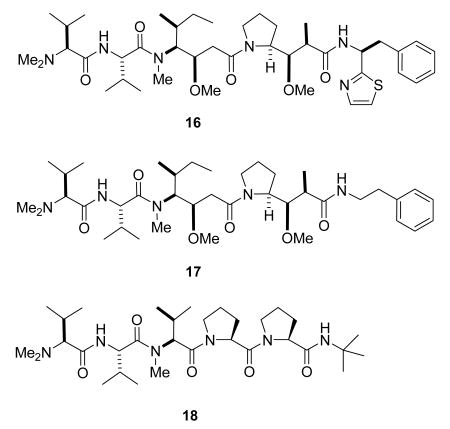
The novel activity of the dolastatins spurred the development of several analogs, and the simplified analog, TZT-1027 (auristatin PE, Soblidotin, 17), was among several analogs prepared for SAR studies.67 It was selected for further development based on its reduced toxicity as compared with dolastatin 10. Clinical results to date are mixed, with antitumor activity observed in some cases but not others.37
A second derivative in clinical development is the dolastatin 15 analog tasidotin (18). Tasidotin is also a tubulin-interactive drug, weakly inhibiting tubulin polymerization to microtubules but strongly suppressing the dynamic instability of microtubules.68 It has completd Phase I trials,69,70 and is currently in Phase II trials under Genzyme.8 Further details of the discovery and development of the dolastatins are available in a comprehensive review.65
Noscapine
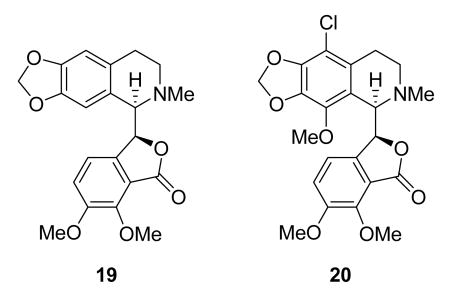
Noscapine (19) is an old compound, being a commonly used antitussive agent without significant side effects,71 but it has recently been found to bind to tubulin and alter its conformation, assembly properties, and microtubule dynamics.72 It shows good oral bioavailability in mice,73 and is active against H460 NSCLC cells in nude mice.74 The analog EM105 (20) is more potent and regresses breast cancer xenografts in nude mice without significant toxicity.75 Noscapine and its analogs are thus interesting lead compounds, and noscapine is in Phase I trials by Cougar Biotechnology.76
Hemiasterlin
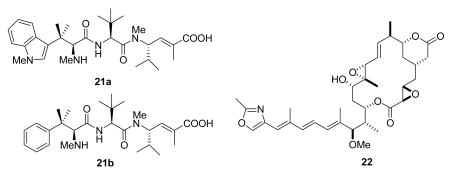
Hemiasterlin (21a) is a tripeptide that was first isolated by Kashman from the sponge Hemiasterella minor, with reported activity against the P388 murine leukemia cell line.77 It was later reisolated by Andersen, who reported that it had antitubulin activity, producing abnormal mitotic spindles at low concentrations and microtubule depolymerization at higher concentrations.78 Synthetic studies identified the phenylalanine analog HTI-286 (21b) as a more accessible and more potent analog,79 and both HTI-28680 and hemiasterlin8 are in clinical trials.81
Rhizoxin
Rhizoxin (22) was isolated in 1984 together with several congeners from the plant pathogenic fungus Rhizopus chinensis.82 It is an inhibitor of tubulin polymerization,83 and is more potent than maytansine against human and murine tumor cells.84 It has been synthesized85 and has been evaluated in clinical trials,86 but has not yet entered clinical use.
Promoters of Tubulin Polymerization
The Taxanes
Paclitaxel (23) was isolated by Wall and Wani in the 1960s from bark of the Pacific yew, Taxus brevifolia, and given the name taxol.87 Its initial discovery was greeted with underwhelming enthusiasm, because of the obvious problems of compound supply and solubility, and because it only showed relatively modest in vivo activity against the then current antileukemic models at the NCI.88 Fortunately, the B16 melanoma solid tumor assay was introduced in the early 1970s, and paclitaxel showed excellent and reproducible activity against this solid tumor, with an increase in life span (ILS) of 126, 32, and 86% in three separate experiments.88 Even with this data the problems of developing paclitaxel loomed large, but fortunately the late Matthew Suffness, who had joined the NCI in 1976, recognized paclitaxel's potential and was instrumental in presenting the case for its development to the NCI Decision Network Committee. This committee approved paclitaxel as a development candidate in 1977.88
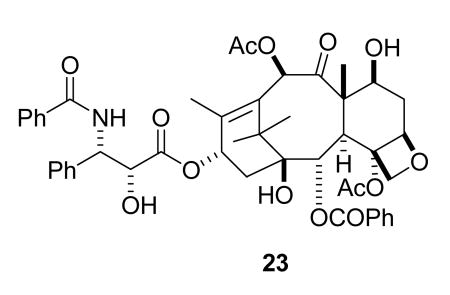
In vivo studies of paclitaxel in the then new human solid tumor xenograft assays in nude mice were carried out in 1978, and these were rewarded when it was found to show strong activity against the MX-1 breast xenograft; this discovery was important in maintaining interest in the compound.88 The final significant finding came with the discovery by Horwitz that paclitaxel promoted the assembly of microtubules;89 this new mechanism was a crucial added factor in raising interest in this compound. The solubility problem was overcome by the development of an emulsion formulation in Cremophor, a polyethoxylated castor oil, which unfortunately caused allergic reactions in some patients. These problems were overcome by premedication with antihistamines and the use of extended intravenous infusions, which were initially for 24 hours but were later reduced to 3 hours.90
Paclitaxel, or taxol as it was then known, was found to have clinical activity against ovarian cancer in 1989,91 and against breast cancer in 1991.92 Further development was taken over by Bristol-Myers Squibb (BMS) in 1991 under a Cooperative Research and Development Agreement (CRADA) with the NCI. BMS was able to trademark the name Taxol® for their formulation of the drug, and the generic name paclitaxel was applied to the chemical compound formerly known as taxol. Taxol® was approved by the FDA in December 1992 for the treatment of refractory breast cancer and refractory ovarian cancer and was launched on the U.S. market by Bristol-Myers Squibb the following year. It was later approved for treatment of breast cancer after failure of combination chemotherapy for metastatic disease, for second-line treatment of AIDS-related Kaposi's sarcoma, and for NSCLC in combination with cisplatin.93 Extensive continuing clinical trials are evaluating combinations of paclitaxel with other drugs for treatment of many other cancers.94,95
The enormous demand for paclitaxel caused by its excellent activity created a significant supply crisis, which was initially solved by aggressive collection of T. brevifolia bark. A viable semisynthetic route was then developed. In this semisynthesis the more readily available 10-deacetylbaccatin III (24), is first converted to 7-triethylsilylbaccatin III (25), and this is coupled with the protected β-lactam 26 in the key step, yielding the protected paclitaxel 27. Final deprotection gives paclitaxel (23) in excellent overall yield (Scheme 1).96 This synthetic route essentially ended the supply crisis. Paclitaxel is now also produced commercially by plant tissue culture methods.97
Scheme 1.

Semisynthesis of paclitaxel
As noted above, paclitaxel acts by promoting the assembly of tubulin into microtubules. This results in the inhibition of the normal dynamic reorganization of the microtubule network that is essential for vital cellular functions, and leads ultimately to cell death by apoptosis. The actual mechanism by which paclitaxel stabilizes the microtubule is still under investigation, but Xiao et al. speculate that “Taxol induces a loss of flexibility in the involved regions that prevents a “roll out” of lateral contacts in microtubules that would otherwise open up their wall.” 98
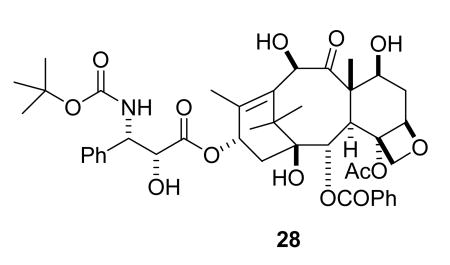
The success of paclitaxel has spurred enormous interest in finding improved analogs as well as improved formulations designed to circumvent the problems associated with the Cremophor formulation, and several analogs are currently in clinical trials. The only analog approved for clinical use to date in the United States is docetaxel (28), a semisynthetic analog developed in France.99 Docetaxel was launched in 1995, and is the drug of choice in treating advanced NSCLC that is refractory to primary therapy.100 It is also used in the treatment of hormone-refractory prostate cancer,101,102 advanced breast cancer,103 and other cancers including head and neck, stomach, and ovarian cancers.104
In addition to paclitaxel and docetaxel, the albumin nanoparticle (nab) formulation of paclitaxel Abraxane™ was approved in 2005 in the United States for the treatment of advanced breast cancer. Since Abraxane™ consists only of albumin-bound paclitaxel nanoparticles, it is free of Cremophor and requires no premedication. A recent review concludes “these studies have demonstrated that nab technology has increased the therapeutic index of paclitaxel compared with the conventional, solvent-based formulation,105 while another review states “Abraxane was safe, effective, induced higher response rate and longer time to progression compared with Taxol® in patients with metastatic breast cancer.”106
Although paclitaxel and docetaxel are the only taxanes currently approved for clinical use, there are many analogs in clinical trials, and the most important of these are summarized briefly below.
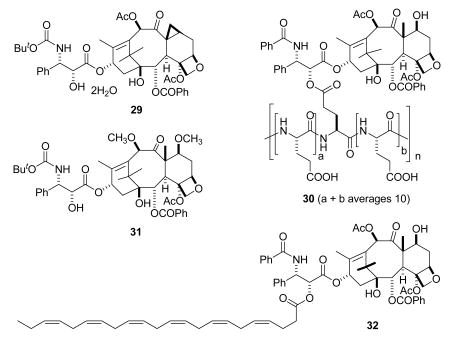
There are four taxanes in Phase III clinical trials. Larotaxel dihydrate (29, Sanofi-Aventis)107 is an interesting derivative of docetaxel in which the methyl group at the C-8 position has formed a cyclopropyl ring; the paclitaxel analog of 29, with a phenyl group replacing the tertiary-butyloxy group in the side chain of 29, was formed on treatment of 7-epi-paclitaxel with DAST.108 Larotaxel is in Phase III trials for treatment of breast and pancreatic cancers, and a report from a Phase II trial indicated that it has a favorable therapeutic index in women with taxane-pretreated metastatic breast cancer.109 Paclitaxel poliglumex (30, Xyotax) is a conjugate of paclitaxel with a biodegradable polyglutamic acid; this feature was designed to increase water solubility and improve its pharmacokinetic profile. It is in Phase III trials by Cell Therapeutics for the treatment of NSCLC and ovarian cancer.110,111 Cabazitaxel (31, XRP-6258, TXD258, Sanofi-Aventis) is a dimethoxy derivative of docetaxel that is in Phase III trials in combination with prednisone for treatment of hormone refractory metastatic prostate cancer.112 It has the advantages that it is not a substrate for P-glycoprotein113 and that it can cross the blood-brain barrier. Taxoprexin (32, DHA-paclitaxel, Luitpold Pharmaceuticals) is a 2′-acyl paclitaxel, and is in Phase III trials for treatment of NSCLC and in Phase II trials for several other cancers. Phase II studies on the compound have been reported.114,115
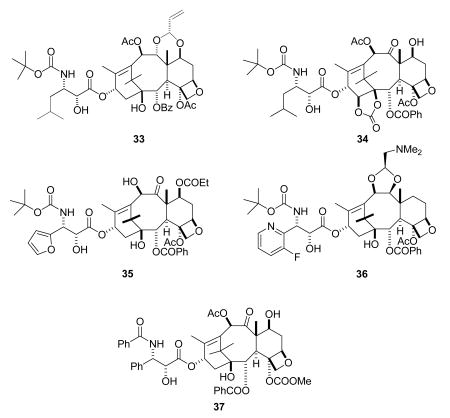
Compounds in Phase II clinical trials include five new compounds and two formulations of paclitaxel. NK-105 is a polymeric micellar macromolecule encapsulated formulation of paclitaxel in phase II clinical trials at Nippon Kayaku for the treatment of solid tumors; it is reported to be a more potent radiosensitizing agent than paclitaxel.116 EndoTAG-1 is a formulation of paclitaxel encapsulated in a positively charged lipid-based complex, and is in phase II clinical trials at MediGene for the treatment of advanced pancreatic cancer.117 TPI-287 (33, NBT-287, Tapestry Pharmaceuticals) is in Phase II trials for treatment of advanced pancreatic cancer and hormone refractory prostate cancer. It is designed to overcome acquired resistance to taxane-based therapies by circumventing the P-glycoprotein drug efflux mechanism, and it is more potent than paclitaxel in paclitaxel-resistant tumors. Results of its Phase I studies have been reported.118,119 Ortataxel (34) was originally developed by Bayer, but the clinical trials encountered severe neutropenia120 and were discontinued. The compound was then licensed exclusively to Spectrum Pharmaceuticals in 2007. It is currently in phase II clinical trials for the treatment of non-small cell lung cancer (NSCLC),121 and it has also shown in vivo activity in animal models against head and neck squamous cell carcinoma (HNSCC).122 Milataxel (35), which was developed by the new company Taxolog, is in Phase II trials under Wyeth.118 One recent report of a Phase II study in colorectal cancer indicated that it had the side effect of neutropenic sepsis, necessitating close surveillance if the drug is to be used at its MTD.123 Tesetaxel (36) was developed by Daiichi Sankyo Co. Ltd. for treatment of colorectal and gastric cancer. It was withdrawn from development in 2006 because of its failure to show clear benefit over existing agents,124 but it was recently licensed by Genta who hope to restart trials.8 BMS-188797 (37) is a simple 4-carbonate derivative of paclitaxel which showed objective responses in four out of sixteen patients in a Phase I trial, including three complete remissions in ovarian and cervical cancer patients;125 a Phase I study of the drug in combination with carboplatin has also been reported.126 Nab™-docetaxel, the nanoparticle albumin bound docetaxel formulation related to Abraxane™, is in early clinical studies at Abraxis BioScience for the treatment of hormone refractory prostate cancer and other solid tumors.127
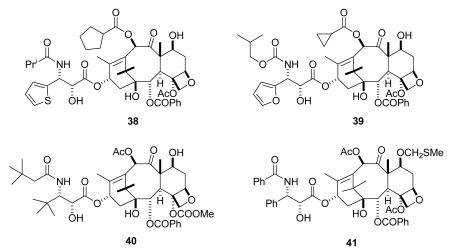
Four compounds that were in Phase I trials, simotaxel (38, Wyeth) and TL-310 (39), the first two compounds from Taxolog, and the Bristol-Myers Squibb analogs BMS-275183 (a 4-carbonate derivative, 40) and BMS-184476 (41), are no longer in clinical trials. Phase II trials on BMS-275183 were halted by BMS, phase I trials on BMS-184476 have been completed,128 phase I trials on simotaxel were halted by Wyeth, and there is no information on TL-310 on clinicaltrials.gov.8
The Epothilones
Epothilones A (42) and B (43) were discovered as antifungal agents in 1986 as metabolites of the myxobacterium Sorangium cellulosum.129 They attracted only moderate scientific interest until the report by Bollag et al. in 1995 that they had the same mechanism of action as paclitaxel, stabilizing the tubulin polymer and causing apoptotic cell death.130 This report triggered a large amount of research on the biology and chemistry of these compounds, which has been reviewed on several occasions.129,131- 133 The progression of the epothilones towards clinical use has also been documented in some recent reviews.134- 137 At this time, one epothilone derivative, the semisynthetic analog ixabepilone (44), has been approved for clinical use in the United States, but several other epothilone derivatives are in advanced clinical trials.
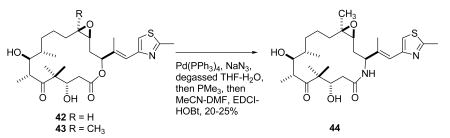
Ixabepilone (Bristol-Myers Squibb) can be prepared by chemical replacement of the lactone oxygen of epothilone B with an amide NH group in a “one pot” reaction involving a π-allylpalladium complex (43 –> 44).138 It was approved by the FDA in 2007 for the treatment of metastatic or locally advanced breast cancer in patients whose tumors are resistant or refractory to anthracyclines, taxanes, and capecitabine. A part of the reason for its effectiveness is that it has a low susceptibility to multiple tumor resistance mechanisms. It also appears to have additive activity with capecitabine.139
Epothilone B (43, patupilone) is in phase III clinical trials at Novartis for the treatment of ovarian cancer. Phase II studies indicated activity in a wide variety of cancers, including taxane-resistant tumors.140 Its activity, as well as that of ixabepilone, against hormone-refractory prostate cancer has been briefly reviewed, and it was concluded “Both patupilone and ixabepilone might be of clinical relevance for patients with HRPC”.141 However, this candidate may have been discontinued, as it is not in Novartis's pipeline nor is it listed on clinicaltrials.gov.8
Sagopilone (ZK-EPO, 45) is a fully synthetic epothilone, in contrast to patupilone, which is a natural fermentation product and ixabepilone which is a modified fermentation product.142 It is in Phase II clinical development at Bayer Schering Pharma for the treatment of recurrent ovarian cancer, metastatic breast cancer, small-cell lung cancer, prostate cancer, and other solid tumors.143 It has good activity in MDR models, probably because it evades the PgP efflux system.142
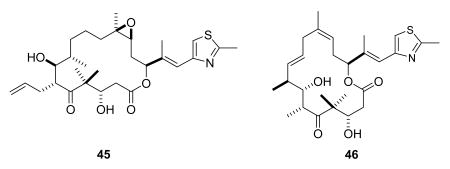
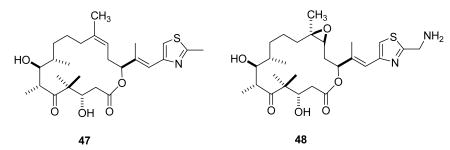
Dehydelone (46, KOS 1584) is an interesting epothilone analog from Kosan based on work done at the Sloan-Kettering Institute for Cancer Research.144 It is in phase I clinical studies for the treatment of advanced or metastatic Stage IIIB/IV NSCLC.145 Epothilone D (47) was entered into clinical trials by Kosan and Roche, but was later dropped in favor of dehydelone and other analogs in the pipeline.8 BMS-310705 (21-aminoepothilone B, 48)146,147 has also most likely been discontinued by BMS, as it is not in BMS's pipeline or listed on clinicaltrials.gov.8
In summary, the epothilones are a promising new class of cancer chemotherapeutic agents, and it is expected that they will play an important role in the treatment of breast cancer, HRPC, and other tumor types. A recent review concluded “The epothilones demonstrate promising antitumor activity in a broad spectrum of taxane-sensitive and -refractory tumors at doses and schedules associated with tolerable side-effects”.135
Other Agents
The epothilones are not the only naturally occurring tubulin polymerization agents to be discovered over the last few years. Three other natural products with the same or very similar activity will be mentioned briefly. Discodermolide (49) is a marine natural product originally isolated from the sponge Discodermia dissoluta,148 and found to have the same effect on tubulin as paclitaxel.149 It was entered into clinical trials by Novartis, and an efficient large-scale synthetic route was developed,150 but sadly it proved to be toxic during its clinical trials and has been dropped.151 In spite of this disappointment, work on the synthesis of analogs continues,152 and it is very possible that a discodermolide derivative may become a clinical candidate in the future.
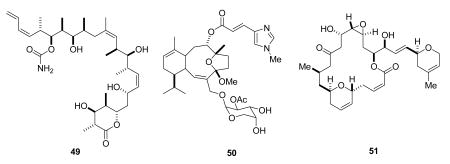
Eleutherobin (50) was initially isolated from a marine soft coral found in the Indian ocean, and was shown to have similar microtubule-stabilizing properties to paclitaxel.153,154 Despite its total synthesis155 and extensive biological studies, it has not yet entered clinical trials.
Laulimalide (51) is also a marine natural product, and was isolated in 1999 from the marine sponge Cacospongia mycofijiensis.156 It stabilizes microtubules in a similar manner to paclitaxel, but it does not bind to the taxoid site on tubulin. It also kills cells resistant to epothilones and paclitaxel, so it is an interesting lead compound.157 It has been synthesized158,159 as have various analogs160,161 but it has not yet entered clinical trials.
Challenges and Future Directions
The importance of tubulin as a target for anticancer drugs has increased significantly since 1990. At that time the only tubulin-interactive drugs available to clinicians were the Vinca alkaloids vinblastine, vincristine, and vindesine. The introduction of paclitaxel into the clinic marked a turning point in the importance of tubulin as a drug target, and opened the door to a flood of new compounds that operate on this protein. The older Vinca alkaloids that work by inhibiting the assembly of tubulin are being supplemented, at least potentially, with the new agents eribulin and combretastatin A-4 phosphate, as well as some new Vinca alkaloid analogs. In the meantime, the tubulin assembly promoter paclitaxel is in the process of being supplemented by several newer analogs, in addition to the established drug docetaxel, and the first of the epothilones is now available, with others almost ready to launch. Other natural product promoters of tubulin assembly are available for development should their preclinical data warrant it. The future thus looks bright for tubulin-interactive agents to play a significant role in the fight against cancer.
Two questions need to be addressed at this point. The first one is “Why are almost all the tubulin-interactive agents natural products or analogs of natural products?” The second is “Can supplies of these scarce materials be assured in the event that they are used clinically on a large scale”?
It is not possible to give a precise answer to the first question, but at least a part of the answer must lie in the fact that most natural products (and all the tubulin-interactive natural products except the combretastatins) are complex compounds with significant stereochemistry. They are thus well adapted to binding to the complex three-dimensional surface of a protein such as tubulin.
The second question can be answered by noting that the problem of the supply of a scarce natural product can be met in several different ways. In the case of paclitaxel, as noted earlier, the major initial supplies came from harvesting yew trees, but this was supplanted, at least for Bristol-Myers Squibb, by a semisynthetic route. As noted above, paclitaxel is also being prepared by plant tissue culture techniques.97 In other cases, such as the halichondrin B derivative eribulin54,55 and discodermolide,150 a total synthesis approach proved to be successful. There is thus reason to hope that a way will be found to produce any natural product or natural product derivative in the quantities needed for clinical use, if its activity profile warrants the investment of resources that would be necessary.
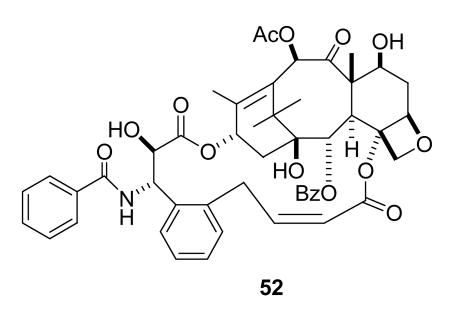
So what are the prospects for the continued discovery of new natural products with tubulin-interactive properties, and for the development of new drugs with these mechanisms of action? There is no doubt that new natural products with tubulin-interactive properties will continue to be discovered, but the development of new analogs of paclitaxel, the epothilones, and other compounds such as discodermolide will also continue to contribute to the development of new drugs. Improvements in targeted drug delivery will also contribute to the success of natural products; an interesting example is the use of colloidal gold nanoparticles in combination with TNF to target paclitaxel to cancer cells.162 Further advances will also come from studies of the detailed interaction between the drug and tubulin, which could lead to the design of simpler synthetic compounds with the requisite tubulin-interactive properties. An example of this approach is the successful design of the paclitaxel analog 52 with improved activity by conformational locking of paclitaxel into the “T-taxol” conformation.163
In summary, it is highly probable that continued studies of naturally occurring tubulin-interactive agents will yield new and improved anticancer agents in the future.
Acknowledgments
The work on paclitaxel from the author's laboratory has been supported most recently by the NIH under grant R01 CA69571 and by a subcontract from CytImmune Inc.: this support is gratefully acknowledged.
References and Not
- 1.This manuscript is based in part on a lecture given at the American Association for Cancer Research Educational Program, Symposium on Natural Products in Anticancer Drug Discovery, San Diego, CA, April 12-16, 2008. A long abstract of the lecture has been published: Kingston DGI. American Association for Cancer Research 99th Annual Meeting Education Book. AACR; Philadelphia: 2008. Tubulin-Interactive Natural Products as Anticancer Agents; pp. 305–313.
- 2.Newman DJ, Cragg GM. J Nat Prod. 2007;70:461–477. doi: 10.1021/np068054v. [DOI] [PubMed] [Google Scholar]
- 3.Butler MS. Nat Prod Rep. 2005;22:162–195. doi: 10.1039/b402985m. [DOI] [PubMed] [Google Scholar]
- 4.Clardy J, Walsh C. Nature. 2004;432:829–837. doi: 10.1038/nature03194. [DOI] [PubMed] [Google Scholar]
- 5.Altmann KH, Gertsch J. Nat Prod Rep. 2007;24:327–357. doi: 10.1039/b515619j. [DOI] [PubMed] [Google Scholar]
- 6.Paterson I, Anderson EA. Science. 2005;310:451–453. doi: 10.1126/science.1116364. [DOI] [PubMed] [Google Scholar]
- 7.Kingston DGI, Newman DJ. IDrugs. 2005;8:990–992. [PubMed] [Google Scholar]
- 8.Butler MS. Nat Prod Rep. 2008;25:475–516. doi: 10.1039/b514294f. [DOI] [PubMed] [Google Scholar]
- 9.Feher M, Schmidt JM. J Chem Inf Comput Sci. 2003;43:218–227. doi: 10.1021/ci0200467. [DOI] [PubMed] [Google Scholar]
- 10.Ortholand JY, Ganesan A. Curr Opin Chem Biol. 2004;8:271–280. doi: 10.1016/j.cbpa.2004.04.011. [DOI] [PubMed] [Google Scholar]
- 11.Jordan MA, Kamath K. Curr Cancer Drug Targets. 2007;7:730–742. doi: 10.2174/156800907783220417. [DOI] [PubMed] [Google Scholar]
- 12.Lowe J, Li H, Downing KH, Nogales E. J Mol Biol. 2001;313:1045–1057. doi: 10.1006/jmbi.2001.5077. [DOI] [PubMed] [Google Scholar]
- 13.Rai SS, Wolff J. J Biol Chem. 1996;271:14707–14711. doi: 10.1074/jbc.271.25.14707. [DOI] [PubMed] [Google Scholar]
- 14.Rao S, Krauss NE, Heerding JM, Swindell CS, Ringel I, Orr GA, Horwitz SB. J Biol Chem. 1994;269:3132–3134. [PubMed] [Google Scholar]
- 15.Rao S, Orr GA, Chaudhary AG, Kingston DGI, Horwitz SB. J Biol Chem. 1995;270:20235–20238. doi: 10.1074/jbc.270.35.20235. [DOI] [PubMed] [Google Scholar]
- 16.Bollag DM, McQueney PA, Zhu J, Hensens O, Koupal L, Liesch J, Goetz M, Lazarides E, Woods CM. Cancer Res. 1995;55:2325–2333. [PubMed] [Google Scholar]
- 17.Noble RL, Beer CT, Cutts JH. Ann N Y Acad Sci. 1958;76:882–894. doi: 10.1111/j.1749-6632.1958.tb54906.x. [DOI] [PubMed] [Google Scholar]
- 18.Svoboda GH, Johnson IS, Gorman M, Neuss N. J Pharm Sci. 1962;51:707–720. doi: 10.1002/jps.2600510802. [DOI] [PubMed] [Google Scholar]
- 19.Gueritte F, Fahy J. In: Anticancer Agents from Natural Products. Cragg GM, Kingston DGI, Newman DJ, editors. CRC Press; Boca Raton, FL: 2005. pp. 123–135. [Google Scholar]
- 20.Rowinsky EK, Tolcher AW. In: Cancer: Principles and Practice of Oncology. 6th. DeVita VT, Hellman S, Rosenberg SA, editors. Lippincott-Raven; Philadelphia: 2001. pp. 431–452. [Google Scholar]
- 21.Dancey J, Steward WP. Anti-Cancer Drugs. 1995;6:625–36. doi: 10.1097/00001813-199510000-00001. [DOI] [PubMed] [Google Scholar]
- 22.Joel S. Cancer Treat Rev. 1995;21:513–25. doi: 10.1016/0305-7372(95)90015-2. [DOI] [PubMed] [Google Scholar]
- 23.Gralla RJ, Gatzemeier U, Gebbia V, Huber R, O'Brien M, Puozzo C. Drugs. 2007;67:1403–1410. doi: 10.2165/00003495-200767100-00003. [DOI] [PubMed] [Google Scholar]
- 24.Aapro MS, Conte P, Gonzalez EE, Trillet-Lenoir V. Drugs. 2007;67:657–667. doi: 10.2165/00003495-200767050-00002. [DOI] [PubMed] [Google Scholar]
- 25.Mano M. Cancer Treat Rev. 2006;32:106–118. doi: 10.1016/j.ctrv.2005.12.008. [DOI] [PubMed] [Google Scholar]
- 26.Jacquesy JC. J Fluor Chem. 2006;127:1484–1487. [Google Scholar]
- 27.Kruczynski A, Barret JM, Etievant C, Colpaert F, Fahy J, Hill BT. Biochem Pharmacol. 1998;55:635–648. doi: 10.1016/s0006-2952(97)00505-4. [DOI] [PubMed] [Google Scholar]
- 28.Bennouna J, Campone M, Delord JP, Pinel MC. Expert Opin Investig Drugs. 2005;14:1259–67. doi: 10.1517/13543784.14.10.1259. [DOI] [PubMed] [Google Scholar]
- 29. [November 26, 2008]; http://www.clinicaltrials.gov/ct2/show/NCT00545766?term=vinflunine.
- 30.McIntyre JA, Castaner J. Drugs Future. 2004;29:574–580. [Google Scholar]
- 31.Ramnath N, Schwartz GN, Smith P, Bong D, Kanter P, Berdzik J, Creaven PJ. Cancer Chemother Pharmacol. 2003;51:227–230. doi: 10.1007/s00280-002-0566-8. [DOI] [PubMed] [Google Scholar]
- 32.Pettit GR, Singh SB, Hamel E, Lin CM, Alberts DS, Garcia KD. Experientia. 1989;45:209–211. doi: 10.1007/BF01954881. [DOI] [PubMed] [Google Scholar]
- 33.Tron GC, Pirali T, Sorba G, Pagliai F, Busacca S, Genazzani AA. J Med Chem. 2006;49:3033–3044. doi: 10.1021/jm0512903. [DOI] [PubMed] [Google Scholar]
- 34.Pettit GR, Rhodes MR. Anticancer Drug Des. 1998;13:183–191. [PubMed] [Google Scholar]
- 35.Chaplin DJ, Horsman MR, Siemann DW. Curr Opin Investig Drugs. 2006;7:522–528. [PubMed] [Google Scholar]
- 36.Hinnen P, Eskens FALM. Br J Cancer. 2007;96:1159–1165. doi: 10.1038/sj.bjc.6603694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Banerjee S, Wang Z, Mohammad M, Sarkar FH, Mohammad RM. J Nat Prod. 2008;71:492–496. doi: 10.1021/np0705716. [DOI] [PubMed] [Google Scholar]
- 38.Kanthou C, Tozer GM. Expert Opin Ther Targets. 2007;11:1443–1457. doi: 10.1517/14728222.11.11.1443. [DOI] [PubMed] [Google Scholar]
- 39.Pettit GR, Singh SB, Niven ML, Hamel E, Schmidt JM. J Nat Prod. 1987;50:119–131. doi: 10.1021/np50049a016. [DOI] [PubMed] [Google Scholar]
- 40.Chan LS, Malcontenti-Wilson C, Muralidharan V, Christophi C. Anti-Cancer Drugs. 2008;19:17–22. doi: 10.1097/CAD.0b013e3282f077a1. [DOI] [PubMed] [Google Scholar]
- 41.Tron GC, Pirali T, Sorba G, Pagliai F, Busacca S, Genazzani AA. J Med Chem. 2006;49:3033–3044. doi: 10.1021/jm0512903. [DOI] [PubMed] [Google Scholar]
- 42.Chaudhary A, Pandeya SN, Kumar P, Sharma PP, Gupta S, Soni N, Verma KK, Bhardwaj G. Mini-Rev Med Chem. 2007;7:1186–1205. doi: 10.2174/138955707782795647. [DOI] [PubMed] [Google Scholar]
- 43.Pinney KG, Jelinek C, Edvardsen K, Chaplin DJ, Pettit GR. In: Anticancer Agents from Natural Products. Cragg GM, Kingston DGI, Newman DJ, editors. CRC Press; Boca Raton, FL: 2005. pp. 23–46. [Google Scholar]
- 44.Ohsumi K, Hatanaka T, Nakagawa R, Fukuda Y, Morinaga Y, Suga Y, Nihei Y, Akiyama Y, Tsuji T. Anti-Cancer Drug Des. 1999;14:539–548. [PubMed] [Google Scholar]
- 45.Hori K, Saito S. Br J Cancer. 2003;89:1334–1344. doi: 10.1038/sj.bjc.6601261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Micheletti G, Poli M, Borsotti P, Martinelli M, Imberti B, Taraboletti G, Giavazzi R. Cancer Res. 2003;63:1534–1537. [PubMed] [Google Scholar]
- 47.Nicholson B, Lloyd GK, Miller BR, Palladino MA, Kiso Y, Hayashi Y, Neuteboom, Saskia TC. Anticancer Drugs. 2006;17:25–31. doi: 10.1097/01.cad.0000182745.01612.8a. [DOI] [PubMed] [Google Scholar]
- 48.Cai SX. Anti-Cancer Drug Discovery. 2007;2:79–101. doi: 10.2174/157489207779561462. Recent Pat. [DOI] [PubMed] [Google Scholar]
- 49.Hirata Y, Uemura D. Pure Appl Chem. 1986;58:701–710. [Google Scholar]
- 50.Pettit GR, Herald CL, Boyd MR, Leet JE, Dufresne C, Doubek DL, Schmidt JM, Cerny RL, Hooper JNA, Rutzler KC. J Med Chem. 1991;34:3339–3340. doi: 10.1021/jm00115a027. [DOI] [PubMed] [Google Scholar]
- 51.Luduena RF, Roach MC, Prasad V, Pettit GR. Biochem Pharmacol. 1993;45:421–427. doi: 10.1016/0006-2952(93)90079-c. [DOI] [PubMed] [Google Scholar]
- 52.Bai RL, Paull KD, Herald CL, Malspeis L, Pettit GR, Hamel E. J Biol Chem. 1991;266:15882–15889. [PubMed] [Google Scholar]
- 53.Aicher TD, Buszek KR, Fang FG, Forsyth CJ, Jung SH, Kishi Y, Matelich MC, Scola PM, Spero DM, Yoon SK. J Am Chem Soc. 1992;114:3162–3164. [Google Scholar]
- 54.Zheng W, Seletsky BM, Palme MH, Lydon PJ, Singer LA, Chase CE, Lemelin CA, Shen Y, Davis H, Tremblay L, Towle MJ, Salvato KA, Wels BF, Aalfs KK, Kishi Y, Littlefield BA, Yu MJ. Bioorg Med Chem Lett. 2004;14:5551–5554. doi: 10.1016/j.bmcl.2004.08.069. [DOI] [PubMed] [Google Scholar]
- 55.Seletsky BM, Wang Y, Hawkins LD, Palme MH, Habgood GJ, DiPietro LV, Towle MJ, Salvato KA, Wels BF, Aalfs KK, Kishi Y, Littlefield BA, Yu MJ. Bioorg Med Chem Lett. 2004;14:5547–5550. doi: 10.1016/j.bmcl.2004.08.068. [DOI] [PubMed] [Google Scholar]
- 56.Jordan MA, Kamath K, Manna T, Okouneva T, Miller HP, Davis C, Littlefield BA, Wilson L. Mol Cancer Ther. 2005;4:1086–1095. doi: 10.1158/1535-7163.MCT-04-0345. [DOI] [PubMed] [Google Scholar]
- 57.Yu MJ, Kishi Y, Littlefield BA. In: Anticancer Agents from Natural Products. Cragg GM, Kingston DGI, Newman DJ, editors. CRC Press; Boca Raton, FL: 2005. pp. 241–265. [Google Scholar]
- 58.Newman S. Curr Opin Invest Drugs. 2007;8:1057–1066. [PubMed] [Google Scholar]
- 59.Atieh DM, Vahdat LT. Breast Diseases: a Year Book Quarterly. Vol. 18. 2007. pp. 21–23. [Google Scholar]
- 60.Pettit GR, Kamano Y, Fujii Y, Herald CL, Inoue M, Brown P, Gust D, Kitahara K, Schmidt JM, Doubek DL, Michael C. J Nat Prod. 1981;44:482–485. doi: 10.1021/np50016a016. [DOI] [PubMed] [Google Scholar]
- 61.Bai R, Pettit GR, Hamel E. Biochem Pharmacol. 1990;39:1941–1949. doi: 10.1016/0006-2952(90)90613-p. [DOI] [PubMed] [Google Scholar]
- 62.Bai R, Pettit GR, Hamel E. J Biol Chem. 1990;265:17141–17149. [PubMed] [Google Scholar]
- 63.Pettit GR, Srirangam JK, Singh SB, Williams MD, Herald DL, Barkbczy J, Kantoci D, Hogan F. J Chem Soc, Perkin Trans. 1996;1:859–863. [Google Scholar]
- 64.Mordant C, Reymond S, Tone H, Lavergne D, Touati R, Hassine BB, Ratovelomanana-Vidal V, Genet JP. Tetrahedron. 2007;63:6115–6123. [Google Scholar]
- 65.Flahive E, Srirangam J. In: Anticancer Agents from Natural Products. Cragg GM, Kingston DGI, Newman DJ, editors. CRC Press; Boca Raton, FL: 2005. pp. 191–213. [Google Scholar]
- 66.Singh R, Mukul S, Joshi P, Rawat DS. Anti-Cancer Agents Med Chem. 2008;8:603–617. [PubMed] [Google Scholar]
- 67.Pettit GR, Srirangam JK, Barkoczy J, Williams MD, Durkin KPM, Boyd MR, Bai R, Hamel E, Schmidt JM, Chapuis JC. Anti-Cancer Drug Des. 1995;10:529–544. [PubMed] [Google Scholar]
- 68.Ray A, Okouneva T, Manna T, Miller HP, Schmid S, Arthaud L, Luduena R, Jordan MA, Wilson L. Cancer Res. 2007;67:3767–3776. doi: 10.1158/0008-5472.CAN-06-3065. [DOI] [PubMed] [Google Scholar]
- 69.Mita AC, Hammond LA, Bonate PL, Weiss G, McCreery H, Syed S, Garrison M, Chu QSC, DeBono JS, Jones CB, Weitman S, Rowinsky EK. Clin Cancer Res. 2006;12:5207–5215. doi: 10.1158/1078-0432.CCR-06-0179. [DOI] [PubMed] [Google Scholar]
- 70.Cunningham C, Appleman LJ, Kirvan-Visovatti M, Ryan DP, Regan E, Vukelja S, Bonate PL, Ruvuna F, Fram RJ, Jekunen A, Weitman S, Hammond LA, Eder JP., Jr Clin Cancer Res. 2005;11:7825–7833. doi: 10.1158/1078-0432.CCR-05-0058. [DOI] [PubMed] [Google Scholar]
- 71.Al-Yahya MA, Hassan MMA. Anal Profiles Drug Subst. 1982;11:407–61. [Google Scholar]
- 72.Ye K, Ke Y, Keshava N, Shanks J, Kapp JA, Tekmal RR, Petros J, Joshi HC. Proc Natl Acad Sci U S A. 1998;95:1601–1606. doi: 10.1073/pnas.95.4.1601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Aneja R, Dhiman N, Idnani J, Awasthi A, Arora SK, Chandra R, Joshi HC. Cancer Chemother Pharmacol. 2007;60:831–839. doi: 10.1007/s00280-007-0430-y. [DOI] [PubMed] [Google Scholar]
- 74.Jackson T, Chougule MB, Ichite N, Patlolla RR, Singh M. Cancer Chemother Pharmacol. 2008;61 doi: 10.1006/s00280-008-0720-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Aneja R, Lopus M, Zhou J, Vangapandu SN, Ghaleb A, Yao J, Nettles JH, Zhou B, Gupta M, Panda D, Chandra R, Joshi HC. Cancer Res. 2006;66:3782–3791. doi: 10.1158/0008-5472.CAN-05-2962. [DOI] [PubMed] [Google Scholar]
- 76. [September 8, 2008]; http://www.cougarbiotechnology.com/docs/122107CougarCB3304PhaseITrialInitiation.pdf.
- 77.Talpir R, Benayahu Y, Kashman Y, Pannell L, Schleyer M. Tetrahedron Lett. 1994;35:4453–4456. [Google Scholar]
- 78.Anderson HJ, Coleman JE, Andersen RJ, Roberge M. Cancer Chemother Pharmacol. 1997;39:223–6. doi: 10.1007/s002800050564. [DOI] [PubMed] [Google Scholar]
- 79.Nieman JA, Coleman JE, Wallace DJ, Piers E, Lim LY, Roberge M, Andersen RJ. J Nat Prod. 2003;66:183–99. doi: 10.1021/np020375t. [DOI] [PubMed] [Google Scholar]
- 80.Andersen RJ, Roberge M. HTI-286, a Synthetic Analog of the Antimitotic Natural Product Hemiasterlin. In: Cragg GM, Kingston DGI, Newman DJ, editors. Anticancer Agents from Natural Products. CRC Press; Boca Raton: 2005. pp. 267–280. [Google Scholar]
- 81.Rawat DS, Joshi MC, Joshi P, Atheaya H. Anticancer Agents Med Chem. 2006;6:33–40. doi: 10.2174/187152006774755519. [DOI] [PubMed] [Google Scholar]
- 82.Iwasaki S, Kobayashi H, Furukawa J, Namikoshi M, Okuda S, Sato Z, Matsuda I, Noda T. J Antibiot. 1984;37:354–362. doi: 10.7164/antibiotics.37.354. [DOI] [PubMed] [Google Scholar]
- 83.Sullivan AS, Prasad V, Roach MC, Takahashi M, Iwasaki S. Cancer Res. 1990;50:4277–80. [PubMed] [Google Scholar]
- 84.Wolpert-deFilippes MK, Adamson RH, Cystic RL, Johns DG. Biochem Pharmacol. 1975;24:751–754. doi: 10.1016/0006-2952(75)90257-9. [DOI] [PubMed] [Google Scholar]
- 85.Mitchell IS, Pattenden G, Stonehouse J. Org Biomol Chem. 2005;3:4412–4431. doi: 10.1039/b507570j. [DOI] [PubMed] [Google Scholar]
- 86.McLeod HL, Murray LS, Wanders J, Setanoians A, Graham MA, Pavlidis N, Heinrich B, ten Bokkel Huinink WW, Wagener DJ, Aamdal S, Verweij J. Br J Cancer. 1996;74:1944–1948. doi: 10.1038/bjc.1996.657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Wani MC, Taylor HL, Wall ME, Coggon P, McPhail AT. J Am Chem Soc. 1971;93:2325–2327. doi: 10.1021/ja00738a045. [DOI] [PubMed] [Google Scholar]
- 88.Suffness M, Wall ME. In: Taxol: Science and Applications. Suffness M, editor. CRC Press, Inc.; Boca Raton, FL: 1995. pp. 3–25. [Google Scholar]
- 89.Schiff PB, Fant J, Horwitz SB. Nature. 1979;277:665–667. doi: 10.1038/277665a0. [DOI] [PubMed] [Google Scholar]
- 90.Arbuck SG, Blaylock BA. In: Taxol: Science and Applications. Suffness M, editor. CRC Press, Inc.; Boca Raton, FL: 1995. pp. 379–415. [Google Scholar]
- 91.McGuire WP, Rowinsky EK, Rosenshein NB, Grumbine FC, Ettinger DS, Armstrong DK, Donehower RC. Ann Intern Med. 1989;111:273–279. doi: 10.7326/0003-4819-111-4-273. [DOI] [PubMed] [Google Scholar]
- 92.Holmes FA, Walters RS, Theriault RL, Forman AD, Newton LK, Raber MN, Buzdar AU, Frye DK, Hortobagyi GN. J Nat Cancer Inst. 1991;83:1797–1805. doi: 10.1093/jnci/83.24.1797-a. [DOI] [PubMed] [Google Scholar]
- 93.Ramalingam S, Belani CP. Expert Opin Pharmacother. 2004;5:1771–1780. doi: 10.1517/14656566.5.8.1771. [DOI] [PubMed] [Google Scholar]
- 94.Ferlini C, Gallo D, Scambia G. Expert Opin Invest Drugs. 2008;17:335–347. doi: 10.1517/13543784.17.3.335. [DOI] [PubMed] [Google Scholar]
- 95.Dubois J. Expert Opin Ther Patents. 2006;16:1481–1496. [Google Scholar]
- 96.Holton RA, Biediger RJ, Boatman PD. In: Taxol: Science and Applications. Suffness M, editor. CRC Press, Inc.; Boca Raton, FL: 1995. pp. 97–121. [Google Scholar]
- 97.Leistner E. Pharm Unserer Zeit. 2005;34:98–103. doi: 10.1002/pauz.200400108. [DOI] [PubMed] [Google Scholar]
- 98.Xiao H, Verdier-Pinard P, Fernandez-Fuentes N, Burd B, Angeletti R, Fiser A, Horwitz SB, Orr GA. Proc Natl Acad Sci USA. 2006;103:10166–10173. doi: 10.1073/pnas.0603704103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Guenard D, Gueritte-Voegelein F, Potier P. Acc Chem Res. 1993;26:160–167. [Google Scholar]
- 100.Davies AM, Lara PN, Mack PC, Gandara DR. Expert Opin Pharmacother. 2003;4:553–65. doi: 10.1517/14656566.4.4.553. [DOI] [PubMed] [Google Scholar]
- 101.Wolff JM. Onkologie. 2003;26:37–40. doi: 10.1159/000076173. [DOI] [PubMed] [Google Scholar]
- 102.Khan MA, Carducci MA, Partin AW. J Urology. 2003;170:1709–1716. doi: 10.1097/01.ju.0000088787.95124.4b. [DOI] [PubMed] [Google Scholar]
- 103.Figgitt DP, Wiseman LR. Drugs. 2000;3:621–651. doi: 10.2165/00003495-200059030-00015. [DOI] [PubMed] [Google Scholar]
- 104.Ramaswamy B, Puhalla S. Drugs Today. 2006;42:265–279. doi: 10.1358/dot.2006.42.4.968648. [DOI] [PubMed] [Google Scholar]
- 105.Gradishar WJ. Expert Opin Pharmacother. 2006;7:1041–1053. doi: 10.1517/14656566.7.8.1041. [DOI] [PubMed] [Google Scholar]
- 106.Henderson IC, Bhatia V. Expert Rev Anticancer Ther. 2007;7:919–943. doi: 10.1586/14737140.7.7.919. [DOI] [PubMed] [Google Scholar]
- 107.Ojima I, Geney R. Curr Opin Invest Drugs. 2003;4:737–740. [PubMed] [Google Scholar]
- 108.Chen SH, Huang S, Wei J, Farina V. J Org Chem. 1993;58:4520–4521. [Google Scholar]
- 109.Dieras V, Limentani S, Romieu G, Tubiana-Hulin M, Lortholary A, Kaufman P, Girre V, Besenval M, Valero V. Ann Oncol. 2008;19:1255–60. doi: 10.1093/annonc/mdn060. [DOI] [PubMed] [Google Scholar]
- 110.Bonomi P. Expert Rev Anticancer Ther. 2007;7:415–422. doi: 10.1586/14737140.7.4.415. [DOI] [PubMed] [Google Scholar]
- 111.Boddy AV. Drugs Future. 2007;32:776–780. [Google Scholar]
- 112. [December 3, 2008]; http://www.clinicaltrials.gov/ct2/show/NCT00417079?term=XRP-6258&rank=1.
- 113.Cisternino S, Bourasset F, Archimbaud Y, Semiond D, Sanderink G, Sherrmann JM. British J Pharmacol. 2003;138:1367–1375. doi: 10.1038/sj.bjp.0705150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Jones RJ, Hawkins RE, Eatock MM, Ferry DR, Eskens FALM, Wilke H, Evans TRJ. Cancer Chemother Pharmacol. 2008;61:435–441. doi: 10.1007/s00280-007-0486-8. [DOI] [PubMed] [Google Scholar]
- 115.Payne M, Ellis P, Dunlop D, Ranson M, Danson S, Schacter L, Talbot D. J Thorac Oncol. 2006;1:984–90. [PubMed] [Google Scholar]
- 116.Negishi T, Koizumi F, Uchino H, Kuroda J, Kawaguchi J, Naito S, Matsumura Y. Br J Cancer. 2006;95:601–606. doi: 10.1038/sj.bjc.6603311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Schuch G. Curr Opin Invest Drugs. 2005;6:1259–1265. [PubMed] [Google Scholar]
- 118.Kingston DGI, Newman DJ. Curr Opin Drug Disc Devel. 2007;10:130–144. [PubMed] [Google Scholar]
- 119.Hwang JJ, Marshall JL, Malik S, Chun H, Ahmed T, Pilia PA. J Clin Oncol. 2006;24:12008. [Google Scholar]
- 120.Baumann K. Pharmazie in Unserer Zeit. 2005;34:110–114. doi: 10.1002/pauz.200400110. [DOI] [PubMed] [Google Scholar]
- 121.Beer M, Lenaz L, Amadori D. J Clin Oncol. 2008 May;26 20:1066. Abstr. [Google Scholar]
- 122.Sano D, Matsuda H, Ishiguro Y, Nishimura G, Kawakami M, Tsukuda M. Oncol Rep. 2006;15:329–334. [PubMed] [Google Scholar]
- 123.Ramanathan Ramesh K, Picus J, Raftopoulos H, Bernard S, Lockhart AC, Frenette G, Macdonald J, Melin S, Berg D, Brescia F, Hochster H, Cohn A. Cancer Chemother Pharmacol. 2008;61:453–8. doi: 10.1007/s00280-007-0489-5. [DOI] [PubMed] [Google Scholar]
- 124.Roche M, Kyriakou H, Seiden M. Curr Opin Invest Drugs. 2006;7:1092–1099. [PubMed] [Google Scholar]
- 125.du Bois A, Jung B, Loehr A, Schaller-Kranz T, Cohen M, Frickhofen N. Br J Cancer. 2006;94:79–84. doi: 10.1038/sj.bjc.6602886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Fishman MN, Garrett CR, Simon GR, Chiappori AA, Lush RM, Dinwoodie WR, Mahany JJ, Dellaportas AM, Cantor A, Gollerki A, Cohen MB, Sullivan DM. Clin Cancer Res. 2006;12:523–528. doi: 10.1158/1078-0432.CCR-05-0928. [DOI] [PubMed] [Google Scholar]
- 127.Hawkins MJ, Soon-Shiong P, Desai N. Adv Drug Deliv Rev. 2008;60:876–85. doi: 10.1016/j.addr.2007.08.044. [DOI] [PubMed] [Google Scholar]
- 128.Sun W, Stevenson JP, Gallagher ML, Vaughn D, Hahn SM, Haller DG, Cohen M, Kopit J, Gallant G, O'Dwyer PJ. Clin Cancer Res. 2003;9:5221–5227. [PubMed] [Google Scholar]
- 129.Hofle G, Reichenbach H. In: Anticancer Agents from Natural Products. Cragg GM, Kingston DGI, Newman DJ, editors. CRC Press; Boca Raton, FL: 2005. pp. 413–450. [Google Scholar]
- 130.Bollag DM, McQueney PA, Zhu J, Hensens O, Koupal L, Liesch J, Goetz M, Lazarides E, Woods CM. Cancer Res. 1995;55:2325–2333. [PubMed] [Google Scholar]
- 131.Altmann KH. Mini Rev Med Chem. 2003;3:149–158. doi: 10.2174/1389557033405269. [DOI] [PubMed] [Google Scholar]
- 132.Watkins EB, Chittiboyina AG, Jung JC, Avery MA. Curr Pharm Des. 2005;11:1615–1653. doi: 10.2174/1381612053764742. [DOI] [PubMed] [Google Scholar]
- 133.Altmann KH, Pfeiffer B, Arseniyadis S, Pratt BA, Nicolaou KC. Chem Med Chem. 2007;2:396–423. doi: 10.1002/cmdc.200600206. [DOI] [PubMed] [Google Scholar]
- 134.Larkin JMG, Kaye SB. Expert Opin Invest Drugs. 2006;15:691–702. doi: 10.1517/13543784.15.6.691. [DOI] [PubMed] [Google Scholar]
- 135.Fumoleau P, Coudert B, Isambert N, Ferrant E. Ann Oncol. 2007;18 5:9–15. doi: 10.1093/annonc/mdm173. [DOI] [PubMed] [Google Scholar]
- 136.Cortes J, Baselga J. The Oncologist. 2007;12:271–280. doi: 10.1634/theoncologist.12-3-271. [DOI] [PubMed] [Google Scholar]
- 137.Harrison M, Swanton C. Expert Opin Investig Drugs. 2008;17:523–546. doi: 10.1517/13543784.17.4.523. [DOI] [PubMed] [Google Scholar]
- 138.Borzilleri RM, Zheng X, Schmidt RJ, Johnson JA, Kim SH, DiMarco JD, Fairchild CR, Gougoutas JZ, Lee FYF, Long BH, Vite GD. J Am Chem Soc. 2000;122:8890–8897. [Google Scholar]
- 139.Conlin A, Fornier M, Hudis C, Kar S, Kirkpatrick P. Nat Rev Drug Discovery. 2007;6:953–954. [Google Scholar]
- 140.Rothermel J, Wartmann M, Chen T, Hohneker J. Sem Oncol. 2003;30:51–55. doi: 10.1016/s0093-7754(03)00125-8. [DOI] [PubMed] [Google Scholar]
- 141.Lee D. Clin Prostate Cancer. 2004;3:80–82. doi: 10.1016/s1540-0352(11)70066-x. [DOI] [PubMed] [Google Scholar]
- 142.Klar U, Buchmann B, Schwede W, Skuballa W, Hoffmann J, Lichtner RB. Angew Chemie Int Ed. 2006;45:7942–7948. doi: 10.1002/anie.200602785. [DOI] [PubMed] [Google Scholar]
- 143.Klar U, Hoffmann J, Giurescu M. Expert Opin Invest Drugs. 2008;17:1735–1748. doi: 10.1517/13543784.17.11.1735. [DOI] [PubMed] [Google Scholar]
- 144.Rivkin A, Yoshimura F, Gabarda AE, Chou TC, Dong H, Tong WP, Danishefsky SJ. J Am Chem Soc. 2003;125:2899–2901. doi: 10.1021/ja029695p. [DOI] [PubMed] [Google Scholar]
- 145. [December 3, 2008]; http://www.clinicaltrials.gov/ct2/show/NCT00651508?term=KOS1584&rank=1.
- 146.Kamath AV, Chang M, Lee FY, Zhang Y, Marathe PH. Cancer Chemother Pharmacol. 2005;56:145–153. doi: 10.1007/s00280-004-0928-5. [DOI] [PubMed] [Google Scholar]
- 147.Kolman A. Curr Opin Invest Drugs. 2004;5:1292–1297. [PubMed] [Google Scholar]
- 148.Gunasekera SP, Gunasekera M, Longley RE. J Org Chem. 1990;55:4912–4915. [Google Scholar]
- 149.ter Haar E, Kowalski RJ, Hamel E, Lin CM, Longley RE, Gunasekera SP, Rosenkranz HS, Day BW. Biochemistry. 1996;35:243–250. doi: 10.1021/bi9515127. [DOI] [PubMed] [Google Scholar]
- 150.Mickel SJ. Pure Appl Chem. 2007;79:685–700. [Google Scholar]
- 151.Shaw SJ. Mini-Rev Med Chem. 2008;8:276–284. doi: 10.2174/138955708783744137. [DOI] [PubMed] [Google Scholar]
- 152.Smith AB, III, Freeze BS. Tetrahedron. 2008;64:261–298. doi: 10.1016/j.tet.2007.10.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Lindel T, Jensen PR, Fenical W, Long BH, Casazza AM, Carboni J, Fairchild CR. J Am Chem Soc. 1997;119:8744–8745. [Google Scholar]
- 154.Long BH, Carboni JM, Wasserman AJ, Cornell LA, Casazza AM, Jensen PR, Lindel T, Fenical W, Fairchild CR. Cancer Res. 1998;58:1111–1115. [PubMed] [Google Scholar]
- 155.Chen XT, Bhattacharya SK, Zhou B, Gutteridge CE, Pettus TRR, Danishefsky SJ. J Am Chem Soc. 1999;121:6563–6579. [Google Scholar]
- 156.Mooberry SL, Tien G, Hernandez AH, Plubrukarn A, Davidson BS. Cancer Res. 1999;59:653–660. [PubMed] [Google Scholar]
- 157.Pryor DE, O'Brate A, Bilcer G, Diaz JF, Wang Y, Wang Y, Kabaki M, Jung MK, Andreu JM, Ghosh AK, Giannakakou P, Hamel E. Biochemistry. 2002;41:9109–9115. doi: 10.1021/bi020211b. [DOI] [PubMed] [Google Scholar]
- 158.Ghosh AK, Wang Y, Kim JT. J Org Chem. 2001;66:8973–8982. doi: 10.1021/jo010854h. [DOI] [PubMed] [Google Scholar]
- 159.Paterson I, De Savi C, Tudge M. Org Lett. 2001;3:3149–3152. doi: 10.1021/ol010150u. [DOI] [PubMed] [Google Scholar]
- 160.Ahmed A, Hoegenauer EK, Enev VS, Hanbauer M, Kaehlig H, Ohler E, Mulzer J. J Org Chem. 2003;68:3026–3042. doi: 10.1021/jo026743f. [DOI] [PubMed] [Google Scholar]
- 161.Wender PA, Hegde SG, Hubbard RD, Zhang L, Mooberry SL. Org Lett. 2003;5:3507–3509. doi: 10.1021/ol035339f. [DOI] [PubMed] [Google Scholar]
- 162.Paciotti GF, Kingston DGI, Tamarkin L. Drug Devel Res. 2006;67:47–54. [Google Scholar]
- 163.Ganesh T, Guza RC, Bane S, Ravindra R, Shanker N, Lakdawala AS, Snyder JP, Kingston DGI. Proc Natl Acad Sci USA. 2004;101:10006–10011. doi: 10.1073/pnas.0403459101. [DOI] [PMC free article] [PubMed] [Google Scholar]