

Abstract
The 4-nitroimidazole PA-824 is active against aerobic and anaerobic Mycobacterium tuberculosis (Mtb) while 5-nitroimidazoles like metronidazole are active against only anaerobic Mtb. We have synthesized analogs of both 4- and 5-nitroimidazoles and explored their antitubercular activities. The nitro group is required for both activities in all compounds. The key determinants of aerobic activity in the 4-nitroimidazoles include the bicyclic oxazine, the lipophilic tail, and the 2-position oxygen. For the 5-nitroimidazoles, neither the corresponding bicyclic analog, nor addition of a lipophilic tail conveyed aerobic activity. Incorporation of a 2-position oxygen atom into a rigid 5-nitroimidazo-oxazine provided the first 5-nitroimidazole with aerobic activity. Across both series, anaerobic and aerobic activities were not correlated and Mtb mutants lacking the deazaflavin-dependent nitroreductase Ddn retained anaerobic sensitivity to some compounds. Aerobic activity appears to be correlated with efficiency as a substrate for Ddn, suggesting a means of structure-based optimization of improved nitroimidazoles.
Introduction
Tuberculosis, caused by Mycobacterium tuberculosis (Mtba), continues to be a major threat to global public health, killing more than 1.5 million people in 2005.1 One major contributing factor towards this mortality is the extended duration of chemotherapy (6-9 months) required to completely cure disease.2 Tuberculous lesions are hypoxic, with a pO2 less than 10 mmHg.3 Hypoxic Mtb does not actively replicate and is significantly less sensitive to the standard antituberculosis drugs that interfere with processes essential for rapid aerobic growth.4 Thus, hypoxia is thought to contribute to the extended duration of therapy and new agents active against these metabolically-altered bacteria are urgently needed.
The nitroimidazoles, such as (S)-2-nitro-6-(4-(trifluoromethoxy)benzyloxy)-6,7-dihydro-5H-imidazo[2,1-b][1,3]oxazine (PA-824, 1)5, are a promising new class of compounds that have shown antitubercular activity under hypoxic conditions and are currently in clinical evaluation. Metronidazole (Mtz, 2-(2-methyl-5-nitro-1H-imidazol-1-yl)ethanol, 2), a 5-nitroimidazole, is widely used for the treatment of diseases caused by anaerobic bacteria.6 It has been known for almost 15 years that under low oxygen conditions Mtb cultures become sensitive to Mtz treatment.7, 8 However, Mtz has no activity against aerobic populations of Mtb and is not used in the treatment of human tuberculosis. 4-Nitroimidazoles based on Mtz were originally synthesized as radiosensitizing agents9 and later found to have significant activity against Mtb.10, 11 From a large number of 2,3-dihydro-6-nitroimidazo[2,1-b]oxazole derivatives, compound 3 (2-ethyl-6-nitro-2,3-dihydroimidazo[2,1-b]oxazole, CGI-17341) emerged as an interesting tool compound with potent in vitro and in vivo activity against Mtb.10 Further work on this class of compounds, led to the discovery of PA-824, a 4-nitroimidazo-oxazine.5 PA-824 and its analogs have shown potent activity against aerobic and anaerobic Mtb, no cross resistance to current TB drugs, and efficacy in the mouse model of TB infection.5, 12-14 PA-824 is currently in clinical development by the Global Alliance for TB Drug Development.15 More recently, a structurally-related 4-nitroimidazo-oxazole, compound 4 ((R)-2-methyl-6-nitro-2-((4-(4-(4-(trifluoromethoxy)phenoxy)piperidin-1-yl)phenoxy)methyl)-2,3-dihydroimidazo[2,1-b]oxazole, OPC-67683), has demonstrated significant antitubercular activity and is also being evaluated in human clinical trials.16 A series of antitubercular and structurally similar nitrofurans have also been reported.17 These compounds appear to be reduced using FGD1 and F420, but through a nitroreductase different from Ddn (Rv3547) used by PA-824.18
Our objective in the current study was to determine the fundamental structure-activity relationship (SAR) of nitroimidazoles as antitubercular compounds, with special emphasis on the structural requirements for aerobic versus anaerobic activities. A better understanding of the SAR would help us to determine if these diverse groups of nitroimidazole compounds have the same cellular mode of action. In particular, we were interested in the differences or similarities in SAR between the 4-nitroimidazo-oxazines (typified by PA-824) and the 5-nitroimidazoles (such as Mtz), and additionally whether the SAR for the aerobic activity would mirror that of the anaerobic activity. To simplify the discussion, we will use the numbering for the imidazole ring throughout the paper. Thus, Mtz has a 5-nitro substituent, while PA-824 and compound 3 are referred to as 4-nitro derivatives.
Chemistry
Simple comparison of the structures of Mtz and PA-824 revealed four key differences: i) the location of the nitro group; ii) the presence or absence of the lipophilic side chain; iii) the electronic nature of the atom at the 2-position of the nitroimidazole; and iv) a monocyclic versus a bicyclic ring structure. We have synthesized a number of compounds which probe the structural space defined by these parameters and then examined their activity against Mtb.
Nitro-isomers of PA-824 and Mtz
We initially prepared the 5-nitro isomer of PA-824 (11 in Scheme 1). The original synthesis of PA-824 and related compounds was published in two US patents several years ago.12, 13 We have improved upon this synthesis and have used this as the basis for much of our synthetic work. The patented synthesis began with 2,4-dinitroimidazole, an explosive. Instead, we used 2-chloro-4-nitroimidazole (5). Condensation of 5 with epoxide 6 gave an isomeric mixture of products. After careful chromatographic separation, alcohols 7a and 7b were isolated in a ratio of 3:1, with a total yield of 86%.19 THP-protection of 5-nitroimidazole 7b furnished protected diol 8 in 70% yield. Cyclization of 8 in the presence of TBAF yielded bicyclic compound 9 which was then deprotected with AcOH to give alcohol 10b. The 4-nitro alcohol 10a was obtained following the same procedures from 7a (not shown). Alkylation of 10b using 4-trifluoromethoxybenzyl bromide with NaH and TBAI gave 5-nitro-824 (11). Interestingly, movement of the nitro group from the 4-position to the 5-position of the imidazole ring dramatically changes the chemical reactivity of these compounds. The yields for each of the reactions leading to compound 11 are significantly lower than those using the corresponding 4-nitro isomers. This is typified by the yield of the final alkylation reaction where PA-824 was obtained in 70% yield,13 while compound 11 was obtained in 13%. The desnitro analog (12) was prepared using the same methodology with 2-nitroimidazole as the starting material (See Supplementary Information).
Scheme 1.

Synthesis of 5-NO2-824 and des-NO2-824.a
aReagents and conditions: (a) 6, K2CO3, EtOH, sealed tube, 70°C, 20 h; (b) DHP, PPTS, CH2Cl2, rt, 2 d; (c) TBAF, THF, rt, 18 h; (d) AcOH, THF, H2O, 60°C, 18 h; (e) NaH, TBAI, 4-trifluoromethoxybenzyl bromide, DMF, −78°C to rt, 18 h. Compounds 10a and 12 were prepared following the same scheme.
To examine the importance of the location of the nitro group in Mtz (2), we also prepared the 4-nitro isomer of this compound (Scheme 2). 2-Methyl-4-nitroimidazole (13) was reacted with 2-bromoethanol and K2CO3 in the presence of TBAI to give 4-nitro-Mtz (14).20
Scheme 2.

Synthesis of analogs to examine the importance of the side chain of nitroimidazoles.a
aReagents and conditions: (a) NaH, CH3I, DMF, −78°C to rt, 18 h; (b) NaH, TBAI, 4-trifluoromethoxybenzyl bromide, DMF, −78°C to rt, 18 h; (c) 2-bromoethanol, K2CO3, TBAI, CH3CN, sealed tube, 75°C, 2 h.
Lipophilic side chain analogs of PA-824 and Mtz
The importance of the lipophilic side chain of PA-824 on aerobic and anaerobic activity was addressed by the compounds shown in Scheme 2. Methylation of alcohol 10a with CH3I and NaH gave the analog (15) with the 4-trifluoromethoxyphenyl side chain removed in 57% yield.21 Simple alkylation of either Mtz (2) or 4-nitro-Mtz (14)22 with 4-trifluoromethoxybenzyl bromide in the presence of NaH and TBAI gave compounds 16 (33%) and 17 (39%), respectively.
Monocyclic analog of PA-824 and bicyclic analogs of Mtz
Compound 20, a ring-opened analog of PA-824 (Scheme 3), was prepared as follows: 2-Chloro-4-nitroimidazole (5) was alkylated using TBS-protected bromoethanol to give compound 18 (25%) in addition to the corresponding 5-nitro isomer (28 (2.3%) in Scheme 5) as a minor product. Substitution of the chlorine, followed by TBS-deprotection in the presence of NaOMe afforded 19 in 61% yield.23 Alkylation of the hydroxyl group in 19 gave rise to a ring-opened version of PA-824 (20) in 58% yield. To give “bicyclic” analogs of Mtz, we envisioned ring-closing metathesis as a key step (Scheme 4).24 Considering the possible conformations of the bicyclic product, both stereoisomers were prepared. Starting from 2-bromo-4-nitroimidazole (21), an epoxide-opening reaction using (R)-vinyloxirane (22) gave an allylic alcohol which was TBS-protected to afford compound 23a (16% in two steps). This procedure also produced the corresponding 4-isomer (e.g., 31 (47%) in Scheme 6). Stille coupling of 23a using tributyl(vinyl)stannane furnished the required diene (24a, 67%).25 Ring-closing metathesis using the second generation Hoveyda-Grubbs' catalyst26 provided the unsaturated bicyclic compound 25a (72%). TBS-protection was required to avoid undesired elimination during the synthesis of 25a. Selective reduction of 25a with Wilkinson's catalyst gave compound 26a in 65% yield.27 TBS-deprotection of 26a with TBAF afforded bicyclic Mtz 27a (69%). The same procedure was followed using (S)-vinyloxirane to obtain the enantiomer 27b.
Scheme 3.

Synthesis of ring-opened analogs of PA-824.a
aReagents and conditions: (a) BrCH2CH2OTBS, K2CO3, MeOH, sealed tube, 75°C, 18 h; (b) NaOMe, MeOH, rt, 24 h; (c) NaH, TBAI, 4-trifluoromethoxybenzyl bromide, DMF, −78°C to rt, 14 h.
Scheme 5.

Synthesis of 2-methoxy-Mtz.a
aReagents and conditions: (a) BrCH2CH2OTBS, K2CO3, MeOH, sealed tube, 75°C, 18 h; (b) NaOMe, MeOH, rt, 15 h; (c) TBAF, THF, rt, 10 min.
Scheme 4.

Synthesis of ”bicyclic” Mtz.a
aReagents and conditions: (a) i) 22, MeOH, sealed tube, K2CO3, MeOH, 70°C, 14 h; ii) TBSCl, imidazole, CH2Cl2, rt, 1 h; (b) tributyl(vinyl)stannane, Pd(PPh3)4, LiCl, DMF, μW, 120°C, 10 min; (c) Hoveyda-Grubbs' II catalyst, CH2Cl2, sealed tube, 60°C, 2.5 h; (d) Wilkinson's catalyst, H2 (g), t-BuOH, THF, rt, 14 h; (e) TBAF, THF, rt, 10 min. 27b was prepared following the same scheme using (S)-vinyloxirane.
Scheme 6.

Synthesis of 2-C-824.a
aReagents and conditions: (a) 22, K2CO3, MeOH, sealed tube, 85°C, 20 h; (b) NaH, TBAI, 4-trifluoromethoxybenzyl bromide, DMF, −78°C to rt, 14 h; (c) tributyl(vinyl)stannane, Pd(PPh3)4, LiCl, DMF, μW, 120°C, 15 min; (d) Grubbs' II catalyst, CH2Cl2, sealed tube, 55°C, 15 h; (e) Wilkinson's catalyst, H2 (g), t-BuOH, THF, rt, 16 h.
2-Position substitutions in each series
To understand the importance of the 2-position substituent on activity, both the 2-methoxy Mtz derivative (30) and the carba analog (35) of PA-824 were made. In Scheme 5, reaction of 2-chloro-4-nitroimidazole (5) with TBS-protected bromoethanol gave the 4- and 5-nitro alkylation products in an 11:1 ratio. Interestingly, preparation of the 5-nitro isomer required use of a protected bromoethanol. The 4- and 5-nitro isomers were separated by careful column chromatography and their structures were confirmed by 2D NOESY experiments and UV λmax differences.28 The 5-nitro isomer (28) was reacted with sodium methoxide to give 29 (58%) in addition to a small amount of 30 (15%). Subsequent TBS-deprotection of 29 with TBAF afforded desired compound 30 in 69% yield. The synthesis of analog 35 is shown in Scheme 6 and follows the synthetic methodology used to prepare compounds 27a and 27b. Alkylation of 2-bromo-4-nitroimidazole (21) furnished allylic alcohol 31. Compound 31 was then alkylated with 4-trifluoromethoxybenzyl bromide to afford 32 in 62% yield to install the side chain of PA-824. 32 underwent Stille coupling, ring-closing metathesis, and selective reduction reactions, furnishing compounds 33, 34, and 35, respectively.
Results and Discussion
A nitro group is essential for both aerobic and anaerobic activities
To assess the importance of the presence of the nitro group we examined the aerobic MIC (minimal inhibitory concentration, defined as the concentration which prevents 99% of bacterial growth) as well as the anaerobic MAC (minimum anaerobicidal concentration, defined as the drug concentration that causes a 90% reduction in bacterial numbers following seven days anaerobic drug exposure) of desnitro-824 (12). Desnitro-824 (12) retained neither the aerobic nor anaerobic activity of the parent compound, indicating that the nitro group is indeed essential for the dual activities of PA-824 (Table 1).
Table 1.
Aerobic, Anaerobic and Enzymatic Activities of Nitroimidazoles
| Number | Name | Structure | wtH37Rv MIC (μM) |
wtH37Rv MAC (μM) |
Class B1 MIC (μM) |
Class C MIC (μM) |
Class C MAC (μM) |
Ddn kcat/Km* |
|---|---|---|---|---|---|---|---|---|
| 4-Nitroimidazoles | ||||||||
| 1 | PA-824 | 0.8 | 8-16 | >100 | >100 | >500 | 0.145 | |
| 12 | Desnitro-824 | >160 | >500 | >100 | >100 | >500 | NS | |
| 10a | 4-NO2-(S)- alcohol |
 |
>100 | 250 | >100 | >100 | 500 | 0.005 |
| 15 | MeO-824 | >125 | 250 | >100 | >100 | 250 | 0.0079 | |
| 20 | Ring-opened- 824 |
6.25 | 250-500 | >100 | >100 | 250 | 0.030 | |
| 17 | 4-NO2-Mtz-824 | >145 | >500 | >100 | >100 | >500 | NS | |
| 14 | 4-NO2-Mtz | >300 | >500 | >100 | >100 | >500 | NS | |
| 34 | 2-C=C-824 | 25 | 62.5 | >100 | >100 | 62.5 | 0.0476 | |
| 35 | 2-C-824 | 25 | 250 | >100 | >100 | 250 | 0.0235 | |
| 5-Nitroimidazoles | ||||||||
| 2 | Mtz | 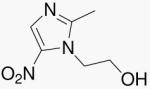 |
>300 | 62.5 | >100 | >100 | 125 | NS |
| 16 | Mtz-824 | >145 | 250 | >100 | >100 | 125 | 0.005 | |
| 30 | 2-MeO-Mtz | 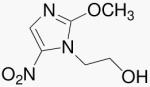 |
>100 | >500 | >100 | >100 | >500 | NS |
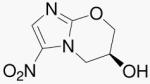
| 10b | 5-NO2-(S)- alcohol |
 |
12.5-25 | 125 | >100 | >100 | 125 | 0.003 |
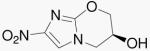
| 11 | 5-NO2-824 | 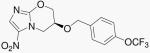 |
4-8 | 31.25 | >100 | >100 | 62.5-125 | 0.0817 |
| 27a | Bicyclic Mtz (R) | 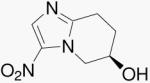 |
>100 | >500 | >100 | >100 | >500 | NS |
| 27b | Bicyclic Mtz (S) | 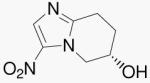 |
>100 | >100 | >100 | >100 | >100 | NS |
NS = not a substrate
SAR of the 4-nitroimidazole series related to PA-824
We approached the SAR of the 4-nitroimidazoles derived from PA-824 systematically as shown in Figure 1. In order to understand the importance of the trifluoromethoxybenzyl ether side chain on the activity of the 4-nitroimidazoles, we prepared compounds 10a and 15. Removal of the side chain to give the simple ‘4-nitro alcohol’ (10a) resulted in a dramatic and complete loss of both aerobic and anaerobic activities. To rule out that this was simply the very different lipophilicities of an ether compared to an alcohol we also explored 15 (the simple methyl ether). Compound 15 likewise showed a dramatic loss of aerobic and anaerobic activities when compared to PA-824. These results are in line with the results from Baker et al.12 who showed that 6-(benzyloxy)-2-nitro-6,7-dihydro-5H-imidazo[2,1-b][1,3]oxazine (i.e., PA-824 without the OCF3) was inactive against M. bovis BCG, while compounds retaining the lipophilic substituent in the para position of the aromatic ring retained aerobic activity.
Figure 1.

Compounds examining the antitubercular SAR of 4-nitroimidazoles.
Compound 20 represents an open ring analog of PA-824 with simple scission of the C-6/C-7 carbon bond of the oxazine ring. As expected, this conservative change retained some of the aerobic activity of the parent compound, PA-824, albeit at a level reduced nearly ten-fold. The anaerobic activity was even more significantly decreased. The response of both activities suggests that the increase in rotational freedom accompanying the loss of the oxazine ring results in a smaller population of active conformers required for recognition by the activating enzyme. Removal of the 2-position oxygen atom from 20 yields compound 17. Interestingly, this change in the electron-donating potential of the 2-substituent leads to a loss in aerobic activity of more than 20-fold, arguing for a very important role of the 2-postion oxygen in determining aerobic activity of the 4-nitroimidazoles. The loss of anaerobic activity of 17 relative to that of Mtz further supports that the 2-oxy substituent is a critical determinant of both aerobic and anaerobic activities in the 4-nitroimidazoles, but not 5-nitroimidazoles.
Removal of the side chain from compound 17 yields a more Mtz-like compound, 14. The sole difference between 14 and Mtz lies in the position of the nitro group. Compound 14 did not display either aerobic or anaerobic activity. This result confirms the importance of the position of the nitro group in Mtz's anaerobic activity. The lack of activity of 14 compared to 17 corroborates the observation that the side chain alone cannot confer aerobic or anaerobic activity in the 4-nitro series.
To probe the importance of the 2-position of the imidazole further, we returned to the bicyclic core structure of PA-824 and replaced the oxazine oxygen with a methylene unit to give compound 35. This modification restores the rotational constraints of the parent compound, PA-824, while eliminating the electron-donating component at the 2-position. As expected, the aerobic activity of compound 35 was reduced more than 30-fold compared to PA-824 and this compound showed negligible anaerobic activity. Interestingly, the olefinic precursor to 35, compound 34, generated as an intermediate following the metathesis reaction, showed comparably low aerobic activity. While the aerobic activity was comparable, the anaerobic activity of 34 was at least four-fold better than its saturated analog. Together, these results indicate that the electronic nature of the 2-position substituent has profound importance for both activities of the 4-nitroimidazoles. Combined with the increase in anaerobic activity seen with the unsaturated carba-analog of PA-824 (34), these results suggest that the SAR associated with aerobic activity and the SAR associated with anaerobic activity are distinct within the 4-nitroimidazoles.
SAR of the 5-nitroimidazoles related to Mtz
We employed a similar feature comparison in attempting to understand the SAR of the antitubercular activities of the 5-nitroimidazoles (Figure 2). Beginning with the lipophilic side chain, we found that compound 16, containing the benzyl ether of PA-824 appended to the Mtz core structure, failed to display any aerobic activity against Mtb, and indeed showed a significant decrease in anaerobic activity relative to that of Mtz. This observation marks a clear distinction between the 4- and 5-nitroimidazole series. The lipophilic side chain is required for both aerobic and anaerobic activity of the 4-nitroimidazoles. Conversely, the presence of the lipophilic side chain did not confer aerobic activity and decreases anaerobic activity in the 5-nitro series.
Figure 2.

Compounds examining the antitubercular SAR of 5-nitroimidazoles.
Building structural elements of PA-824 into Mtz, compound 30 has a 2-position oxygen atom. While this feature is essential for high aerobic and anaerobic activities in the 4-nitro series, this modification alone is not sufficient to convey either activity in the 5-nitro series. Indeed, 30 lost anaerobic activity relative to the parent, Mtz.
We were intrigued to note the biological activity data from compound 10b, the “bicyclic” analog of Mtz containing the 2-position oxygen atom. Compound 10b unexpectedly showed aerobic activity and weak anaerobic activity. The rigid bicyclic structure imparts both aerobic and anaerobic activity compared with compound 30. Further, this result is contrary to that found for the 4-nitro isomer (10a), which was inactive (Table 1).
The addition of the side chain from PA-824 to compound 10b yields compound 11. This change improves both the aerobic and anaerobic activity of 10b. Compound 11 can also be viewed as the 5-nitro isomer of PA-824 and is only slightly less active compared to that parent.
To understand the influence of the 2-position oxygen atom alone, we prepared compounds 27a/b, conformationally rigid, bicyclic forms of Mtz. This change gave one of the most striking results in this study. Comparing compounds 10b and 27a/b, we find that replacing only the 2-position oxygen atom from compound 10b with a methylene results in the loss of both aerobic and anaerobic activities. This result indicates that, for 5-nitroimidazo-oxazines, the 2-position oxygen atom is an essential requirement for both activities. As seen above, this was not found to be the case with 4-nitroimidazo-oxazines. Compound 35, with a 2-position methylene, retained both aerobic and anaerobic activities.
The relationship between aerobic and anaerobic SAR
Considering all the compounds together, we can deduce that the structural requirements for aerobic and anaerobic activity for the 4- and 5-nitroimidazoles are fundamentally different. To explore this further we performed MIC and MAC determinations against both wild type Mtb and defined Mtb mutants that were selected for resistance to PA-824. PA-824 is activated by the nitroreductase Ddn (Rv3547) at the expense of cofactor F420. Mutants resistant to this drug fall into three classes. Class A mutants have lost the F420-dependent glucose-6-phosphate dehydrogenase Fgd1.29 Class B mutants have disruptions in the biosynthetic genes for co-factor F420. Class C mutants have lesions in Ddn (Rv3547), the protein that reductively activates these pro-drugs at the expense of reduced F420.30 Aerobically, all classes of mutants show complete cross-resistance to all of the compounds with aerobic activity described in this study, suggesting that the aerobic SAR was the result of reduction of these compounds only by Ddn (Table 1). Anaerobically, however, compound 11 (5-nitro-824) and compound 34 (the olefin precursor to the 2C analog of PA-824) both showed significant killing of Class C mutants. This result suggests that the anaerobic activity of these two compounds was unrelated to activation by this particular nitroreductase.
Kinetics of activation of nitroimidazoles by Ddn
In order to better differentiate the activities of these compounds we also directly examined the efficiency of these compounds as substrates of the F420-dependent nitroreductase Ddn (Rv3547). Table 1 shows the kcat/Km values of these compounds, including that for PA-824. These data clearly show that PA-824 is the best substrate for Ddn, while Mtz is not a substrate at all. In general, the activity of these compounds as substrates of Ddn is mostly proportional to their observed aerobic killing activity. To a limited extent both 4- and 5-nitroimidazoles are substrates for Ddn, although bicyclic 4-nitroimidazoles appear to be superior. The SAR associated with aerobic activity in these nitroimidazoles is likely to reflect their interaction with Ddn, suggesting that these compounds could be further optimized, informed by the structure of their interaction with that receptor. Anaerobic activity, however, appears to be more complicated than simple efficiency as a substrate for Ddn.28
Conclusion
This study suggests that, even though the 4-nitroimidazoles represented by PA-824 are chemically related to 5-nitroimidazoles such as Mtz, the mechanism of cellular toxicity of these molecules differs substantially. By exploring the common elements of both series of molecules we were able to both gain aerobic activity (10b) in the 5-nitroimidazoles and lose aerobic activity in the 4-nitro series (34 and 35). This work has elucidated some key structural features that are important for aerobic activity, most notably the preference for the 4-nitroimidazole, the nature of the substituent at the 2-position of the imidazole ring, a hydrophobic substituent at the 6-position of the oxazine ring and the conformationally rigid bicyclic system. By correlating these structural changes with whole cell activity against wild-type and defined mutants resistant to PA-824, we were able to establish which structures made genuine contributions to understanding SAR. By assessing the efficiency of these compounds as substrates for Ddn, the nitroreductase that activates these pro-drugs, we were able to further delineate on-target from off-target compounds. As antibacterial development programs gradually shift towards more whole-cell based screening and lead optimization programs, these results illustrate the ease with which multiple target effects can confuse and mislead medicinal chemists. The improved understanding of the aerobic SAR resulting from these studies should facilitate the rational design and synthesis of even more potent analogs of PA-824.
Experimental Section
Anhydrous solvents and reagents were purchased from Sigma-Aldrich and used as received unless otherwise stated. Melting points were obtained on an Electrothermal 9100 apparatus and are uncorrected. LC-MS analysis was conducted on an Agilent 1100 series HPLC with attached Agilent quadrupole mass analyzer model G1946 D SL with electrospray ionization in positive ion mode. LC chromatography used a Phenomenex Luna C18(2) column (2 × 50 mm, 3 μm) with a water/acetonitrile (each with 0.1% (v/v) formic acid) gradient using a flow rate of 0.3 mL/min. UV detection was with an Agilent Diode Array Detector model G1315A spectrometer at 270 and 310 nm. Proton (1H) and carbon (13C) NMR spectra were recorded at 300 and 75.5 MHz, respectively, on a Varian Gemini spectrometer, using TMS or the solvent peak as an internal standard. Column chromatography was conducted using either silica gel (Geduran, 60, mesh 40-63 μm) or prepacked RediSep columns (Teledyne Isco, Inc., Lincoln, NE USA) on an Isco CombiFlash Optix10 instrument. Elemental analyses were performed by Atlantic Microlab, Inc. (Norcross, GA USA). HRMS analyses were performed at the W.M. Keck Foundation Biotechnology Resource Laboratory (Yale University, New Haven, CT USA) or at the National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK), NIH. Isoniazid, Rifampicin, Mtz and methylene blue were obtained from Sigma-Aldrich. PA-824 was prepared as described in US patent 5,668,127. All stocks were made in 20 mM DMSO. Compound 5 was purchased from TimTec (Newark DE USA).
(S)-1-(tert-Butyldimethylsilyloxy)-3-(2-chloro-4-nitro-1H-imidazol-1-yl)propan-2-ol (7a) and (S)-1-(tert-Butyldimethylsilyloxy)-3-(2-chloro-5-nitro-1H-imidazol-1-yl)propan-2-ol (7b)
A mixture of 5 (2.4 g, 16.2 mmol), 6 (4.6 g, 24.4 mmol) and anhydrous K2CO3 (0.34 g, 2.4 mmol), in absolute EtOH (16 mL) was heated in a sealed tube at 70°C for 20 h. The solvent and excess 6 were then removed under reduced pressure. CH2Cl2 (50 mL) was added to the residue, and filtered through a bed of Celite® (2 cm thick). The filtrate was then concentrated in vacuo to give a brown oil, which was purified via column chromatography (silica gel, 25% to 50% EtOAc in hexanes) to give 7a as a white crystalline solid (3.9 g, 66%). Mp = 116.5-118.5°C, 1H NMR (CDCl3) δ 0.10 (s, 6 H), 0.90 (s, 9 H), 2.74 (d, br, J = 5.0 Hz, 1 H, alcohol-H), 3.61 (dd, J = 13.5, 3.7 Hz, 1 H), 3.71 (dd, J = 13.5, 4.2 Hz, 1 H); 13C NMR (CDCl3) δ −5.7, 17.9, 25.6, 50.6, 63.9, 69.8, 122.6, 132.5, 144.8; LC-MS m/z 336.4 [M + H+]; and 7b as a colorless oil (1.1 g, 20%). 1 H NMR (CDCl3) δ −0.13 (s, 3 H), 0.04 (s, 3 H), 0.87 (s, 9 H), 3.60-3.64 (m, 2 H), 3.99-4.25 (m, 3 H), 7.80 (s, 1 H); 13C NMR (CDCl3) δ −4.9, 18.0, 25.8, 50.8, 63.8, 70.7, 122.4, 132.9, 145.6; LC-MS m/z 336.4 [M + H+].
1-((2S)-3-(tert-Butyldimethylsilyloxy)-2-(tetrahydro-2H-pyran-2-yloxy)propyl)-2-chloro-5-nitro-1H-imidazole (8)
A mixture of 7b (0.78 g, 2.3 mmol), DHP (0.43 mL, 4.6 mmol) and PPTS (0.88 g, 3.5 mmol) in CH2Cl2 (20 mL) was stirred under inert atmosphere at rt for 2 days. Saturated NaHCO3 aqueous solution (20 mL) was added to the reaction mixture. CH2Cl2 layer was separated, and the aqueous layer extracted with CH2Cl2 (2 × 15 mL). The organic extracts and the original CH2Cl2 layer were combined, washed with brine (30 mL), and dried over MgSO4. The solvent was then removed under reduced pressure to give a pale brown oil, which was purified via column chromatography (silica gel, EtOAc/hexanes = 3/1) to give 8 (a mixture of two diastereomers) as a colorless oil (0.68 g, 70%). 1H NMR (CDCl3) δ −0.12 (d, J = 4.5 Hz, 3 H), −0.04 (d, J = 1.8 Hz, 3 H), 0.85 (s, 4.5 H), 0.86 (s, 4.5 H), 1.50-1.83 (m), 3.25-3.31 (m, 1 H), 3.44 (dd, J = 12.3, 2.1 Hz, 1 H), 3.49-3.56 (m, 1 H), 3.63 (dd, J = 10.0, 7.0 Hz, 1 H), 3.73-3.88 (m, 2 H), 3.99-4.15 (m, 3 H), 4.20-4.32 (m, 1 H), 4.54-4.59 (m, 1 H), 7.84 (s, 0.5 H), 7.90 (s, 0.5 H). LC-MS m/z 419.5 [M + H+].
(S)-6,7-Dihydro-3-nitro-6-(tetrahydro-2H-pyran-2-yloxy)-5H-imidazo[2,1-b][1,3]oxazine (9)
TBAF (1.0 M in THF, 3.0 mL, 3.0 mmol) was added dropwise to a stirred solution of 8 (0.70 g, 1.6 mmol) in anhydrous THF (10 mL) at room temperature. Then the reaction mixture was further stirred at room temperature under inert atmosphere for 18 h. The solvent was removed under reduced pressure. Aqueous NaHCO3 saturated solution (10 mL) was added to the brown syrupy residue, and the mixture was extracted with CH2Cl2 (3 × 10 mL). The organic extracts were combined, washed with brine, dried over MgSO4 and the solvent was removed under reduced pressure to give a pale brown syrup, which was purified via column chromatography (silica gel, EtOAc:Hex = 3:1 to straight EtOAc) to give 9 as a white crystalline solid (0.10 g, 23%). Mp = 118.0-119.1°C; 1H NMR (CDCl3) δ showed a mixture of diastereomers; LC-MS m/z 270.0 [M + H+].
(S)-6,7-Dihydro-3-nitro-5H-imidazo[2,1-b][1,3]oxazin-6-ol (10b)
A mixture of 9 (0.22 g, 0.81 mmol), glacial acetic acid (4.0 mL, 69 mmol), tetrahydrofuran (2.0 mL) and H2O (1.0 mL) was heated at 60°C for 18 h. The solvent and extra reagents were removed under reduced pressure. The residue was triturated with CH2Cl2 to give 10b as a yellow crystalline solid (0.12 g, 75%). Mp = 174.0-175.2 °C; 1H NMR (CD3OD) δ 3.76 (dd, J = 12.9, 3.6 Hz, 1 H), 3.98 (dd, J = 12.9, 3.0 HZ, 1 H), 4.24 (dd, J = 10.2, 6.6 Hz, 1 H), 4.40 (dd, J = 10.2, 9.0 Hz, 1 H), 5.44 (dddd, J = 12.6, 9.6, 6.6, 3.3 Hz, 1 H), 7.85 (s, 1 H); 13C (CD3OD) δ 46.2, 62.9, 89.8, 115.4; LC-MS m/z 185.9 [M + H+].
(S)-6-(4-(Trifluoromethoxy)benzyloxy)-6,7-dihydro-3-nitro-5H-imidazo[2,1-b][1,3]oxazine (11)
At −78°C under argon atmosphere, NaH (60% dispersion in mineral oil, 9.5 mg, 0.24 mmol) was added to a stirred solution of 10b (40.0 mg, 0.21 mmol), 4-trifluromethoxybenzyl bromide (66.0 mg, 0.26 mmol) and tetrabutylammonium iodide (3.7 mg, 0.01 mmol) in anhydrous DMF (5 mL). The mixture was then warmed to rt, and further stirred at rt for 18 h. The reaction was quenched with methanol (0.5 mL). The solvent was removed under reduced pressure. CH2Cl2 (10 mL) was added to the residue and filtered to remove the inorganic salts. The CH2Cl2 solution was then concentrated in vacuo. The yellow gum residue was purified via column chromatography (0-2% MeOH in CH2Cl2) to give a yellow solid, which was further purified via preparative TLC (eluted with 5% MeOH in CH2Cl2) to give 11 as a yellow crystalline solid (10 mg, 13%). [α]D20 = +7.4 (c 0.37, MeOH); 1H NMR (CDCl3) δ 3.78 (dd, J = 11.1, 3.3 Hz, 1 H), 3.89 (dd, J = 11.1, 3.9 Hz, 1 H), 4.27 (dd, J = 9.9, 6.6 Hz, 1 H), 4.34 (dd, J = 9.9, 8.7 Hz, 1 H), 4.60 (s, 2 H), 5.39-5.47 (m, 1 H), 7.18-7.31 (m, 1,4-disubstituted pattern, 4 H), 7.54 (s, 1 H); 13C (CDCl3) δ 45.5, 69.4, 73.2, 85.3, 112.7, 121.3, 129.3, 135.8, 147.5, 149.2, 156.7; LC-MS m/z 360.1 [M + H+].
2-(2-Methyl-4-nitro-1H-imidazol-1-yl)ethanol (14)
A mixture of 2-methyl-4-nitroimidazole (13, 254 mg, 2.0 mmol), 2-bromoethanol (170 μL, 2.4 mmol), potassium carbonate (0.55 g, 4.0 mmol), tetrabutylammonium iodide (15 mg, 0.04 mmol) and acetonitrile (2 mL) was stirred at 75°C for 2 h. Silica gel (2 g) was added and the solvent was removed under reduced pressure. The residue was purified twice via column chromatography (0-5% MeOH in CH2Cl2) to give a white solid (76 mg, 22% yield). Mp = 126.7–127.8 °C (lit. 130-131 °C)22; 1H NMR (CD3OD) δ 2.44 (s, 3 H), 3.84 (t, J = 5.1 Hz, 2 H), 4.12 (t, J = 5.1 Hz, 2 H), 8.07 (s, 1 H).
(S)-6,7-Dihydro-6-methoxy-2-nitro-5H-imidazo[2,1-b][1,3]oxazine (15)
NaH (60% dispersion in mineral oil, 24 mg, 0.60 mmol) was added to a stirred solution of 10a (93.0 mg, 0.50 mmol) and CH3I (40.5 μL, 0.65 mmol) in anhydrous DMF (2 mL) at −78°C. The reaction mixture was then warmed to room temperature and stirred under an inert atmosphere for 18 h. The reaction was quenched with methanol (1 mL). Silica gel was added to the mixture, and the solvent was removed under reduced pressure. The residue was purified via column chromatography (silica gel, 2-5% MeOH in CHCl3) to give 15 as a yellow solid (57 mg, 57%). Mp: 80.0-80.9°C; 1H NMR (CDCl3) δ 3.48 (s, 3 H), 3.91-3.96 (m, 1 H), 4.14 (dt, J = 12.9, 2.4 Hz, 1 H), 4.21 (dd, J = 12.9, 3.9 Hz, 1 H), 4.34 (d, br, J = 12.0 Hz, 1 H), 4.63 (ddd, J =12.0, 3.6, 2.1 Hz, 1 H), 7.42 (s, 1 H); 13C NMR (CDCl3) δ 47.6, 56.7, 68.3, 69.7, 117.4, 148.3; LC-MS m/z 200.1 [M + H+].
1-(2-(4-(Trifluoromethoxy)benzyloxy)ethyl)-2-methyl-5-nitro-1H-imidazole (16)
NaH (60% dispersion in mineral oil, 44 mg, 1.1 mmol) was added to a stirred mixture of 1 (Mtz, 171 mg, 1.0 mmol), 4-trifluoromethoxybenzyl bromide (306 mg, 1.2 mmol) and TBAI (18 mg, 0.05 mmol) in anhydrous DMF (5 mL) at −78°C. The reaction mixture was then warmed to rt, and stirred at room temperature under Ar for 18 h. The reaction was quenched with MeOH (10 drops). The solvent was removed under reduced pressure. H2O was added to the residue. The mixture was extracted with CH2Cl2 (3 × 30 mL). The organic extracts were combined, washed with H2O, and dried over MgSO4. The solvent was then removed under reduced pressure to give a brown oil, which was purified via preparative TLC (eluted with 5% MeOH in CH2Cl2) to give 16 as yellow solid (113 mg, 33%). Mp = 64.5-65.5°C; 1H NMR (CDCl3) δ 2.50 (s, 3H), 3.79 (t, J = 4.8 Hz, 2H), 4.42 (s, 2H), 4.51 (t, J = 4.8 Hz, 2H), 7.13-7.19 (m, 4H), 7.94 (s, 1H); 13C NMR (CDCl3) δ 14.8, 46.7, 68.9, 72.6, 121.2, 128.9, 133.3, 136.3, 149.0, 151.9; LC-MS m/z 346.1 [M + H+].
1-(2-(4-(Trifluoromethoxy)benzyloxy)ethyl)-2-methyl-4-nitro-1H-imidazole (17)
NaH (60% dispersion in mineral oil, 40 mg, 1.0 mmol) was added to a stirred mixture of 14 (140 mg, 0.82 mmol), 4-trifluoromethoxybenzyl bromide (255 mg, 1.0 mmol) and TBAI (18 mg, 0.05 mmol) in anhydrous DMF (5 mL) at −78°C. The reaction mixture was then warmed to room temperature and stirred under Ar for 18 h. The reaction was quenched with MeOH (2 drops). The solvent was removed under reduced pressure. H2O (30 mL) was added to the residue. The mixture was extracted with CH2Cl2 (3 × 25 mL). The organic extracts were combined, washed with H2O (2 × 100 mL), and dried over MgSO4. The solvent was then removed under reduced pressure to give a brown solid, which was purified via preparative TLC (eluted with EtOAc/hexanes = 3/1) to give 17 as a yellow solid (111 mg, 39%). Mp = 86.9-88.5°C; 1H NMR (CDCl3) δ 1.93 (s, 3 H), 3.24 (t, J = 5.0 Hz, 2 H), 3.62 (t, J = 5.0 Hz, 2 H), 4.00 (s, 2 H), 6.66-6.77 (m, 1,4-disubstituted benzene pattern); 13C NMR (CDCl3) δ 13.2, 47.2, 68.5, 72.5, 120.5, 121.1, 129.0, 136.0, 145.4, 146.4, 148.8; LC-MS m/z 346.1 [M + H+].
1-(2-(tert-Butyldimethylsilyloxy)ethyl)-2-chloro-4-nitro-1H-imidazole (18) and 1-(2-(tert-Butyldimethylsilyloxy)ethyl)-2-chloro-5-nitro-1H-imidazole (28)
A solution of 5 (255 mg, 1.73 mmol), BrCH2CH2OTBS (605 mg, 2.53 mmol), and K2CO3 (99 mg, 0.72 mmol) in MeOH (5 mL) was heated in a sealed tube at 75°C for 18 h. The reaction mixture was quenched with H2O, and extracted with EtOAc (2x). The combined organic layers were dried with MgSO4, filtered, and concentrated. Chromatography on silica gel (EtOAc/hexanes = 1/1) afforded 18 as a white solid (130 mg, 0.424 mmol, 25%) and 28 as a white solid (19 mg, 0.061 mmol, 3.5%). 18: Mp = 87.5-88.6°C; 1H NMR (CDCl3) δ −0.07 (s, 6H), 0.78 (s, 9H), 3.86 (t, J = 4.8 Hz, 2H), 4.13 (t, J = 4.8 Hz, 2H), 7.83 (s, 1H); 13C NMR (CDCl3) δ −5.9, 17.9, 25.5, 50.0, 60.8, 121.9, 132.2, 145.2; HRMS (ESMS) calcd for C11H21ClN3O3Si [M + H+] 306.1041, found 306.1035; presence of 2D-NOESY correlationships: aromatic H with 2 CH2s; λmax = 300 nm. 28: Mp = 47.0-47.8 °C; 1H NMR (CDCl3) δ −0.07 (s, 6H), 0.80 (s, 9H), 3.89 (t, J = 5.3 Hz, 2H), 4.60 (t, J = 5.3 Hz, 2H), 7.91 (s, 1H); 13C NMR (CDCl3) δ −5.8, 18.0, 25.6, 49.2, 61.1, 132.1, 138.8, 139.1; HRMS (ESMS) calcd for C11H21ClN3O3Si [M + H+] 306.1041, found 306.1035; absence of 2D-NOESY correlationships: aromatic H with 2 CH2s; λmax = 310nm.
2-(2-Methoxy-4-nitro-1H-imidazol-1-yl)ethanol (19)
To a solution of 18 (438 mg, 1.43 mmol) in MeOH (10 mL) was added NaOMe (619 mg, 11.5 mmol) at room temperature. The reaction mixture was stirred at room temperature for 24 h and concentrated. Chromatography on silica gel (MeOH/CH2Cl2 = 1/20) afforded 19 as a white solid (164 mg, 0.874 mmol, 61%). Mp = 132-133°C; 1H NMR (MeOH-d4) δ 3.79 (t, J = 5.3 Hz, 2H), 3.97 (t, J = 5.3 Hz, 2H), 4.07 (s, 3H), 7.85 (s, 1H); 13C NMR (MeOH-d4) δ 48.7, 58.6, 61.0, 120.0, 143.5, 152.9; HRMS (ESMS) calcd for C6H10N3O4 [M + H+] 188.0671, found 188.0688.
1-(2-(4-(Trifluoromethoxy)benzyloxy)ethyl)-2-methoxy-4-nitro-1H-imidazole (20)
A mixture of 19 (19.6 mg, 0.105 mmol), 4-trifluoromethoxybenzyl bromide (40.1 mg, 0.157 mmol), and nBu4NI (4.6 mg, 0.012 mmol) in DMF (1 mL) was prepared under argon atmosphere. To this mixture 60% NaH (5.6 mg, 0.14 mmol) was added at −78°C. The reaction mixture was stirred at −78°C for 30 min and then at room temperature for 14 h, quenched with H2O, and extracted with EtOAc (2x). The combined organic layers were dried with MgSO4, filtered, and concentrated. Chromatography on silica gel (EtOAc/hexanes = 2/1) afforded 20 as a slightly yellow oil (22 mg, 0.061 mmol, 58%). 1H NMR (CDCl3) δ 3.67 (t, J = 5.0 Hz, 2H), 3.99 (t, J = 5.0 Hz, 2H), 4.04 (s, 3H), 4.48 (s, 2H), 7.14 (d, J = 8.4 Hz, 2H), 7.23 (d, J = 8.4 Hz, 2H), 7.57 (s, 1H); 13C NMR (CDCl3) δ 44.8, 57.7, 67.5, 72.2, 117.9, 118.6, 120.9, 122.0, 128.8, 135.9, 142.5, 148.6, 150.7; HRMS (ESMS) calcd for C14H15F3N3O5 [M + H+] 362.0964, found 362.0972.
(R)-2-Bromo-1-(2-(tert-butyldimethylsilyloxy)but-3-enyl)-5-nitro-1H-imidazole (23a) and (R)-1-(2-Bromo-4-nitro-1H-imidazol-1-yl)but-3-en-2-ol (31)
A mixture of 21 (2.0 g, 10.4 mmol), (R)-vinyloxirane (22, 1.00 g, 13.5 mmol), and K2CO3 (560 mg, 4.05 mmol) in MeOH (10 mL) was heated in a sealed tube at 70°C for 14 h. The reaction mixture was quenched with H2O and extracted with EtOAc (3x). The combined organic layers were dried with anhydrous MgSO4, filtered, and concentrated. To the crude organic mixture (2.55 g) in CH2Cl2 (50 mL) was added TBSCl (2.93 g, 19.5 mmol) and imidazole (1.33 g, 19.5 mol) at room temperature. The reaction mixture was stirred at rt for 1 h, quenched with H2O, and extracted with CH2Cl2 (2x). The combined organic layers were dried with anhydrous MgSO4, filtered, and concentrated. Chromatography on silica gel (EtOAc/hexanes = 1/2-1/1) afforded (R)-23a (611 mg, 1.62 mmol, 16%) as a slightly yellow oil in addition to the 4-NO2 desTBS product (31, 1.25 g, 4.76 mmol, 48%) as a viscous yellow oil. (R)-23a: [α]D20 = −26.5 (c 1.89, CHCl3); 1H NMR (CDCl3) δ 0.01 (s, 6H), 0.83 (s, 9H), 3.90-4.00 (m, 2H), 4.93-4.98 (m, 1H), 5.26 (dd, J = 17.3, 1.4 Hz, 1H), 5.44 (dd, J = 10.4, 1.4 Hz, 1H), 5.98 (ddd, J = 17.3, 10.4, 6.0 Hz, 1H), 7.92 (s, 1H); 13C NMR (CHCl3) δ −5.75, −5.67, 18.0, 25.6, 62.5, 64.2, 119.5, 120.4, 120.7, 131.4, 147.3; HRMS (ESMS) calcd for C13H23BrN3O3Si [M + H+] 376.0692, found 376.0638. 31: [α]D20 = −6.86 (c 1.69, CHCl3); 1H NMR (CDCl3) δ 3.95-4.03 (m, 1H), 4.16 (dd, J = 14.4, 3.3 Hz, 1H), 4.49-4.57 (m, 1H), 5.27 (d, J = 10.5 Hz, 1H), 5.39 (d, J = 17.1 Hz, 1H), 5.86 (ddd, J = 16.8, 10.8, 6.0 Hz, 1H), 7.98 (s, 1H); 13C NMR (CDCl3) δ 53.8, 71.2, 118.9, 120.7, 123.3, 135.9, 146.6; HRMS (ESMS) calcd for C7H9N3O3Br [M + H+] 261.9827, found 261.9829.
(S)-2-Bromo-1-(2-(tert-butyldimethylsilyloxy)but-3-enyl)-5-nitro-1H-imidazole (23b)
This compound was prepared in the same manner as 23a, using (S)-vinyloxirane. Yield: 23b (15%) and the 4-NO2-desTBS product (45%). For compound 23b: [α]D20 = +23.7 (c 0.58, CHCl3); 1H NMR (CDCl3) δ −0.04 (s, 6H), 0.79 (s, 9H), 3.88-3.98 (m, 2H), 4.90-4.98 (m, 1H), 5.23 (d, J = 17.3, 1H), 5.40 (d, J = 10.4, 1H), 5.94 (ddd, J = 17.3, 10.4, 6.0 Hz, 1H), 7.92 (s, 1H); 13C NMR (CDCl3) δ −5.9, −5.9, 17.9, 25.5, 62.4, 64.1, 119.5, 120.4, 120.6, 131.3, 147.2; HRMS (ESMS) calcd for C13H22BrN3O3Si [M + H+] 376.0692, found 376.0704.
(R)-1-(2-(tert-Butyldimethylsilyloxy)but-3-enyl)-5-nitro-2-vinyl-1H-imidazole (24a)
A mixture of (R)-23a (300 mg, 0.797 mmol), tributylvinylstannane (0.255 mL, 0.874 mmol), Pd(PPh3)4 (37 mg, 0.032 mmol), and LiCl (33 mg, 0.80 mmol) in DMF (2 mL) was heated under microwave at 120°C for 10 min. The reaction mixture was concentrated under vacuum. Chromatography on silica gel (EtOAc/hexanes = 1/2) afforded (R)-24a (144 mg, 0.446 mmol, 56%) as a slightly yellow oil. [α]D20 = −13.7 (c 0.37, CHCl3); 1H NMR (CDCl3) δ −0.06 (s, 3H), −0.05 (s, 3H), 0.78 (s, 9H), 3.87 (dd, J = 10.8, 6.6 Hz, 1H), 3.97 (dd, J = 10.8, 4.1 H, 1H), 4.84-4.90 (m, 1H), 5.18 (dd, J = 17.4, 1.5 Hz, 1H), 5.39 (dd, J = 10.4, 1.5 Hz, 1H), 5.56 (dd, J = 10.8, 1.5 Hz, 1H), 5.95 (ddd, J = 17.4, 10.8, 5.7 Hz, 1H), 6.36 (dd, J = 17.1, 1.5 Hz, 1H), 6.55 (dd, J = 17.1, 10.8 Hz, 1H), 7.85 (s, 1H); 13C NMR (CHCl3) δ −5.84, −5.81, 17.9, 25.5, 60.3, 64.7, 118.6, 120.1, 121.3, 123.2, 132.0, 144.9, 147.1; HRMS (ESMS) calcd for C15H26N3O3Si [M + H+] 324.1743, found 324.1745.
(S)-1-(2-(tert-Butyldimethylsilyloxy)but-3-enyl)-5-nitro-2-vinyl-1H-imidazole (24b)
This compound was prepared in the same manner as (R)-24a. Yield: (S)-24b (53%) as a slightly yellow oil. [α]D20 = +14.6 (c 2.23, CHCl3); 1H NMR (CDCl3) δ −0.04 (s, 3H), −0.03 (s, 3H), 0.80 (s, 9H), 3.85-3.91 (m, 2H), 4.82-4.92 (m, 1H), 5.20 (dd, J = 17.4, 1.5 Hz, 1H), 5.41 (dd, J = 10.5, 1.5 Hz, 1H), 5.59 (dd, J = 10.5, 1.5 Hz, 1H), 5.91-6.02 (m, 1H), 6.39 (dd, J = 17.1, 1.5 Hz, 1H), 6.52-6.61 (m, 1H), 7.86 (s, 1H); 13C NMR (CHCl3) δ −5.79, −5.75, 18.0, 25.5, 60.3, 64.7, 118.6, 120.2, 121.3, 123.3, 132.0, 144.9, 147.2; HRMS (ESMS) calcd for C15H26N3O3Si [M + H+] 324.1743, found 324.1749.
(R)-6-(tert-Butyldimethylsilyloxy)-3-nitro-5,6-dihydroimidazo[1,2-a]pyridine (25a)
To a solution of (R)-24a (81 mg, 0.25 mmol) in CH2Cl2 (10 mL) was added Hoveyda-Grubbs 2nd generation catalyst (22 mg, 0.035 mmol). The reaction mixture was stirred at 60°C for 2.5 h. Chromatography on preparative TLC (EtOAc/Hx = 1/1) afforded (R)-25a (54 mg, 0.18 mmol, 72%) as a slightly brown solid. [α]D20 = −17.8 (c 0.48, CHCl3); Mp = 106-108°C; 1H NMR (CDCl3) δ 0.09 (d, J = 0.9 Hz, 6H), 0.92 (s, 9H), 3.50 (dd, J = 10.5, 9.0 Hz, 1H), 4.11 (dd, J = 10.5, 4.2 Hz, 1H), 4.82-4.87 (m, 1H), 6.76 (s, 2H), 8.06 (s, 1H); 13C NMR (CHCl3) δ −5.3, −5.2, 18.4, 26.0, 63.9, 66.4, 118.0, 122.9, 132.3, 138.9, 153.7; HRMS (ESMS) calcd for C13H22N3O3Si [M + H+] 296.1430, found 296.1423.
(S)-6-(tert-Butyldimethylsilyloxy)-3-nitro-5,6-dihydroimidazo[1,2-a]pyridine (25b)
This compound was prepared in the same manner as (R)-25a. Yield: (S)-25b (72%) as a slightly brown solid. [α]D20 = +19.9 (c 1.36, CHCl3); Mp = 108-109°C; 1H NMR (CDCl3) δ 0.09 (d, J = 0.9 Hz, 6H), 0.92 (s, 9H), 3.50 (dd, J = 10.5, 9.0 Hz, 1H), 4.11 (dd, J = 10.5, 4.2 Hz, 1H), 4.82-4.87 (m, 1H), 6.76 (s, 2H), 8.06 (s, 1H); 13C NMR (CHCl3) δ −5.6, −5.5, 18.1, 25.7, 63.3, 66.2, 117.8, 122.5, 138.8, 150.3, 153.7; HRMS (ESMS) calcd for C13H22N3O3Si [M + H+] 296.1430, found 296.1429.
(R)-6-(tert-Butyldimethylsilyloxy)-3-nitro-5,6,7,8-tetrahydroimidazo[1,2-a]pyridine (26a)
A solution of (R)-25a (58 mg, 0.20 mmol) in a t-BuOH (1.5 mL) and THF (0.5 mL) was prepared and degassed by freezing and thawing (3x). To the mixture was added Wilkinson's catalyst (17 mg, 0.018 mmol) and degassed by freezing and thawing (1x). Under a hydrogen balloon atmosphere the reaction mixture was stirred at rt for 14 h. Chromatography on preparative TLC (EtOAc/hexanes = 1/1) afforded (R)-26a (34 mg, 0.11 mmol, 57%) as a slightly yellow solid in addition to the starting material ((R)-25a, 22 mg, 0.074 mmol, 37%). [α]D20 = 0.00 (c 0.31, CHCl3); Mp = 68.0-68.9°C; 1H NMR (CDCl3) δ 0.04 (s, 3H), 0.06 (s, 3H), 0.88 (s, 9H), 2.23-2.34 (m, 1H), 2.64-2.76 (m, 1H), 2.82-3.06 (m, 2H), 3.66 (dd, J = 10.8, 7.5 Hz, 1H), 3.89 (dd, J = 10.8, 3.6 Hz, 1H), 4.39-4.47 (m, 1H), 7.81 (s, 1H); 13C NMR (CHCl3) δ −5.6, −5.5, 18.4, 23.1, 25.7, 27.9, 60.1, 65.4, 115.7, 128.6, 132.0, 152.9; HRMS (ESMS) calcd for C13H24N3O3Si [M + H+] 298.1587, found 298.1594.
(S)-6-(tert-Butyldimethylsilyloxy)-3-nitro-5,6,7,8-tetrahydroimidazo[1,2-a]pyridine (26b)
This compound was prepared in the same manner as (R)-26a. Yield: (S)-26b (55%) as a slightly yellow solid in addition to the starting material ((S)-25b, 12 mg, 0.039 mmol, 42%). [α]D20 = +0.01 (c 0.75, CHCl3); Mp = 67.2-68.0°C; 1H NMR (CDCl3) δ 0.03 (s, 3H), 0.05 (s, 3H), 0.88 (s, 9H), 2.23-2.34 (m, 1H), 2.63-2.75 (m, 1H), 2.84-3.02 (m, 2H), 3.66 (dd, J = 10.8, 7.5 Hz, 1H), 3.89 (dd, J = 10.8, 3.6 Hz, 1H), 4.39-4.47 (m, 1H), 7.81 (s, 1H); 13C NMR (CHCl3) δ −5.61, −5.56, 18.1, 23.0, 25.7, 27.9, 60.0, 65.4, 115.7, 150.5, 152.8; HRMS (ESMS) calcd for C13H24N3O3Si [M + H+] 298.1587, found 298.1595.
(R)-5,6,7,8-Tetrahydro-3-nitroimidazo[1,2-a]pyridin-6-ol (27a)
To a solution of (R)-26a (22 mg, 0.074 mmol) in THF (1 mL) was added 1.0 M TBAF in THF (0.10 mL, 0.10 mmol) at rt. The reaction mixture was stirred at rt for 10 min. Chromatography on preparative TLC (MeOH/EtOAc = 1/9) afforded (R)-27a (9.4 mg, 0.051 mmol, 69%) as a white solid. [α]D23 = −10.7 (c 0.43, MeOH); Mp = 163.5-164.1°C; 1H NMR (MeOH-d4) δ 2.34-2.45 (m, 1H), 2.66-2.78 (m, 1H), 2.83-3.02 (m, 2H), 3.66 (dd, J = 12.0, 6.3 Hz, 1H), 3.92 (dd, J = 11.6, 3.5 Hz, 1H), 4.47-4.54 (m, 1H), 8.09 (s, 1H); 13C NMR (MeOH-d4) δ 23.9, 29.1, 61.9, 64.6, 117.8, 151.2, 155.2; HRMS (ESMS) calcd for C7H10N3O3 [M + H+] 184.0722, found 184.0724.
(S)-5,6,7,8-Tetrahydro-3-nitroimidazo[1,2-a]pyridin-6-ol (27b)
This compound was prepared in the same manner as 27a. Yield: (S)-27b (91%) as a white solid. [α]D20 = +8.3 (c 0.29, MeOH); Mp = 159.8-161.0°C; 1H NMR (MeOH-d4) δ 2.34-2.46 (m, 1H), 2.63-2.80 (m, 1H), 2.82-3.02 (m, 2H), 3.66 (dd, J = 12.0, 6.3 Hz, 1H), 3.92 (dd, J = 11.7, 3.3 Hz, 1H), 4,47-4.53 (m, 1H), 8.09 (s, 1H); 13C NMR (MeOH-d4) δ 24.0, 29.1, 61.9, 64.9, 117.8, 151.2, 155.2; HRMS (ESMS) calcd for C7H10N3O3 [M + H+] 184.0722, found 184.0723.
1-(2-(tert-Butyldimethylsilyloxy)ethyl)-2-methoxy-5-nitro-1H-imidazole (29)
To a solution of 28 (29 mg, 0.095 mmol) was added NaOMe (50 mg, 0.93 mmol) at rt. The reaction mixture was stirred at rt for 15 h and concentrated. Chromatography on silica gel (EtOAc/Hexanes = 1/1) afforded 29 as a slightly yellow oil (17 mg, 0.055 mmol, 58%) in addition to 30 (2.7 mg, 0.014 mmol, 15%). 1H NMR (CDCl3) δ −0.07 (s, 6H), 0.80 (s, 9H), 3.89 (t, J = 5.3 Hz, 2H), 4.60 (t, J = 5.3 Hz, 2H), 7.91 (s, 1H); 13C NMR (CDCl3) δ −5.8, 18.0, 25.6, 46.2, 57.3, 61.1, 131.0, 135.3, 155.4; HRMS (ESMS) calcd for C12H24N3O4Si [M + H+] 302.1536, found 302.1538.
2-(2-Methoxy-5-nitro-1H-imidazol-1-yl)ethanol (30)
To a solution of 29 (17 mg, 0.055 mmol) in THF (1 mL) was added 1.0 M TBAF in THF (0.070 mL, 0.070 mmol) at rt. The reaction mixture was stirred at rt for 10 min and concentrated. Chromatography with preparative TLC gave 30 as a white solid (7.1 mg, 0.038 mmol, 69%). Mp = 99.0-99.8°C; 1H NMR (CDCl3) δ 3.91 (t, J = 5.6 Hz, 2H), 4.15 (s, 3H), 4.40 (t, J = 5.6 Hz, 2H), 7.78 (s, 1H); 13C NMR (CDCl3) δ 46.3, 57.9, 61.3, 131.5, 135.5, 155.4; LC-MS m/z 188.1 [M + H+].
1-((R)-2-(4-(Trifluoromethoxy)benzyloxy)but-3-enyl)-2-bromo-4-nitro-1H-imidazole (32)
A mixture of 31 (166 mg, 0.633 mmol), 4-trifluoromethoxybenzyl bromide (250 mg, 0.95 mmol), and nBu4NI (23 mg, 0.063 mmol) in DMF (5 mL) was prepared under argon atmosphere. To this mixture 60% NaH (30 mg, 0.76 mmol) was added at −78°C. The reaction mixture was stirred at 0°C for 30 min and then at rt for 14 h, quenched with H2O, and extracted with EtOAc (2x). The combined organic layers were dried with MgSO4, filtered, and concentrated. Chromatography on silica gel (EtOAc/hexanes = 3/4) afforded 32 as a slightly yellow oil (171 mg, 0.392 mmol, 62%). [α]D20 = −18.9 (c 1.95, CHCl3); 1H NMR (CDCl3) δ 4.26-4.40 (m, 3H), 4.48 (d, J = 12.0 Hz, 1H), 4.78 (d, J = 12.0 Hz, 1H), 5.58-5.63 (m, 2H), 5.87-5.98 (m, 1H), 7.31 (d, J = 8.7 Hz, 2H), 7.38 (d, J = 8.7 Hz, 2H), 8.09 (s, 1H); 13C NMR (CDCl3) δ 52.3, 69.4, 77.6, 118.4, 120.4, 120.7, 121.1, 121.9, 122.9, 128.8, 133.0, 135.8, 146.6, 148.46, 148.48; HRMS (ESMS) calcd for C15H14BrF3N3O4 [M + H+] 436.0120, found 436.0132.
1-((R)-2-(4-(Trifluoromethoxy)benzyloxy)but-3-enyl)-4-nitro-2-vinyl-1H-imidazole (33)
A mixture of 32 (741 mg, 1.70 mmol), tributyl(vinyl)stannane (0.55 mL, 1.9 mmol), Pd(PPh3)4 (79 mg, 0.068 mmol), and LiCl (72 mg, 1.70 mmol) in DMF (5 mL) was prepared. The reaction mixture was heated under microwave at 120 °C for 15 min. The reaction mixture was concentrated under vacuum. Chromatography on silica gel (EtOAc/hexanes = 1/3-1/1) afforded 33 as a slightly yellow oil (590 mg, 1.54 mmol, 91%). [α]D20 = −13.0 (c 0.59, CHCl3); 1H NMR (CDCl3) δ 3.96-4.13 (m, 3H), 4.25 (d, J = 12.0 Hz, 1H), 4.57 (d, J = 12.3 Hz, 1H), 5.37-5.46 (m, 2H), 5.56 (d, J = 10.8 Hz, 1H), 5.65-5.76 (d, J = 10.8 Hz, 1H), 6.37 (dd, J = 17.1, 1.5 Hz, 1H), 6.53 (dd, J = 17.1, 11.1 Hz, 1H), 7.08-7.17 (m, 4H), 7.74 (s, 1H); 13C NMR (CDCl3) δ 50.8, 69.6, 78.5, 118.6, 120.9, 121.0, 121.2, 121.4, 122.0, 123.2, 129.0, 133.4, 135.7, 144.8, 146.9, 148.76, 148.78; HRMS (ESMS) calcd for C17H17F3N3O4 [M + H+] 384.1171, found 384.1167.
(R)-6-(4-(Trifluoromethoxy)benzyloxy)-5,6-dihydro-2-nitroimidazo[1,2-a]pyridine (34)
A mixture of 33 (560 mg, 1.54 mmol) and Grubbs' 2nd generation catalyst (130 mg, 0.15 mmol) in CH2Cl2 (5 mL) was heated in a sealed tube at 55°C for 15 h. The reaction mixture was concentrated under vacuum. Chromatography on silica gel (EtOAc/hexanes = 1/1) afforded 34 as a gray-white solid (320 mg, 0.901 mmol, 59%). [α]D20 = −189.6 (c 0.51, CHCl3); Mp = 101.7-102.8°C; 1H NMR (CDCl3) δ 4.14-4.22 (m, 1H), 4.34-4.42 (m, 2H), 4.59 (s, 2H), 6.43-6.48 (m, 1H), 6.65 (d, J = 10.2 Hz, 1H), 7.14 (d, J = 8.4 Hz, 2H), 7.30 (d, J = 8.7 Hz, 2H), 7.77 (s, 1H); 13C NMR (CDCl3) δ 48.3, 67.6, 69.7, 118.5, 120.2, 120.7, 120.9, 121.9, 129.0, 130.2, 135.8, 141.0, 147.3, 148.7; HRMS (ESMS) calcd for C15H13F3N3O4 [M + H+] 356.0858, found 356.0851.
(R)-6-(4-(Trifluoromethoxy)benzyloxy)-5,6,7,8-tetrahydro-2-nitroimidazo[1,2-a]pyridine (35)
A solution of 34 (144 mg, 0.405 mmol) in THF:tBuOH (2mL:6mL) was degassed by freezing and thawing (3x). To this mixture was added Wilkinson's catalyst (72 mg, 0.078 mmol) at rt. The reaction mixture was degassed by freezing and thawing (1x). The reaction mixture was stirred at rt under a H2 (g) balloon for 16 h, and concentrated under vacuum. Chromatography on silica gel (EtOAc/hexanes = 1/1) afforded 35 as a yellow oil (73 mg, 0.20 mmol, 50%). [α]D20 = −13.2 (c 1.8, CHCl3); 1H NMR (CDCl3) δ 1.95-2.06 (m, 1H), 2.27-2.36 (m, 1H), 2.84-3.08 (m, 2H), 4.07-4.19 (m, 3H), 4.55 (d, J = 12.0 Hz, 1H), 4.63 (d, J = 11.7 Hz, 1H), 7.16 (d, J = 8.7 Hz, 2H), 7.31 (d, J = 8.7 Hz, 2H), 7.61 (s, 1H); 13C NMR (CDCl3) δ 19.7, 24.2, 49.6, 69.6, 69.8, 118.6, 118.7, 121.0, 122.0, 128.8, 136.0, 144.1, 147.2, 148.7; HRMS (ESMS) calcd for C16H15F3N3O6 [M + HCO2−] 402.0913, found 402.0929.
Mtb MIC Assay
A stock culture of Mtb H37Rv (ATCC 27294) was grown to OD 0.5 in Middlebrook 7H9 broth (Difco) supplemented with 0.05% Tween 80, 0.2% glycerol and albumin/NaCl/glucose (ADC) complex. The culture was diluted 1:1000 in 7H9-based medium before aliquoting 50 μL into each well of a 96-well plate. The inhibitors were dissolved in DMSO to make stock solutions of 50 μmol/mL. Inhibitors were added to the first row of wells of the 96-well plate with 100 μL 7H9-based medium. After pipette mixing and using a multi-channel pipette, 50 μL was removed from each well in the first row and added to the second row. 2-Fold dilution in this manner was carried out to give 8 dilutions of each inhibitor (1000 μM – 7.8 μM). The plates were incubated for 2 weeks at 37°C and the MIC99 values were read macroscopically using an inverted plate reader. Each measurement was made three independent times.
Minimum Anaerobicidal concentration estimation experiment
For oxygen depletion assays, an early log phase Mtb culture in Glycerol free Dubos broth was diluted 100-fold and 20 mL were transferred to tubes (Pyrex 16 × 125 mm culture tubes) to maintain a head space ratio of 0.5 as described previously.31 The tubes were sealed with paraplast and incubated for 20 days under uniform stirring at 180 rpm using magnetic stirring bars. 1.5 μg/mL of methylene blue was added to a reference tube to visualize oxygen depletion. 100 μL of Mtb NRP-2 stage cells were exposed to various concentrations of drugs in a 96-well microplate. Two-fold drug dilutions in DMSO (1.95-500 μM) and a DMSO control were used. Handling of NRP-2 cells was done in a Vinyl Anaerobic Chamber (Coy Laboratories, Michigan) fitted with a Coy Model 10 gas analyzer and a vacuum air lock chamber. The anaerobic chamber was maintained under 90% nitrogen and 10% hydrogen. 96-Well plates were placed in a Type A Bio-bag anaerobic chamber (Becton Dickinson, Maryland) along with an oxygen indicator strip and incubated at 37°C for 7 days. After drug exposure, the cells were washed thrice with fresh Dubos broth and serially-diluted by two-fold dilutions. The minimal anaerobicidal concentration (MAC) against Mtb was estimated by measuring the number of viable bacilli in each dilution after drug treatment as follows: 5 μL of cell suspension from each well is spotted in a single well of a 7H11 agar-containing 96-well plate. After 3 weeks of incubation at 37°C, the MAC was read as the concentration of drug causing a 90% reduction in visible growth.
Expression and purification of Mtb-Rv3547 in E. coli
The Mtb-Rv3547 genomic PCR fragment was amplified using XbaI-FP (5- cga ccc ttg gtc tct aga atc agc gtc atg ccg -3′) and D3-RP (5′- tgg ctt cac gtt cgg caa gct tgg gaa cgg tca -3′). PCR fragment digested XbaI and HindIII was cloned in a pMALc2x vector digested with the same restriction enzymes for expression of Mtb-Rv3547 with the N-terminal MBP tag. Rv3547 DNA sequencing in pMAL-c2x-Rv3547 was confirmed using malE-FP (NEB #S12375) and M13 seq (NEB# S12115) primers. MBP-tagged Rv3547 was reproducibly expressed in E. coli Turbo cells (NEB). Protein induction at 37 °C with 300 μM IPTG yielded Rv3547-MBP protein in the cytosol. The fusion protein was purified to homogeneity using amylose resin affinity column chromatography (NEB # E8021) as per the manufacturer's instructions.
Nitroreductase assay
F420 purified from Methanobacterium thermoautotrophicum Marburg was a kind gift from Lacy Daniels, Texas A & M University, Kingsville, Texas. Reduced F420 (F420H2) was prepared using a recombinant Mtb glucose-6-phosphate dehydrogenase as described,29 followed by heat inactivation of FGD1 at 55°C water bath for 15 minutes and ultra-filtration (Centricon 10 kDa). Reduction of 1 or other nitroimidazoles by Rv3547 was monitored by oxidation of F420H2 at 400nm using a Cary UV spectrophotometer at 25°C. Initial rates were calculated based on gain of absorbance at 400 nm due to oxidation of F420 (ε400 = 25.71 mM−1 cm−1). Reactions were performed in phosphate buffer pH 7.4, 50μM F420, 0.6μM Rv3457 and varying amounts of PA-824 and analogs. Apparent steady-state kinetic parameters, Km and Vmax, were obtained by fitting initial rate data to the Michaelis-Menton equation.
Supplementary Material
Image 1.

Acknowledgments
Funding for this work was provided by the Division of Intramural Research, NIAID, NIH. We thank Dr. Lacy Daniels (Texas A&M University Health Science Center) for F420, Mr. Michael Goodwin (TRS, NIAID, NIH), Mr. John Lloyd (NIDDK, NIH), and Mr. Wesley White (NIDDK, NIH) with assistance in obtaining analytical data.
Footnotes
Abbreviations: FGD1, F420-dependent glucose-6-phosphate dehydrogenase; MAC, minimum anaerobicidal concentration; MIC, minimum inhibitory concentration; MBP, maltose-binding protein; Mtb, Mycobacterium tuberculosis; Mtz, metronidazole; SAR, structure-activity relationship; TB, tuberculosis; TBS or TBDMS, tert-butyldimethylsilyl; THP, tetrahydro-2H-pyran; TBAF, tetrabutylammonium fluoride; TBAI, tetrabutylammonium iodide; 2D NOESY, two-dimensional nuclear Overhauser effect spectroscopy.
Supporting Information Available: Experimental procedures for the preparation of desnitro-824 (12) and purity information for target compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.WHO International Fact sheet N°104. 2007 http://www.who.int/mediacentre/factsheets/fs104/en/index.html.
- 2.Duncan K, Barry CE. Prospects for new antitubercular drugs. Current Opinion in Microbiology. 2004;7:460–465. doi: 10.1016/j.mib.2004.08.011. [DOI] [PubMed] [Google Scholar]
- 3.Via LE, Lin PL, Ray SM, Carrillo J, Allen SS, Eum SY, Taylor K, Klein E, Manjunatha U, Gonzales J, Lee EG, Park SK, Raleigh JA, Cho SN, McMurray DN, Flynn JL, Barry CE., 3rd. Tuberculous granulomas are hypoxic in guinea pigs, rabbits, and nonhuman primates. Infect Immun. 2008;76:2333–40. doi: 10.1128/IAI.01515-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Boshoff HI, Barry CE., 3rd. Tuberculosis - metabolism and respiration in the absence of growth. Nat Rev Microbiol. 2005;3:70–80. doi: 10.1038/nrmicro1065. [DOI] [PubMed] [Google Scholar]
- 5.Stover CK, Warrener P, VanDevanter DR, Sherman DR, Arain TM, Langhorne MH, Anderson SW, Towell JA, Yuan Y, McMurray DN, Kreiswirth BN, Barry CE, Baker WR. A small-molecule nitroimidazopyran drug candidate for the treatment of tuberculosis. Nature. 2000;405:962–6. doi: 10.1038/35016103. [DOI] [PubMed] [Google Scholar]
- 6.Barry CE, Boshoff HIM, Dowd CS. Prospects for clinical introduction of nitroimidazole antibiotics for the treatment of tuberculosis. Current Pharmaceutical Design. 2004;10:3239–3262. doi: 10.2174/1381612043383214. [DOI] [PubMed] [Google Scholar]
- 7.Dick T, Lee BH, Murugasu-Oei B. Oxygen depletion induced dormancy in Mycobacterium smegmatis. FEMS Microbiol Lett. 1998;163:159–64. doi: 10.1111/j.1574-6968.1998.tb13040.x. [DOI] [PubMed] [Google Scholar]
- 8.Wayne LG, Sramek HA. Metronidazole is bactericidal to dormant cells of Mycobacterium tuberculosis. Antimicrob Agents Chemother. 1994;38:2054–8. doi: 10.1128/aac.38.9.2054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Agrawal KC, Bears KB, Sehgal RK, Brown JN, Rist PE, Rupp WD. Potential radiosensitizing agents. Dinitroimidazoles. J Med Chem. 1979;22:583–6. doi: 10.1021/jm00191a025. [DOI] [PubMed] [Google Scholar]
- 10.Ashtekar DR, Costa-Perira R, Nagrajan K, Vishvanathan N, Bhatt AD, Rittel W. In vitro and in vivo activities of the nitroimidazole CGI 17341 against Mycobacterium tuberculosis. Antimicrob Agents Chemother. 1993;37:183–6. doi: 10.1128/aac.37.2.183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nagarajan K, Shankar RG, Rajappa S, Shenoy SJ, Costa-Pereira R. Nitroimidazoles XXI 2,3-dihydro-6-nitroimidazo [2,1-b] oxazoles with antitubercular activity. European Journal of Medicinal Chemistry. 1989;24:631–633. [Google Scholar]
- 12.Baker WRS,C, Keeler EL. US Patent 5,668,127 Nitroimidazole antibacterial compounds and methods of use thereof. 1997
- 13.Baker WRS,C, Keeler EL. US Patent 6,087,358 Nitro-[2,1-b]imidazopyran compounds and antibacterial uses thereof. 2000
- 14.Lenaerts AJ, Gruppo V, Marietta KS, Johnson CM, Driscoll DK, Tompkins NM, Rose JD, Reynolds RC, Orme IM. Preclinical testing of the nitroimidazopyran PA-824 for activity against Mycobacterium tuberculosis in a series of in vitro and in vivo models. Antimicrob Agents Chemother. 2005;49:2294–301. doi: 10.1128/AAC.49.6.2294-2301.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. http://www.tballiance.org/home/home.php.
- 16.Sasaki H, Haraguchi Y, Itotani M, Kuroda H, Hashizume H, Tomishige T, Kawasaki M, Matsumoto M, Komatsu M, Tsubouchi H. Synthesis and antituberculosis activity of a novel series of optically active 6-nitro-2,3-dihydroimidazo[2,1-b]oxazoles. J Med Chem. 2006;49:7854–60. doi: 10.1021/jm060957y. [DOI] [PubMed] [Google Scholar]
- 17.Tangallapally RP, Yendapally R, Lee RE, Hevener K, Jones VC, Lenaerts AJ, McNeil MR, Wang Y, Franzblau S. Synthesis and evaluation of nitrofuranylamides as novel antituberculosis agents. J Med Chem. 2004;47:5276–83. doi: 10.1021/jm049972y. [DOI] [PubMed] [Google Scholar]
- 18.Hurdle JG, Lee RB, Budha NR, Carson EI, Qi J, Scherman MS, Cho SH, McNeil MR, Lenaerts AJ, Franzblau SG, Meibohm B, Lee RE. A microbiological assessment of novel nitrofuranylamides as anti-tuberculosis agents. J Antimicrob Chemother. 2008;62:1037–45. doi: 10.1093/jac/dkn307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yang C-C, Goldberg IH. Synthesis of 1-([18O2]-2-nitro-1-imidazolyl)-3-methoxy-2-propanol ([18O2]-misonidazole) Journal of Labelled Compounds and Radiopharmaceuticals. 1989;27:423–434. [Google Scholar]
- 20.Liu Z, C. Hengchang, Cao Shengli, Li Runtao. Solid-liquid phase-transfer catalytic method for N-alkylation of nitroimidazole. Synthetic Communications. 1993;23:2611–2615. [Google Scholar]
- 21.Massip S, Guillon J, Bertarelli D, Bosc JJ, Leger JM, Lacher S, Bontemps C, Dupont T, Muller CE, Jarry C. Synthesis and preliminary evaluation of new 1-and 3-[1-(2-hydroxy-3-phenoxypropyl)]xanthines from 2-amino-2-oxazolines as potential A(1) and A(2A) adenosine receptor antagonists. Bioorganic & Medicinal Chemistry. 2006;14:2697–2719. doi: 10.1016/j.bmc.2005.11.050. [DOI] [PubMed] [Google Scholar]
- 22.Liu Z, Chen H, Cao S, Li R. Solid-liquid phase-transfer catalytic method for N-alkylation of nitroimidazole. Synthetic Communications. 1993;23:2611–2615. [Google Scholar]
- 23.Mani NS, Jablonowski JA, Jones TK. A scalable synthesis of a histamine H-3 receptor antagonist. Journal of Organic Chemistry. 2004;69:8115–8117. doi: 10.1021/jo040225i. [DOI] [PubMed] [Google Scholar]
- 24.Gracias V, Gasiecki AF, Djuric SW. Synthesis of fused bicyclic imidazoles by sequential van Leusen/ring-closing metathesis reactions. Organic Letters. 2005;7:3183–3186. doi: 10.1021/ol050852+. [DOI] [PubMed] [Google Scholar]
- 25.Lang P, Gerez C, Tritsch D, Fontecave M, Biellmann JF, Burger A. Synthesis of 8-vinyladenosine 5 ′-di- and 5 ′-triphosphate: evaluation of the diphosphate compound on ribonucleotide reductase. Tetrahedron. 2003;59:7315–7322. [Google Scholar]
- 26.Cossy J, BouzBouz S, Hoveyda AH. Cross-metathesis reaction. Generation of highly functionalized olefins from unsaturated alcohols. Journal of Organometallic Chemistry. 2001;624:327–332. [Google Scholar]
- 27.Jourdant A, Gonzalez-Zamora E, Zhu J. Wilkinson's catalyst catalyzed selective hydrogenation of olefin in the presence of an aromatic nitro function: a remarkable solvent effect. J Org Chem. 2002;67:3163–4. doi: 10.1021/jo025595q. [DOI] [PubMed] [Google Scholar]
- 28.Hoffer M, Toome V, Brossi A. Nitroimidazoles .2. Synthesis and Reactions of Iodonitroimidazoles. Journal of Heterocyclic Chemistry. 1966;3:454–7. [Google Scholar]
- 29.Manjunatha UH, Boshoff H, Dowd CS, Zhang L, Albert TJ, Norton JE, Daniels L, Dick T, Pang SS, Barry CE., 3rd. Identification of a nitroimidazo-oxazine-specific protein involved in PA-824 resistance in Mycobacterium tuberculosis. Proc Natl Acad Sci U S A. 2006;103:431–6. doi: 10.1073/pnas.0508392103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Singh R, Manjunatha UH, Boshoff H, Ha YH, Niyomrattanakit P, Ledwidge R, Dowd CS, Lee IY, Kim P, Zhang L, Kang S, Keller TH, Jiricek J, Barry CE., 3rd. Bicyclic nitroimidazoles act as intracellular NO donors and kill non-replicating Mycobacterium tuberculosis. Science. 2008 in press. [Google Scholar]
- 31.Wayne LG. In: Mycobacterium tuberculosis Protocols. Parish T, Stoker NG, editors. Humana Press; New Jersey: 2001. pp. 247–270. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.