

Abstract

Hydroboration of alkenes with pyridine iodoborane followed by treatment with pinacol/NaOH affords monoalkyl pinacol boronates in moderate to good yield. Dialkylborinic acid derivatives are formed competitively, especially in the case of terminal alkenes. This side reaction can be minimized by using excess of pyridine iodoborane. More hindered alkenes give the best results.
In a prior report,1 the pyridine iodoborane reagent 1 was shown to react with alkenes to afford hydroboration products at room temperature. A tentative mechanistic proposal was advanced as illustrated in Scheme 1 for the case of β-methylstyrene, involving initial conversion from 1 to 4 by nucleophilic displacement of iodide to generate the π-complex ion pair 2 and subsequent hydroboration via the 4-center interaction shown in 3. We were interested in extending this chemistry to the preparation of pinacol boronates. Indeed, treatment of β-methylstyrene with 1.5 equiv of 1 (2 h, rt) followed by reaction with excess pinacol/NaOH afforded the isolable boronate 5 in high yield (15:1 regioisomer ratio in favor of the indicated isomer by NMR assay). Evidently, a second hydroboration event from the monoalkylborane stage 4 is slow compared to the step from 1 to 4. This is consistent with increased steric hindrance near boron in the adduct 4 compared to the starting reagent 1 if the same nucleophilic displacement mechanism were to operate. Efficient formation of 5 was anticipated because similar selectivity had been inferred for a variety of alkenes in our prior study,1a based on ESMS assays and results from oxidative workup. However, our attempts to prepare pinacol boronates from less hindered alkenes have encountered a different scenario than was expected from the earlier work.1b A re-evaluation of the hydroboration process has been necessary to correctly define the ratio of mono- vs. di-alkylborane products, and a re-optimized procedure has been developed in the context of conversion of alkenes to monoalkyl pinacolboronates as described below.
Scheme 1.

The same hydroboration sequence using 1.5 equiv of 1 that was so effective for the preparation of 5 was tested with α-methylstyrene. After several tries, it became clear that conversion to 6 using the original procedure is inefficient (Table 1, entry 1; ca. 30% pinacol boronate isolated) because the reaction affords a substantial amount of a byproduct. Pure samples of the byproduct have not been obtained due to decomposition during chromatography, but structure 7 (two diastereomers) is proposed based on ESMS data (positive ion detection, m/z = 289.2, M + Na) and a methine signal in the 1H NMR spectrum (broad multiplet at δ 2.99 ppm). This signal is distinct from the corresponding methine signal for 6 (δ 3.07 ppm), and provides a basis to estimate the product ratios in Table 1. Alternative structures that still contain a boronpyridine bond were ruled out due to the presence of the same mass peak and NMR signal from hydroborations using 2,6-lutidine•BH2I, Me2S•BH2I, or Me2S•BH3 (entries 9–11) in place of 1. Furthermore, the reagents used in entries 9–11 always favored 7 vs. 6 while the pyridine iodoborane 1 favored 6. The proposed dialkyl borinate structure 7 was further supported by showing that the ratio of 6:7 increases as the relative amount of reagent 1 increases. A substantial excess of 1 was required for acceptable yields of 6 (entries 5–7). Lower temperatures (entries 2–4) gave better ratios of 6:7, but hydroboration was slow and afforded lower isolated yields of 6.2
Table 1.
Conversion of •-methylstyrene to 6 using 1(a)
| Entry | Equiv | Reagent | Yield of 6 | Ratio 6:7 |
|---|---|---|---|---|
| 1 | 1.5 | 1 | 30%(b) | 1.2:1 |
| 2 | 1.5 | 1 | 40%(c) | 1.1:1 |
| 3 | 1.5 | 1 | 31%(d) | 1.6:1 |
| 4 | 1.5 | 1 | 17%(e) | 5.2:1 |
| 5 | 2 | 1 | 60% | 2.6:1 |
| 6 | 3 | 1 | 62% | 3.8:1 |
| 7 | 6 | 1 | 80% | >10:1 |
| 8 | 10 | 1 | 82% | ND |
| 9 | 1.5 | 2,6-lutidine-BH2I | 17% | 1:2.9 |
| 10 | 2 | Me2SBH2I | 6% | 1:5.6 |
| 11 | 2 | Me2SBH3 | 19% | 1:1.8 |
HB in DCM at RT, 2 h, quenching at the indicated temperature with aqueous NaOH, followed by stirring with pinacol/NaOH, 15 h unless noted.
average of three experiments (27-32%)
HB, 18 h at 0 °C
HB, 18 h at -20 °C
HB, 18 h at -40 °C
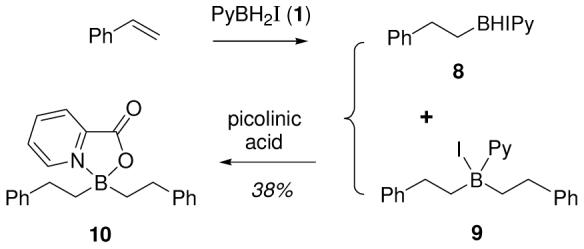
Given the difficulty of isolating the dialkyl borinic acid 7 and analogous structures, further evidence was needed to confirm the assignment. Derivatization with picolinic acid to form an oxazaborolidinone was promising,3 but the presence of two stereocenters in 7 and an additional stereocenter at boron in the desired heterocycle prompted the investigation of a simpler substrate. Thus, treatment of styrene with 1 under conditions biased to promote the formation of 9 relative to 8 (1 equiv of 1, rt; Scheme 2), followed by quenching and heating with picolinic acid (aqueous THF/EtOH) allowed isolation of 10 (38%) by chromatography. This relatively stable compound was fully characterized, leaving no doubt that dialkylborane derivatives are the byproducts formed using 1 for hydroboration.
Scheme 2.
Attention was now focused on several representative alkenes to better define the level of steric hindrance needed for successful conversion into the monoalkyl pinacol boronates 11. Terminal alkenes were the most problematic, resulting in low yields (Table 2, entries 1–4) or requiring a large excess of 1 to achieve moderate yields (entry 5). In all cases, the indicated pinacol boronate regioisomer was the dominant product observed by NMR assay, but traces of a methyl doublet at 0.96 ppm were detected in the spectrum for the pinacol boronate obtained in entry 5 that may correspond to the regioisomeric hydroboration product (≤2%). The 1,2-disubstituted alkenes were better behaved (entries 6–8), but the yield of 11 was low in the case of norbornene unless 6 equiv of 1 was used (entries 6,7). Cyclohexene (entry 8) proved to be better behaved than norbornene and gave good results without the need for a large excess of 1. With the trisubstituted 1-phenylcyclohexene, a good yield was also obtained although a longer reaction time was needed (15 h, entry 9).4
Table 2.
Conversion of alkenes to pinacolboronates 11 using 1
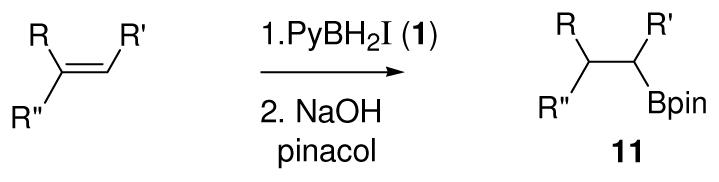 | ||||
|---|---|---|---|---|
| Entry | R, R’ | R” | Equiv 1 (PyBH2I) |
Yield(a),(b) |
| 1 | nC8H17, H | H | 2 | 40% |
| 2 | nC10H21, H | H | 2 | 40% |
| 3 | Ph, H | H | 2 | 50% |
| 4 | BzO(CH2)4, H | H | 2 | 26% |
| 5 | BzO(CH2)4, H | H | 6 | 55% |
| 6 | norbornene | H | 2 | 47% |
| 7 | norbornene | H | 6 | 68% |
| 8 | -C4H8- | H | 1.5 | 67% |
| 9 | -C4H8- | Ph | 2 | 67%(c) |
HB in DCM at RT, 2 h, followed by stirring with pinacol and NaOH (2-fold and 3-fold excess, respectively, relative to 1), 15 h, unless noted; isolated yields.
The pinacol boronates from entries 1-3 and 6-9 have been described previously and were identified by NMR comparisons; see Supporting Information for references.
HB required 15 h to go to completion.
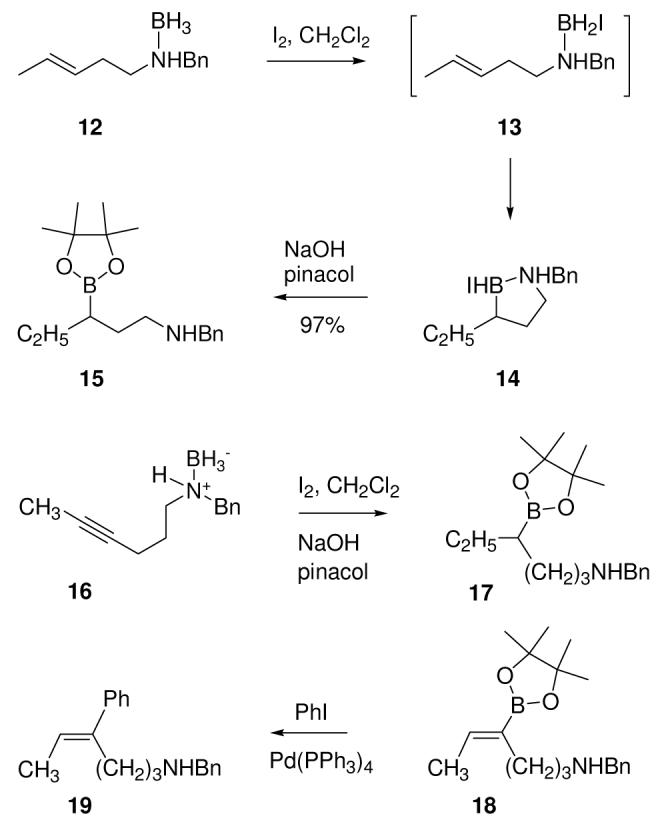
With the direct formation of pinacol boronates demonstrated from the intermediates obtained using 1, it was also of interest to see if a similar procedure would allow derivatization of the analogous intermediates from the internal hydroboration of unsaturated amine boranes (Scheme 3).5 Treatment of 12 with iodine to generate 13 and 14 followed by pinacol/NaOH gave 15 in excellent yield. However, an attempt to extend this chemistry to an alkynylamine substrate afforded a mixture of saturated as well as the expected unsaturated pinacol boronates. Thus, activation of 16 with iodine followed by pinacol/NaOH gave an inseparable mixture of 17 and 18 according to 1H NMR and ESMS evidence (17: δ 0.92 ppm, t, J = 7.1 Hz; ESMS m/z = 318.2 amu; 18: δ 1.72, d, J = 6.9 Hz, 6.42, q, J = 6.9 Hz; ESMS m/z = 316.2 amu). The relative amount of 17 corresponds qualitatively to the amount of I2 used (50 mol% I2, 18:82 18:17; 30 mol% I2, 41:59 18:17; 10 mol% I2, 77:23 18:17), but the pinacol boronates could not be separated or purified due to partial decomposition during chromatography. The formation of an alkenyliodoborane intermediate is supported by the formation of the Molander-Suzuki coupling product 196 (ca. 25 % overall) from a similar experiment using 10 mol% I2 for activation of 16 followed by treatment with KHF2 and coupling with p-nitroiodobenzene/PdCl2(dppf). These results implicate a second hydroboration event as the reason for the apparent reduction,7 but do not clarify the role of iodine stoichiometry.
Scheme 3.
Studies described here define the optimum conditions for conversion of alkenes into monoalkylborane products using the activated pyridine borane reagent 1. Also presented is a method for the formation of pinacol boronates directly from hydroboration mixtures. Alternative reagents for hydroboration are known that allow conversion to monoalkylboronic acid derivatives, including the recently developed Snieckus di(isopropylprenyl)borane8 as well as haloboranes9 or catechol borane under metal catalysis.10 Hydroboration using PyBH2I (1) is a simple alternative that readily provides purifiable pinacol boronates in most cases and works best for the more hindered 1,2-di- or tri-substituted alkenes where competition by the second hydroboration stage is disfavored.
Experimental Section
Pinacol boronate ester 5
A solution of pyridine borane (330 μL, 3.3 mmol) in CH2Cl2 (15 mL) was cooled to 0 °C. Iodine (419 mg, 1.65 mmol) was added in several portions and the solution was stirred at 0 °C until gas evolution ceased. The solution was warmed to room temperature and β-methylstyrene (285 μL, 2.2 mmol) was added. Stirring was continued until the reaction was complete based on TLC assay (2 h). The solution was cooled to 0 °C, sodium hydroxide (5 mL, 1M) was added, the mixture was warmed to room temperature, and then a solution of pinacol (440 mg, 3.7 mmol) in CH2Cl2 (5 mL) was added and stirred for 15 h. The resulting mixture was added to H2O (10 mL), extracted with ether, dried (Na2SO4) and concentrated (aspirator). The crude products were purified by flash chromatography using 2% ether in hexanes to yield 520 mg (96%) of 510b as a colorless oil; 1H NMR (400 MHz, CDCl3) δ 7.28 (m, 4H), 7.17 (m, 1H), 2.28 (t, J = 8.3 Hz, 1H), 1.99 (m, 1H), 1.78 (m, 1H), 1.24 (m, 12H), 0.91 (t, J = 7.9 Hz, 3H). A minor doublet (ca. 8-10% relative to the 0.91 triplet) at δ 0.97 was also resolved, tentatively assigned to the inseparable regioisomeric pinacol boronate; 13C NMR (100 MHz, CDCl3; only the major regioisomer 5 detected) δ 143.4, 128.4, 128.2, 125.1, 83.2, 34.4, 25.9, 24.7, 24.6, 14.0.
Pinacol boronate ester 6 (Table 1, Entry 6)
A solution of pyridine borane (660 μL, 6.6 mmol) in CH2Cl2 (15 mL) was cooled to 0 °C. Iodine (838 mg, 3.3 mmol) was added in several portions and the solution was stirred at 0 °C until gas evolution ceased. The solution was warmed to room temperature and α-methylstyrene (285 μL, 2.2 mmol) was added. Stirring was continued until the reaction was complete based on TLC (2 h). The solution was cooled to 0 °C, sodium hydroxide (10 mL, 1M) was added, warmed to room temperature, and then a solution of pinacol (880 mg, 7.4 mmol) in CH2Cl2 (10 mL) was added and stirred for 15 h. The same workup and purification as described above gave 334 mg (62%) of 610b as a colorless oil 1H NMR (400 MHz, CDCl3) δ 7.30 (m, 4H), 7.19 (m, 1H), 3.10 (m, 1H), 1.31 (d, J = 7.4 Hz, 3H), 1.18 (m, 14H); 13C NMR (100 MHz, CDCl3) δ 149.2, 128.2, 126.6, 125.7, 83.0, 35.8, 24.8, 24.7, 21.3 (C attached to quadrupole B not observed).
Procedures for Tables 1 and 2
For Table 1, Entries 1, 2, 5, 9, 10, and 11 and for Table 2, entries, 1 – 4, 6, 8, and 9, the procedure for formation of 5 was followed. For Table 1, Entries 3 & 4, after stirring was completed, the solution was transferred by cannula into sodium hydroxide (5 mL, 1 M) at 0 °C.
For Table 1, Entry 7, and Table 2, Entries 5 and 7, the solution was cooled to 0 °C, sodium hydroxide (15 mL, 1M) was added, warmed to room temperature, and then a solution of pinacol (1.32 g, 11.1 mmol) in CH2Cl2 (15 mL) was added and stirred for 15 h.
For Table 1, Entry 8, the solution was cooled to 0 °C, sodium hydroxide (30 mL, 1M) was added, warmed to room temperature, and then a solution of pinacol (2.64 g, 22.2 mmol) in CH2Cl2 (30 mL) was added and stirred for 15 h.
Oxazaborolidine 10
A solution of pyridine borane (220 μL, 2.2 mmol) in CH2Cl2 (5 mL) was cooled to 0 °C. Iodine (280 mg, 1.1 mmol) was added and the solution was stirred at 0 °C until gas evolution ceased. The solution was warmed to room temperature and styrene (252 μL, 2.2 mmol) was added. After stirring 2 h, the solution was cooled to 0 °C and methanol (6.5 mL) was added very slowly and the solvent was removed under vacuum. The solid residue was dissolved in THF (3 mL) and then 2-picolinic acid (295 mg, 2.4 mmol), water (6 mL), and ethanol (6 mL) were added and the solution was brought to reflux (15 h). The solution was concentrated, and the residual solid was partitioned between CH2Cl2 and water and the water layer was extracted with CH2Cl2, dried (Na2SO4), and concentrated. The crude products were purified by flash chromatography using 2/1 ether/hexanes to yield 142 mg (38%) of 10 as a colorless oil. IR (CDCl3) 1737 cm-1; 1H NMR (400 MHz, CDCl3) δ 8.20 (m, 3H), 7.61 (m, 1H), 7.15 (m, 4H), 7.06 (m, 6H), 2.62 (m, 2H), 2.18 (m, 2H), 1.28 (m, 2H), 1.00 (m, 2H); 13C NMR (100 MHz, CDCl3) δ 163.5, 145.4, 143.8, 142.1, 140.3, 128.1, 128.0, 127.8, 125.1, 123.4, 30.7, 24.2; 11B NMR (128 MHz, CDCl3) * 10.1; ESMS m/z (relative intensity) 366.1 (M+Na, 100%); HRMS m/z calcd for C22H22BNO2 (M + Na) 366.1641, found 366.1624.
Pinacol boronate 15
A solution of iodine (51 mg, 0.20 mmol) in CH2Cl2 (6.3 mL) was added slowly to amine borane 12 (73 mg, 0.39 mmol) in CH2Cl2 (6.3 mL) and stirred for 2 h. The solution was cooled to 0 °C, and NaOH (1.25 mL, 1M) was added followed by pinacol (188 mg, 1.6 mmol) in CH2Cl2 (2.2 mL) and the mixture was stirred overnight. The solution was added to water (10 mL), extracted with ether, dried over Na2SO4, and concentrated. The residue was placed under high vacuum (0.1 torr) to remove excess pinacol and gave 114 mg (97% recovery, >95% major component by NMR assay) of 15 as a colorless oil, not purified further due to instability to silica gel chromatography; IR (CDCl3 ) 2975 cm-1; 1H NMR (300 MHz, CDCl3) δ 7.27 (m, 5H), 3.72 (m, 2H), 2.61 (m, 2H), 1.87 (s, 1H), 1.61 (m, 1H), 1.50 (m, 1H), 1.46 (m, 1H), 1.30 (m, 1H), 1.15 (m, 13H), 0.86 (t, J = 7.4 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 139.4, 128.4, 128.2, 127.1, 82.0, 53.3, 48.4, 30.7, 25.0, 24.9, 24.2, 13.9; 11B NMR (160 MHz, CDCl3) δ 29.8; ESMS m/z (relative intensity) 304.2 (M+H, 100%); HRMS calcd for C18H30BNO2 (M+) 303.2370, found 303.2367.
Supplementary Material
ACKNOWLEDGMENT
This work was supported by NIH (GM067146)
References
- 1(a).Clay JM, Vedejs E. J. Am. Chem. Soc. 2005;127:5766. doi: 10.1021/ja043743j. Errors in some of the NMR and ESMS data and interpretation have been found, and statements regarding clean formation of RBHI(Py) and RBF3K are wrong see Clay JM, Karatjas AG, Vedejs E.J. Am. Chem. Soc 2008130, Additions and Corrections.
- 2.Increased reaction time at -40 °C did not improve the yield.
- 3.Baker SJ, Akama T, Zhang Y-K, Sauro V, Pandit C, Singh R, Kully M, Khan J, Plattner JJ, Benkovic SJ, Lee V, Maples KR. Bioorg. Med. Chem. Lett. 2006;16:5963. doi: 10.1016/j.bmcl.2006.08.130. [DOI] [PubMed] [Google Scholar]
- 4.3-Methyl-1-cyclohexene gave pinacolboronates in 68% yield using 2 equiv 1), but the mixture of isomers could not be separated. Cyclohexadiene yielded 31% of the B-(cyclohexen-3-yl) pinacolboronate with 1 equiv of 1 followed by the standard workup and chromatography.
- 5.Scheideman M, Shapland P, Vedejs E. J. Am. Chem. Soc. 2003;125:10502. doi: 10.1021/ja034655m. [DOI] [PubMed] [Google Scholar]
- 6.Scheidemann M. Ph. D. Thesis. University of Michigan; 2005. [Google Scholar]
- 7.Isolation of alcohols from alkyne hydroboration followed by oxidative cleavage has been attributed to the formation of 1,1-diboro intermediates and subsequent nucleophile-induced deboronation: Brown HC, Zweifel G. J. Am. Chem. Soc. 1961;83:3834.
- 8.Kalinin AV, Scherer S, Snieckus V. Angew. Chemie Int. Ed. 2003;42:3399. doi: 10.1002/anie.200351312. [DOI] [PubMed] [Google Scholar]
- 9.Josyula KVB, Gao P, Hewitt C. Tetrahedron Lett. 2003;44:7789. and references therein.
- 10(a).Review: Crudden CM, Edwards D. Eur. J. Org. Chem. 2003:4695.Cipot J, Vogels CM, McDonald R, Westcott SA, Stradiotto M. Organometallics. 2006;25:5965.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.