

Abstract
Cytochromes P450 are ubiquitous heme-containing enzymes that catalyze a wide range of reactions in nature including many oxidation reactions. The active oxidant species in P450 enzymes are widely thought to be iron(IV)-oxo porphyrin radical cations, termed Compound I species, but these intermediates have not been observed under turnover conditions. We prepared Compounds I of the mammalian hepatic P450 enzyme CYP2B4 and three mutants (E301Q, T302A, and F429H) by laser flash photolysis of the Compound II species that, in turn, were prepared by reaction of the resting enzymes with peroxynitrite. The PN treatment resulted in a small amount of nitration of the P450 as determined by mass spectrometry, but no change in reactivity of the P450 in a test reaction. CYP2B4 Compound I oxidized benzphetamine to norbenzphetamine in high yield in bulk studies. In direct kinetic studies of benzphetamine oxidations, Compounds I displayed saturation kinetics with similar binding equilibrium constants (Kbind) for each. The first-order oxidation rate constants (kox) were comparable for Compounds I of CYP2B4, the E301Q mutant, and the T302A mutant, whereas the kox for Compound I of the F429H mutant was reduced by a factor of two. CYP119 Compound I, studied for comparison purposes, reacted with benzphetamine with a binding constant that was nearly an order of magnitude smaller than that of CYP2B4 but a rate constant that was similar. Substrate binding constants for P450 Compound I are important for controlling overall rates of oxidation reactions, and the intrinsic reactivities of Compounds I from various P450 enzymes are comparable.
The cytochrome P450 (CYP or P450) enzymes function as catalysts for many biological reactions including oxidation reactions of high-energy carbon-hydrogen bonds that produce alcohol products or intermediates.1 P450s are heme-containing enzymes with thiolate from a protein cysteine as the fifth ligand to iron. In the human, broad spectrum P450s in the liver oxidize drugs, pro-drugs and other xenobiotics,2 and over-expression of highly specific P450s is linked to a variety of disease states, including breast and prostate cancers and liver disease.3–5 These two properties have generated great interest in P450s from both the pharmaceutical and medicinal perspectives.
The nature of the iron-oxygen species formed in P450 enzymes has been a focus of study since the first reports of these enzymes in the 1960s. Heme-containing peroxidase and catalase enzymes react with hydrogen peroxide to form iron(IV)-oxo porphyrin radical cations termed Compounds I,6 and similar species have long been assumed to be the active oxidants in P450s. Most P450s are activated in nature by the sequence reduction, oxygen binding, a second reduction, and two protonation steps,1 but many are known to react with hydroperoxy compounds in “shunt”reactions that produce active oxidants. Despite that similarity between peroxidases and P450s, attempts to detect active oxidants in P450 enzymes by rapid mixing studies have met with limited success,7–10 and the P450cam hydroperoxy-iron transient produced under cryogenic reduction conditions reacted upon annealing without accumulation of Compound I.11 The result is that no kinetic studies of reactions by P450 Compounds I have been obtained from rapid mixing or freeze-quench methods.
Because of the difficulty in producing P450 Compound I in mixing experiments, we developed an alternative entry to these intermediates. Laser flash photolysis (LFP) of iron(IV)-oxo neutral porphyrin species, known as Compounds II in biological contexts, resulted in electron photo-ejection reactions that gave iron(IV)-oxo porphyrin radical cations.12 The method was applied for generation of Compound I of CYP119, a soluble P450 from a thermophile,13 which permitted kinetic studies of Compound I epoxidation and hydroxylation reactions.14
In the present work, we report use of the photo-oxidation method for production and kinetic studies of Compounds I of CYP2B4 and three of its mutants (Figure 1). CYP2B4, an hepatic phenobarbital-induced P450 from rabbit originally known as P450LM2, was the first mammalian P450 to be solubilized from microsomal membranes,15 and the present work reports the direct detection of Compound I from a P450 that is membrane bound in nature. We studied oxidations of the anti-obesity drug benzphetamine by Compounds I of wild-type CYP2B4 and the E301Q, T302A, and F429H mutants (Figure 1), and, for comparison, Compound I of CYP119.14 The results provide insight into the general reactivity of P450 Compounds I and demonstrate that substrate reactivities are controlled largely by binding constants as opposed to differences in the inherent reactivities of Compounds I.
Figure 1.

Stereo view of CYP2B4 active site showing the heme and positions of mutated residues (pdb:1SUO). Iron is the green ball in the center of heme, the negatively charged surfaces of E301 and T302 are shown in red, positively charged surfaces are in blue, and neutral surfaces are in white. The figure was generated with DS viewer Pro (Accelrys, Inc. San Diego CA).
Results and Discussion
Production of Compounds I of CYP2B4 and its mutants via Compounds II involved the reactions in Figure 2. Reaction of the resting enzyme with peroxynitrite (PN) gave the Compound II species.16 Photo-ejection of an electron from Compound II12 with concomitant loss of a proton gave Compound I.17 Compound I bound substrate reversibly and oxidized substrate with a first-order rate constant to give oxidized product.
Figure 2.

Reactions for production of P450 Compound I via electron photo-ejection. The oval represents the porphyrin macrocycle in heme.
Peroxynitrite oxidations of resting heme-containing enzymes are interesting in part because they can be involved in reactions of the enzymes in nature. The PN reaction(s) are complex and give Compound II species for several, but not all, heme enzymes.18 When Compound II is formed, the reaction sequence is thought to involve oxo-transfer from PN to the ferric enzyme to give a formal iron(V)-oxo species followed by fast reduction of this species by the nitrite by-product in the active site in competition with escape of nitrite. Not all P450s react with PN to give Compound II, but the production of Compound II was reported for P450BM3 (CYP102)19 and CYP11916 as well as the related heme-thiolate protein chloroperoxidase (CPO).13,20
Upon reaction of the resting enzymes in buffer with peroxynitrite (PN), the UV-visible spectrum of the Soret band shifted from λmax = 418 nm for the resting enzyme to λmax = 433 nm for Compound II, which persisted when PN was present and decayed after the PN was depleted. A typical spectrum and kinetic behavior, which resemble the spectra and behavior found with CYP119,16 are shown in the Supporting Information (Figure S1).
PN is known to nitrate tyrosine groups and has been reported to nitrate several P450 enzymes,19,21 but the amounts of PN used in those studies were greater than the amount used here. We conducted two studies to evaluate the effect of PN on CYP2B4, one to demonstrate nitration of the P450 by PN, and another to determine if the enzyme activity was altered by the nitration. In both cases, CYP2B4 in buffer was treated with PN under conditions similar to those used for production of Compound I, but Compound II was allowed to decay without the photolysis step. The CYP2B4 obtained after the PN reaction was then compared to a sample of the enzyme that was not treated.
In the first study, CYP2B4 was analyzed by HPLC-mass spectrometry with electrospray ionization before and after treatment with PN by a method recently described.21 Typical results are shown in Figure 3. Both the untreated and PN-treated CYP2B4 enzymes eluted in narrow bands reflecting good homogeneity. The theoretical mass of the PN-treated enzyme was 55713.5 Da, and the experimentally measured value was 55723 Da, an “error”in accuracy of only 0.02%. The PN-treated sample had a mass of 55877 Da. On repeated analyses, the standard deviation in measured masses were 5.7 and 7.4 Da for the untreated and PN-treated protein, respectively. The increased mass of the PN-treated enzyme was 154 Da corresponding to the addition of 3.4 nitro groups (45 Da each) per protein on average. From studies of P450s treated with higher concentrations of PN,21 we assume that nitration occurred mainly on phenol groups of tyrosine residues.
Figure 3.

HPLC-mass spectrometry results. (A) Total ionization as a function of HPLC retention times. (B) Ionization in the range m/z 50,000 to 60,000 for the peaks eluting at 32 minutes, the maxima are at m/z = 55,723 (before PN) and m/z = 55,877 (after PN).
In the second study, the ability of CYP2B4 to catalyze oxidations under turnover conditions was investigated with untreated enzymes and samples that had been treated with PN. The mechanistic probe trans-2-phenylmethylcyclopropane22 was oxidized by untreated CYP2B4 and PN-treated enzyme under turnover conditions to give known products. The total yields of products were similar for both enzymes (121 ± 2 nmol for PN-treated enzyme and 125 ± 1 nmol for untreated enzyme), and, importantly, the product ratios were the same for both PN-treated and untreated enzymes. Details of the probe experiments are in the Supporting Information. Thus, the effect of PN treatment in regard to altering the catalytic activity of CYP2B4 was inconsequential.
The production of CYP2B4 Compound I was accomplished by laser flash photolysis (LFP) of Compound II in a manner similar to that previously reported for studies with CYP119.14 LFP of the mixture containing Compound II with 355 nm laser light gave the spectral results shown in Figure 4. The LFP difference spectrum (dashed line) exhibited a predominant bleaching at λ = 433 nm and growth in the region between λ = 375 to 410 nm. Addition of the spectrum of CYP2B4 Compound II (dotted line, scaled to minimize the bleaching in the Q-band region) to the difference spectrum gave the spectrum of the Compound I (solid line).
Figure 4.

UV-visible spectrum of CYP2B4 Compound I. The LFP experiment gave an observed difference spectrum (dashed line) that was combined with a scaled spectrum of Compound II (dotted line) to give the Compound I spectrum (solid line).
Before the UV-visible spectrum of a P450 Compound I was measured, the prediction was that the Soret band from these species would be centered at about 367 nm based on the spectrum of Compound I of another heme-thiolate enzyme, chloroperoxidase (CPO), reported in 1980 by Hager.23 Egawa and co-workers subsequently demonstrated that the Soret band from CPO Compound I actually contained two overlapping transitions at 365 nm and 415 nm.24 The two peaks in CPO Compound I are obvious in its second derivative spectrum,14 and the second derivative spectrum of CYP119 Compound I contains the same two peaks as in CPO Compound I.14 The second derivative spectrum of CYP2B4 Compound I also contains the same two transitions (Supporting Material, Figure S2), and the close similarity of the various Compound I spectra supports the assignment of the new species as CYP2B4 Compound I.
No rate constants for reactions of P450 Compounds I were reported before the recent study with CYP119 Compound I.14 In the present work, we measured kinetics for the oxidations of the anti-obesity drug benzphetamine (1) (Figure 5), which is known to be oxidized by CYP2B4 predominantly at the N-methyl group to give the N-demethylated product norbenzphetamine (3). The oxidation reaction is thought to proceed by oxidation of the N-methyl group to give hemi-aminal intermediate 2 that hydrolyzes to 3 with release of formaldehyde.25
Figure 5.
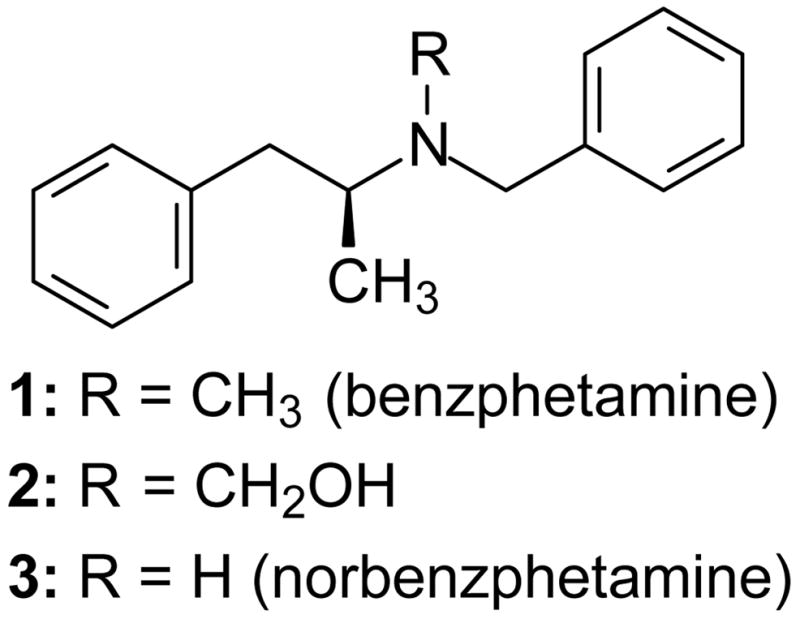
Benzphetamine and its oxidation products hydroxybenzphentamine and norbenzphetamine.
The production of norbenzphetamine in the photochemical process was confirmed in bulk photolysis experiments at 0 °C designed to test for turnover in the photo-oxidation sequence. Relative to CYP2B4, the reaction mixtures contained 50 equivalents of PN and 150 equivalents of benzphetamine, and the samples were irradiated with 15 pulses of light. Following the photolyses, the UV-visible spectra were the same as that of the resting enzyme. Analyses for norbenzphetamine by HPLC as described previously25 indicated that 64 ± 1 nmol (2 runs) of norbenzphetamine was produced, giving a yield of 1800% based on enzyme or 120% based on enzyme per light pulse. In control reactions, the yields of norbenzphetamine were 3.6 ± 0.2 nmol (light omitted, 2 runs), 0.6 nmol (PN omitted), and 0.4 nmol (CYP2B4 omitted). Norbenzphetamine is an impurity in samples of benzphetamine at the level of 0.4 nmol for the sample size used.
The high yields of norbenzphetamine in the bulk photolyses studies demonstrate that the CYP2B4 enzyme turns over successfully in the PN-photolysis sequence. CYP2B4 Compound I formed upon photolysis of Compound II reacts rapidly with benzphetamine (see below), giving resting enzyme that can react again with PN to give Compound II. From the rate constants for Compound II formation and for the reaction of Compound I with benzphetamine (see below), the turnover is complete in less that 0.1 second, which is the minimum irradiation time for the lamp used in the bulk photolysis reaction. Thus, a portion of the enzyme was expected to turn over within the duration of one light pulse resulting in overall turnovers that exceeded 100% per light pulse.
Kinetic studies were accomplished by generating Compounds I from CYP2B4 and its mutants in the presence of benzphetamine in varying amounts. A solution containing the resting enzyme and benzphetamine and a solution of PN were mixed to give Compound II in a stopped-flow mixing unit, the sample was irradiated with 355 nm laser light after a delay of 5 s, and the rates of reactions of Compound I were measured at 430 nm, where reaction of Compound I results in an increase in absorbance. Figure S3 in Supporting Material shows typical results. The traces were solved for double exponential kinetics. The major process changed as a function of substrate concentration, whereas the minor process was relatively constant in the absence or presence of substrate. At ambient temperature, the rate constant for decay of CYP2B4 Compound I at 22 °C was 4–5 s−1, similar to the rate constant for decay of CYP119 Compound I in the absence of substrate,14 and Compounds I from the three CYP2B4 mutants decayed with similar rate constants.
Detailed results of kinetic studies of oxidations of benzphetamine by Compounds I from CYP2B4, the three CYP2B4 mutants, and CYP119 are listed in Table 1. The observed rate constants increased as a function of substrate concentration, and plots of the rate constants against substrate concentration clearly displayed saturation kinetics (Figure 6). Saturation kinetics are described by Eq 1 where kobs is the observed rate constant, k0 is the background rate constant in the absence of substrate, Kbind is the substrate binding constant, kox is the first-order rate constant for the oxidation reaction, and [Sub] is the concentration of substrate. Solution of the data according to Eq 1 gave the results shown as solid line fits in Figure 6. Values for the equilibrium binding constant (Kbind) and the oxidation rate constant (kox) are listed in Table 2.
Table 1.
Observed rate constants for reactions of benzphetamine with Compounds I.a
| Enzyme | Bnz conc (mM)b | kobs (s−1)c |
|---|---|---|
| CYP2B4 | 0 | 4.4 ± 0.4 |
| 0.075 | 20.8 ± 2.9 | |
| 0.15 | 34.1 ± 3.0 | |
| 0.225 | 37.5 ± 4.8 | |
| 0.30 | 40.9 ± 4.8 | |
| 0.45 | 44.3 ± 3.2 | |
| 0.60 | 42.6 ± 2.7 | |
| CYP2B4 E301Q | 0 | 2.8 ± 0.2 |
| 0.075 | 21.7 ± 2.9 | |
| 0.15 | 32.3 ± 3.2 | |
| 0.225 | 38.2 ± 2.7 | |
| 0.30 | 39.5 ± 2.0 | |
| 0.45 | 42.2 ± 3.0 | |
| 0.60 | 45.4 ± 3.4 | |
| CYP2B4 T302A | 0 | 2.3 ± 0.2 |
| 0.075 | 16.4 ± 3.5 | |
| 0.15 | 26.4 ± 3.0 | |
| 0.225 | 32.6 ± 4.5 | |
| 0.30 | 35.1 ± 4.0 | |
| 0.45 | 38.7 ± 3.8 | |
| 0.60 | 39.5 ± 2.9 | |
| CYP2B4 F429H | 0 | 2.8 ± 0.2 |
| 0.075 | 10.5 ± 1.5 | |
| 0.15 | 15.2 ± 1.9 | |
| 0.30 | 19.6 ± 3.2 | |
| 0.60 | 21.6 ± 2.8 | |
| CYP119 | 0 | 2.9 ± 0.3 |
| 0.3 | 9.9 ± 1.3 | |
| 0.6 | 15.9 ± 2.0 | |
| 0.9 | 18.7 ± 1.0 | |
| 1.5 | 22.4 ± 1.3 |
Reactions at 22 ± 1 °C in 50 mM phosphate buffer (pH 7.4).
Concentration of benzphetamine.
Observed rate constant with error at 1σ.
Figure 6.

Rate constants for reactions of Compounds I with benzphetamine: CYP2B4 (black), CYP2B4 E301Q (green), CYP2B4 T302A (red), CYP2B4 F429H (blue), and CYP119 (pink). The solid lines were generated from the kinetic parameters in Table 2.
Table 2.
Binding constants and first-order rate constants for oxidations of benzphetamine by P450 Compounds I.a
| Enzyme | Kbind (M−1) | kox (s−1) |
|---|---|---|
| CYP2B4 | 9000 ± 2000 | 48.4 ± 3.2 |
| CYP2B4 E301Q | 8900 ± 1000 | 50.6 ± 1.6 |
| CYP2B4 T302A | 6500 ± 900 | 48.4 ± 2.3 |
| CYP2B4 F429H | 7100 ± 900 | 23.8 ± 1.1 |
| CYP119 | 1030 ± 170 | 32.5 ± 2.7 |
For reactions at 22 °C in 50 mM potassium phosphate buffer (pH 7.4). Errors are at 1σ.
| (1) |
The results in Table 2 are the first directly measured rate constants for an oxidation by Compounds I of a mammalian P450 and its mutants. The kinetic effects of mutations have been studied with a number of P450s, but previous studies involved reactions under turnover conditions where the mutation can affect both the efficiency of formation of the oxidant(s) and the rate of substrate oxidation once the oxidant(s) are formed. Perhaps the most noteworthy finding in this work, therefore, is that the mutations of Glu301 and Thr302 on the I-helix to Gln and Ala, respectively, had almost no effect on the rates of benzphetamine oxidation, whereas mutation of Phe429 to His had a pronounced effect on the rate constant for oxidation.
Residues E301 and T302 in CYP2B4 are on the I-helix located on the distal side of the heme where the oxo moiety is formed and where the substrate is bound (Figure 1). The Glu and Thr residues in this location are highly conserved in P450s and are involved in the protonation reactions necessary to give the active oxidants.11,26 The T302A mutation in a truncated version of the CYP2B4 enzyme, CYPΔ2B4, resulted in major changes in rates of reactions,27 in regioselectivity in oxidations,22,28 and in kinetic isotope effects.29 Similar changes in reactivities were reported for CYPΔ2E1 and its T303A mutant,22,28–30 for P450cam and its T252A mutant,11,31–33 and for P450BM3 and its T268A mutant.34
In the present study, Compounds I from CYP2B4 and its T302A mutant displayed the same oxidation rate constant for reactions with benzphetamine and similar binding constants. Therefore, the large differences in reactivities found previously under turnover conditions for the various P450s and their Thr→Ala mutants apparently are due to changes in the rates or amounts of Compound I produced in the wild type and mutant pairs and not from changes in the reactivity of the various Compounds I. A case has been made that a second active electrophilic oxidant can be formed in P450 enzymes,35 a hydroperoxy-iron or iron-complexed hydrogen peroxide, and the results of the present work strongly support such a model. For example, large changes in kinetic isotope effects,29 in the overall efficiency of the reactions,22 and in the amounts of hydrogen peroxide released36,37 by CYPΔ2B4 and its T302A mutant are likely to reflect changes in the percentages of the two oxidants formed and not changes in the reactivities of Compounds I.
Unlike the E301Q and T302A mutations, which were expected to have little effect on the rates of reactions of Compounds I once formed, the mutation of Phe429 to His was expected to have a kinetic effect. Phe429 is located on the proximal side of the heme adjacent to Cys436, which provides the thiolate ligand to iron (Figure 1). The new histidine residue in the F429H mutant might hydrogen bond to the thiolate, as suggested for the related Phe→His mutation in P450BM3.38 Such hydrogen bonding would reduce the electron density of the thiolate and increase the positive charge on iron, which, in turn, will strengthen the iron-oxygen bond of Compound I in the F429H mutant. Thus, the iron-oxygen bond in Compound I of CYP2B4 F429H was predicted to be stronger than the iron-oxygen bond in Compound I of the wild-type enzyme in advance of our experiments, and a decrease in the rate constants for reactions of Compound I from this mutant was expected.
The results for oxidation of benzphetamine by CYP119 Compound I provide two important insights. The similarity of the oxidation rate constant for CYP119 Compound I with those of Compounds I of CYP2B4 and its mutants suggests that the reported rate constants for oxidations of substrates by CYP119 Compound I14 are typical values and not greatly affected because the enzyme is optimized for reactivity in a thermophile. By extrapolation, other rate constants determined for CYP119 Compound I hydroxylations and epoxidations are typical values. Especially noteworthy in that regard is the rate constant for CYP119 Compound I hydroxylation of unactivated C-H bonds in lauric acid, which is kox = 0.8 s−1 at ambient temperature.14 Apparently that is a typical rate constant for oxidation of a high energy C-H bond by a P450 Compound I.
The second insight involves the overall kinetic effects of the Compound I binding constants. The reduced binding constant for benzphetamine by CYP119 Compound I in comparison to those for Compounds I of CYP2B4 and its mutants demonstrates a method by which the overall reactivity of a P450 enzyme can be tuned. Specifically, the oxidation rate constants for Compounds I of these much different P450 enzymes apparently have little variation, but changes in the substrate binding constants will have a major effect on rates of oxidation when the substrates are present at low enough concentrations to avoid saturation kinetics. Attempts to predict the reactivities of various P450 Compound I species should focus on substrate binding constants, which can be quite variable, as well as the small differences in inherent reactivities of the Compound I oxidants.
The results with CYP2B4 represent the first kinetic studies of reactions of Compound I from a mammalian hepatic P450 enzyme. The rate constants indicate that, whereas P450 Compounds I are more reactive than model iron(IV)-oxo porphyrin radical cations,39 they are not highly reactive. This observation is in marked contrast to the high reactivity of P450 oxidants implicated in some other studies. For example, Compounds I of P450 enzymes have not been readily observed in rapid mixing, stopped-flow, or freeze-quench studies, and cryo-reduction studies of P450cam11 suggest that the active oxidant formed under those conditions reacts with camphor with a rate constant that might exceed 1000 s−1 at ambient temperature because the oxidant does not accumulate to detectable levels. The paradoxical conclusion that Compounds I are not highly reactive whereas at least some P450 oxidants are highly reactive suggests that an oxidant species more reactive than Compound I might be formed in some P450 reactions, perhaps a perferryl-oxo, or iron(V)-oxo, species such as those tentatively identified as highly reactive transients from ligand cleavage reactions of corrole or porphyrin bound iron complexes.40,41 The P450 oxidant reactivity paradox will require more study before it is resolved, but we note that P450 Compound I can effect difficult oxidations even if the reactivity is not great because a large binding constant for substrate in the activated enzyme can compensate for modest reactivity of the oxidant. The CYP2B4 Compound I oxidation of benzphetamine is not especially fast, but the oxidation is accomplished efficiently due to a favorable binding constant.
In summary, Compounds I of CYP2B4 and three of its mutants were produced by laser flash photolysis photo-oxidation of Compounds II formed by reactions of the resting enzymes with peroxynitrite. The kinetics of the Compound I oxidations of benzphetamine reveal that mutations of Glu301 and Thr302 to Gln and Ala, respectively, had little effect on the oxidation rate constants, whereas mutation of Phe429 to His resulted in a two-fold reduction in the oxidation rate constant, presumably due to hydrogen bonding of His to the thiolate ligand to iron from Cys436. P450-catalyzed oxidations appear to be sensitive to the ability of the enzyme and its mutants to form Compounds I, but there is little variation in the rate constants for reactions of Compounds I once they are formed. Substrate binding constants for Compounds I can vary dramatically, however, and are critically important in determining the overall efficiencies of P450 oxidations.
Experimental Section
Materials
CYP2B4 and its mutants42,43 and CYP11913,44,45 were prepared and purified as previously described. The enzymes used in this study had high R/Z ratios with (ASoretMax/A280) > 1.5. Sodium peroxynitrite (PN) solutions were prepared by the method of Uppu and Pryor.46 A sample of (S,S)-2-phenylcylopropylmethane and samples of the products from enzyme-catalyzed oxidations of this substrate were prepared as previously reported.47,48
Production of Compounds I
The method used was similar to that reported for CYP119.13,14 In a typical reaction, 10 μM CYP2B4 in 100 mM potassium phosphate buffer (pH 7.0) was mixed with an equal volume of 0.25 mM PN solution in a stopped-flow mixing unit affixed to an LFP kinetic unit at ambient temperature. The final solution after mixing was 50 mM potassium phosphate buffer (pH 7.4), and the final concentration of P450 was 5 μM. Formation of the Compound II species was monitored by signal growth at 430 nm. Approximately 5 s after mixing, the mixture was irradiated with 355 nm light (third harmonic of a Nd-YAG laser, ca. 5 mJ delivered in 7 ns). A UV-visible difference spectrum was produced by monitoring monochromatic light at 5 nm intervals with a photomultiplier tube. A spectrum of the Compound II species was added to the difference spectrum to give the Compound I spectrum.
Kinetic Studies
The above method was modified such that benzphetamine was present at a desired concentration in the initial enzyme solution. After irradiation, the signal change at 430 nm was monitored. The kinetic traces were solved for double exponential signal growth which consisted of a substrate concentration-dependent term and a substrate concentration-independent term, where the substrate concentration-dependent process was 80–90% of the total. The concentration-dependent rate constants were treated according to Eq 1 in the text.
Products in Bulk Photolysis Reactions
A solution of 3.5 nmol of CYP2B4, 50 equivalents of PN, and 150 equivalents of benzphetamine in 200 μL of 100 mM potassium phosphate buffer (pH 7.0) was prepared at 0 °C in a cuvette held in a temperature-controlled cuvette holder. Approximately 10 s after mixing, the sample was irradiated with 15 pulses of light delivered from a pulsed mercury lamp with a cut-off filter that passed 320–490 nm light (1 J per pulse, 0.1 s pulse duration, 0.5 s between pulses). Following irradiation, the mixture had a UV-visible spectrum similar to that of the resting enzyme. The mixture was extracted with methylene chloride, and the product mixture was analyzed by HPLC using a standard curve to determine the amount of norbenzphetamine as previously described.25
Effect of PN treatment on CYP2B4
A. Nitration of CYP2B4
The reaction was conducted as described above for preparation of Compound I with the exception that the sample was not irradiated. Compound II decayed fully within ca. 120 s as determined by UV-visible spectroscopy. The sample was concentrated and analyzed by HPLC using electrospray ionization in a manner similar to that reported.21
B. CYP2B4-catalyzed oxidation of (S,S)-2-phenylcyclopropylmethane
The oxidation reactions were conducted by the general methods described previously.22,48 Samples of CYP2B4 were treated with 25 equivalents of PN, which was allowed to decay. The samples of CYP2B4 thus treated and untreated samples (0.5 nmol of P450) and 1.0 nmol of P450 reductase in DLPC microsomes with NADPH in 50 mM phosphate buffer (pH 7.0) were allowed to react with the substrate at 10 °C for 30 min, and the products were worked up. Yields of oxidation products were determined by GC with flame-ionization detection, and products were identified by GC-mass spectral comparison to authentic samples. The Supporting Material has additional information.
Supplementary Material
Spectra of CYP2B4 resting enzyme and Compound II, second derivative spectra of Compounds I, representative kinetic traces, details of test reactions of untreated and PN-treated CYP2B4. This material is available free of charge via the Internet at http://pubs.acs.org.
Acknowledgments
This work was supported by the National Institutes of Health (GM48722, M.N.; CA16954, P.F.H.; GM35533, L.W.).
Contributor Information
Lucy Waskell, Email: waskell@med.umich.edu.
Paul F. Hollenberg, Email: phollen@umich.edu.
Martin Newcomb, Email: men@uic.edu.
References
- 1.Ortiz de Montellano PR, editor. Cytochrome P450 Structure, Mechanism, and Biochemistry. 3. Kluwer; New York: 2005. [Google Scholar]
- 2.Guengerich FP. In: Cytochrome P450 Structure, Mechanism, and Biochemistry. 3. Ortiz de Montellano PR, editor. Kluwer; New York: 2005. pp. 377–530. [Google Scholar]
- 3.Scripture CD, Sparreboom A, Figg WD. Lancet Oncol. 2005;6:780–789. doi: 10.1016/S1470-2045(05)70388-0. [DOI] [PubMed] [Google Scholar]
- 4.Rodriguez-Antona C, Ingelman-Sundberg M. Oncogene. 2006;25:1679–1691. doi: 10.1038/sj.onc.1209377. [DOI] [PubMed] [Google Scholar]
- 5.Lieber CS. Drug Metabol Rev. 2004;36:511–529. doi: 10.1081/dmr-200033441. [DOI] [PubMed] [Google Scholar]
- 6.Sono M, Roach MP, Coulter ED, Dawson JH. Chem Rev. 1996;96:2841–2887. doi: 10.1021/cr9500500. [DOI] [PubMed] [Google Scholar]
- 7.Egawa T, Shimada H, Ishimura Y. Biochem Biophys Res Commun. 1994;201:1464–1469. doi: 10.1006/bbrc.1994.1868. [DOI] [PubMed] [Google Scholar]
- 8.Spolitak T, Dawson JH, Ballou DP. J Biol Chem. 2005;280:20300–20309. doi: 10.1074/jbc.M501761200. [DOI] [PubMed] [Google Scholar]
- 9.Schünemann V, Jung C, Terner J, Trautwein AX, Weiss R. J Inorg Biochem. 2002;91:586–596. doi: 10.1016/s0162-0134(02)00476-2. [DOI] [PubMed] [Google Scholar]
- 10.Jung C, Schünemann V, Lendzian F, Trautwein AX, Contzen J, Galander M, Böttger LH, Richter M, Barra AL. Biol Chem. 2005;386:1043–1053. doi: 10.1515/BC.2005.120. [DOI] [PubMed] [Google Scholar]
- 11.Davydov R, Makris TM, Kofman V, Werst DE, Sligar SG, Hoffman BM. J Am Chem Soc. 2001;123:1403–1415. doi: 10.1021/ja003583l. [DOI] [PubMed] [Google Scholar]
- 12.Zhang R, Chandrasena REP, Martinez E, II, Horner JH, Newcomb M. Org Lett. 2005;7:1193–1195. doi: 10.1021/ol050296j. [DOI] [PubMed] [Google Scholar]
- 13.Newcomb M, Zhang R, Chandrasena REP, Halgrimson JA, Horner JH, Makris TM, Sligar SG. J Am Chem Soc. 2006;128:4580–4581. doi: 10.1021/ja060048y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sheng X, Horner JH, Newcomb M. J Am Chem Soc. 2008;120:13310–13320. doi: 10.1021/ja802652b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lu AYH, Coon MJ. J Biol Chem. 1968;243:1331–1332. [PubMed] [Google Scholar]
- 16.Newcomb M, Halgrimson JA, Horner JH, Wasinger EC, Chen LX, Sligar SG. Proc Natl Acad Sci USA. 2008;105:8179–8184. doi: 10.1073/pnas.0708299105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sheng X, Zhang H, Hollenberg PF, Newcomb M. Biochemistry. 2009;48:0000–0000. doi: 10.1021/bi802279d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mehl M, Daiber A, Herold S, Shoun H, Ullrich V. Nitric Oxide. 1999;3:142–152. doi: 10.1006/niox.1999.0217. [DOI] [PubMed] [Google Scholar]
- 19.Daiber A, Herold S, Schöneich C, Namgaladze D, Peterson JA, Ullrich V. Eur J Biochem. 2000;267:6729–6739. doi: 10.1046/j.1432-1033.2000.01768.x. [DOI] [PubMed] [Google Scholar]
- 20.Gebicka L, Didik J. J Inorg Biochem. 2007;101:159–164. doi: 10.1016/j.jinorgbio.2006.09.007. [DOI] [PubMed] [Google Scholar]
- 21.Lin HL, Myshkin E, Waskell L, Hollenberg PF. Chem Res Toxicol. 2007;20:1612–1622. doi: 10.1021/tx700220e. [DOI] [PubMed] [Google Scholar]
- 22.Toy PH, Newcomb M, Coon MJ, Vaz ADN. J Am Chem Soc. 1998;120:9718–9719. [Google Scholar]
- 23.Palcic MM, Rutter R, Araiso T, Hager LP, Dunford HB. Biochem Biophys Res Commun. 1980;94:1123–1127. doi: 10.1016/0006-291x(80)90535-5. [DOI] [PubMed] [Google Scholar]
- 24.Egawa T, Proshlyakov DA, Miki H, Makino R, Ogura T, Kitagawa T, Ishimura Y. J Biol Inorg Chem. 2001;6:46–54. doi: 10.1007/s007750000181. [DOI] [PubMed] [Google Scholar]
- 25.Zhang HM, Im SC, Waskell L. J Biol Chem. 2007;282:29766–29776. doi: 10.1074/jbc.M703845200. [DOI] [PubMed] [Google Scholar]
- 26.Nagano S, Poulos TL. J Biol Chem. 2005;280:31659–31663. doi: 10.1074/jbc.M505261200. [DOI] [PubMed] [Google Scholar]
- 27.Roberts ES, Pernecky SJ, Alworth WL, Hollenberg PF. Arch Biochem Biophys. 1996;331:170–176. doi: 10.1006/abbi.1996.0295. [DOI] [PubMed] [Google Scholar]
- 28.Vaz ADN, McGinnity DF, Coon MJ. Proc Natl Acad Sci USA. 1998;95:3555–3560. doi: 10.1073/pnas.95.7.3555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Newcomb M, Aebisher D, Shen RN, Chandrasena REP, Hollenberg PF, Coon MJ. J Am Chem Soc. 2003;125:6064–6065. doi: 10.1021/ja0343858. [DOI] [PubMed] [Google Scholar]
- 30.Newcomb M, Shen R, Choi SY, Toy PH, Hollenberg PF, Vaz ADN, Coon MJ. J Am Chem Soc. 2000;122:2677–2686. [Google Scholar]
- 31.Imai M, Shimada H, Watanabe Y, Matsushimahibiya Y, Makino R, Koga H, Horiuchi T, Ishimura Y. Proc Natl Acad Sci USA. 1989;86:7823–7827. doi: 10.1073/pnas.86.20.7823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Martinis SA, Atkins WM, Stayton PS, Sligar SG. J Am Chem Soc. 1989;111:9252–9253. [Google Scholar]
- 33.Jin SX, Makris TM, Bryson TA, Sligar SG, Dawson JH. J Am Chem Soc. 2003;125:3406–3407. doi: 10.1021/ja029272n. [DOI] [PubMed] [Google Scholar]
- 34.Yeom H, Sligar SG, Li HY, Poulos TL, Fulco AJ. Biochemistry. 1995;34:14733–14740. doi: 10.1021/bi00045a014. [DOI] [PubMed] [Google Scholar]
- 35.Newcomb M, Hollenberg PF, Coon MJ. Arch Biochem Biophys. 2003;409:72–79. doi: 10.1016/s0003-9861(02)00445-9. [DOI] [PubMed] [Google Scholar]
- 36.Chandrasena REP, Vatsis KP, Coon MJ, Hollenberg PF, Newcomb M. J Am Chem Soc. 2004;126:115–126. doi: 10.1021/ja038237t. [DOI] [PubMed] [Google Scholar]
- 37.Vatsis KP, Peng HM, Coon MJ. J Inorg Biochem. 2002;91:542–553. doi: 10.1016/s0162-0134(02)00438-5. [DOI] [PubMed] [Google Scholar]
- 38.Ost TWB, Miles CS, Munro AW, Murdoch J, Reid GA, Chapman SK. Biochemistry. 2001;40:13421–13429. doi: 10.1021/bi010716m. [DOI] [PubMed] [Google Scholar]
- 39.Pan ZZ, Zhang R, Newcomb M. J Inorg Biochem. 2006;100:524–532. doi: 10.1016/j.jinorgbio.2005.12.022. [DOI] [PubMed] [Google Scholar]
- 40.Harischandra DN, Zhang R, Newcomb M. J Am Chem Soc. 2005;127:13776–13777. doi: 10.1021/ja0542439. [DOI] [PubMed] [Google Scholar]
- 41.Pan ZZ, Zhang R, Fung LWM, Newcomb M. Inorg Chem. 2007;46:1517–1519. doi: 10.1021/ic061972w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hanna IH, Teiber JF, Kokones KL, Hollenberg PF. Arch Biochem Biophys. 1998;350:324–332. doi: 10.1006/abbi.1997.0534. [DOI] [PubMed] [Google Scholar]
- 43.Saribas AS, Gruenke L, Waskell L. Protein Expression Purif. 2001;21:303–309. doi: 10.1006/prep.2000.1377. [DOI] [PubMed] [Google Scholar]
- 44.McLean MA, Maves SA, Weiss KE, Krepich S, Sligar SG. Biochem Biophys Res Commun. 1998;252:166–172. doi: 10.1006/bbrc.1998.9584. [DOI] [PubMed] [Google Scholar]
- 45.Maves SA, Sligar SG. Protein Sci. 2001;10:161–168. doi: 10.1110/ps.17601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Uppu RM, Pryor WA. Anal Biochem. 1996;236:242–249. [PubMed] [Google Scholar]
- 47.Fu H, Newcomb M, Wong CH. J Am Chem Soc. 1991;113:5878–5880. [Google Scholar]
- 48.Atkinson JK, Hollenberg PF, Ingold KU, Johnson CC, Le Tadic MH, Newcomb M, Putt DA. Biochemistry. 1994;33:10630–10637. doi: 10.1021/bi00201a009. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Spectra of CYP2B4 resting enzyme and Compound II, second derivative spectra of Compounds I, representative kinetic traces, details of test reactions of untreated and PN-treated CYP2B4. This material is available free of charge via the Internet at http://pubs.acs.org.