

Abstract
On-bead screening of one-bead-one-compound (OBOC) libraries provides a powerful method for the rapid identification of active compounds against molecular or cellular targets. However, on-bead screening is susceptible to interference from nonspecific binding, which results in biased screening data and false positives. In this work, we have found that a major source of nonspecific binding is derived from the high ligand loading on the library beads, which permits a macromolecular target (e.g., a protein) to simultaneously interact with multiple ligands on the bead surface. To circumvent this problem, we have synthesized a phosphotyrosyl (pY)-containing peptide library on spatially segregated TentaGel microbeads, which feature a 10-fold reduced peptide loading on the bead surface but a normal peptide loading in the bead interior. The library was screened against a panel of 10 Src homology 2 (SH2) domains including those of Csk and Fyn kinases and adaptor protein SLAP, and the specific recognition motif(s) was successfully identified for each of the domains. In contrast, when the SH2 domains were screened against a control library that contained unaltered (high) ligand loading at the bead surface, six of them exhibited varying degrees of sequence biases, ranging from minor perturbation in the relative abundance of different sequences to the exclusive selection of false positive sequences that have no measurable affinity to the target protein. These results indicate that reduction of the ligand loading on the bead surface represents a simple, effective strategy to largely eliminate the interference from nonspecific binding, while preserving sufficient amounts of materials in the bead interior for compound identification. This finding should further expand the utility of OBOC libraries in biomedical research.
Keywords: Combinatorial library, nonspecific binding, one-bead-one-compound library, partial Edman degradation, reduced ligand loading, spatial segregation, Src homology 2 domain
Introduction
On-bead screening of one-bead-one-compound (OBOC) combinatorial libraries has been widely practiced to identify the binding ligands of macromolecular receptors (e.g., proteins),1 the preferred substrates of enzymes,2 and catalysts for chemical transformations.3 The popularity of OBOC libraries is fueled by the fact that large OBOC libraries are readily accessible through the “split-and-pool” synthesis method4 and a large number of beads/compounds can be screened simultaneously (and thus rapidly) against a target of interest (e.g., up to 107 beads can be screened in a few hours). However, on-bead screening of OBOC libraries does pose some significant technical challenges. First, it necessitates post-screening identification of “hit” compounds individually. For peptide and peptoid libraries, this challenge has largely been overcome with the advent of enabling technologies such as partial Edman degradation/mass spectrometry (PED/MS)5 and tandem mass spectrometry (MS/MS).6 For small-molecule libraries, hit identification is still problematic despite of the availability of several ingenious encoding strategies.7 Second, on-bead screening against macromolecular targets is highly susceptible to interference from nonspecific interactions between the target molecule and the bead surface (which are defined as any interactions other than the intended interaction), resulting in biased screening data or in some cases, complete failure of the screening experiment. For example, during our previous work to profile the sequence specificity of Src homology 2 (SH2)8-10 and PDZ domains,11 we found that some of the protein domains selected peptide sequences that are rich in positively charged residues (Arg, Lys, and His). When the selected peptides were individually synthesized and tested for binding to their “cognate” protein domains, it was clear that the positively charged residues contributed little to the binding affinity. In some cases, the selected peptides failed to bind to the target protein at all. This problem appears to be especially prevalent and severe for protein domains that have intrinsically low affinity to their specific ligands (e.g., PDZ domains).11 Other investigators have reported similar problems.1d, 12 It was generally assumed that the false positives and sequence biases were caused by some type(s) of nonspecific interactions between the target proteins and the bead surfaces, but the nature of the nonspecific interactions has never been fully investigated. In this work, we have found that a major source of the nonspecific interactions is the high ligand loading on the commercially available resins commonly used for the OBOC libraries. To address this problem, we have synthesized OBOC peptide libraries on spatially segregated beads, which contain a reduced ligand density on the bead surface but a high loading in the bead interior. The resulting libraries were screened against several previously problematic SH2 domains and the specific binding motif(s) was successfully determined for each of the SH2 domains with little interference from the nonspecific interactions.
Results and Discussion
Possible Origin of Nonspecific Interactions
We felt that the high ligand loading on the library beads might be a source of the nonspecific interactions, as high ligand density on a surface makes it possible for a target protein to simultaneously interact with multiple ligand molecules; the resulting avidity may rival or even exceed the affinity of a specific protein-ligand interaction. The TentaGel resin commonly used for OBOC libraries (90-μm beads) has a loading capacity of ∼0.3 mmol/g, which corresponds to a ligand concentration of ∼100 mM on the resin. At such a high ligand density, a negatively charged protein (or a negatively charged region on the protein surface) may simultaneously bind to several adjacent, positively charged peptides through charge-charge interactions (Figure 1a). Similarly, a positively charged protein may bind to a bead displaying negatively charged ligands, or a protein containing a hydrophobic surface may interact with multiple nonpolar ligands on a bead. Interactions of this type would lead to the formation of false positive beads. Alternatively, the target protein may be engaged in specific interactions with an immobilized ligand, but other regions of the protein surface that are outside the specific ligand-binding site may interact with neighboring ligands on the same bead (Figure 1b). This would bias the screening towards sequences that are capable of such nonspecific interactions (e.g., positively charged ligands when the target protein is negatively charged). We envision that both types of nonspecific binding should be eliminated (or at least greatly reduced) if the ligand loading on the beads were to be decreased (e.g., by 10−100-fold) so that a target protein cannot interact with more than one ligand molecule (Figure 1c). The reduced ligand concentration (1−10 mM) should have little effect on the specific interaction, as it is still well above the dissociation constants of most biologically relevant interactions (which are typically in the nM to μM range). Unfortunately, a simple reduction of the ligand loading by 10−100-fold would complicate the subsequent hit identification, as the amount of compounds carried by a single bead (1−10 pmol) would be insufficient for the analytical techniques currently available for compound identification.
Figure 1.
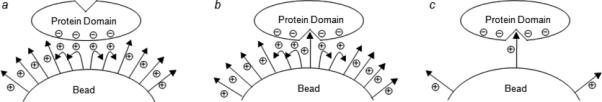
Scheme showing the different mechanisms by which a macromolecular target (e.g., protein) binds to a bead containing immobilized low-molecular-weight ligands (e.g., peptides). (a) Nonspecific binding of a protein containing a negatively charged surface patch (either near or remote from the specific ligand-binding site) to a bead containing positively charged, high-density ligands via multidentate charge-charge interactions (avidity effect), giving rise to false positive beads. (b) Biased binding of a protein to a bead containing a high loading of low-affinity ligands and the weak specific interaction is enhanced by charge-charge interactions between the protein and neighboring ligands. (c) Binding of a protein to a reduced-density bead through high-affinity, specific interaction between the protein and a single immobilized ligand molecule.
Design and Synthesis of Low-Density (LD) pY Peptide Library
We chose to test our hypothesis on SH2 domains, which are ∼100-amino acid modular domains frequently found in signaling proteins.13 SH2 domains promote protein-protein interactions by recognizing specific pY peptide motifs and their sequence specificity is primarily determined by 2 to 3 amino acid residues flanking the pY residue (usually positions −2 to +3 relative to pY, which is designated as position 0). One of our ongoing projects is to systematically determine the recognition motifs of the 119 human SH2 domains through screening of combinatorial peptide libraries and subsequently use the specificity information to identify their in vivo protein partners. In our previous work,1e, 9, 10 all SH2 domains were screened against a conventional pY peptide library, TAXXpYXXXLNBBRM-resin [high-density (HD) library, where X represents the 18 proteinogenic amino acids (except for Cys and Met) plus norleucine (Nle) and L-α-aminobutyric acid (Abu) as Met and Cys surrogates, respectively; B is β-alanine],14 synthesized on 90-μm TentaGel beads (0.3 mmol/g). Although the specific recognition motifs were successfully identified from the library for most of the tested SH2 domains, we have also encountered a significant number of problematic proteins. For example, screening of the HD library against the SH2 domain of the Csk kinase resulted in sequences rich in arginine at the pY-1, pY+1, and pY+2 positions (Figure 2a).9 When one of the selected peptides, R(Abu)pYRSILN, was synthesized and tested for binding to the Csk SH2 domain, it had only modest affinity (KD = 4.1 μM). Studies from other laboratories have reported that the Csk SH2 domain strongly prefers Ala, Ser, and Thr at the pY+1 position (but not Arg).15, 16 Screening of the HD library against the SH2 domain of Fyn, a member of the Src family kinases, gave exclusively arginine- and lysine-rich sequences that failed to bind to the SH2 domain when tested individually (vide infra). We hypothesized that the positively charged sequences were selected because they were engaged in nonspecific interactions with the negatively charged regions on the SH2 domains (Csk and Fyn SH2 domains have pI values of 6.0 and 8.8, respectively) (Figure 1a and b).
To test this hypothesis and determine the recognition motifs of these difficult protein domains, we designed a second pY peptide library with reduced ligand loading (LD library). The LD library is identical to the HD library except that each bead in the LD library is spatially segregated into two different layers; the inner core carries a normal peptide loading (∼0.3 mmol/g), while the surface layer has a 10-fold reduced ligand density (Scheme 1). The library was synthesized on TentaGel S NH2 resin (90 μm, ∼2.86 μ 106 beads/g, ∼100 pmol/bead). Bead segregation was achieved by using the method of Lam and co-workers.17 Briefly, TentaGel beads that had been pre-equilibrated in water were quickly suspended in 55:45 (v/v) dichloromethane/diethyl ether containing 0.4 equiv of Nα-t-butoxycarbonyl-phenylalanine N-hydroxysuccinimide ester (Boc-Phe-OSu), 0.1 equiv of Nα-fluorenylmethoxycarbonyl-methionine N-hydroxysuccinimide ester (Fmoc-Met-OSu), and 0.5 equiv of diisopropylethylamine (DIPEA). As the surface layer gradually re-equilibrated in the organic solvents, the free amino groups in this layer became acylated with Boc-Phe and Fmoc-Met. The amines in the inner layer did not react during this period because they remained in the aqueous phase and were inaccessible to the acylating agents. When the organic solvents eventually reached the inner layer, the acylating agents (total 0.5 equiv) had already been exhausted. The internal amines (0.5 equiv) were subsequently acylated with Fmoc-Met-OH. The library beads were sequentially treated with trifluoroacetic acid and acetic anhydride to permanently block the majority of the surface amines. After that, the library was synthesized by the split-and-pool method4 and standard Fmoc/HBTU chemistry. During library synthesis, the actual peptide loading in the surface layer was determined by treating small aliquots of the resin after synthetic steps b and c (Scheme 1) with piperidine and quantifying the amounts of Fmoc group released, as well as measuring the amount of free amine after step d (by ninhydrin test). It was found that the peptide loading in the surface layer was ∼10-fold lower than that of the bead interior. This indicates that Fmoc-Met-OSu reacted with the resin-bound amine more slowly or was hydrolyzed faster than Boc-Phe-OSu did.
Scheme 1.

Synthesis of Reduced-Density pY Peptide Librarya
Sequence Specificity of the Csk SH2 Domain
The LD library was screened against the Csk SH2 domain using an on-bead enzyme-linked assay.1e Binding of the biotinylated SH2 domain to a bead recruits a streptavidin-alkaline phosphatase (SA-AP) conjugate to the bead. Upon subsequent treatment of 5-bromo-4-chloro-3-indolyl phosphate (BCIP), a positive bead becomes turquoise colored. Screening of the Csk SH2 domain (0.5 μM) against a total of 120 mg of the library (∼300,000 beads) resulted in 60 intensely colored beads, which were removed from the library and individually sequenced by the PED/MS method5 to give 50 complete sequences (Table 1). A comparison of these sequences with those previously selected from the HD pY library showed significant differences at all positions except for the pY+3 position (Figure 2). For the sequences derived from the HD library, Arg and Lys are among the most frequently selected amino acids at positions from pY-2 to pY+2. This is not the case for peptides selected from the LD library. At the pY+1 position, the Csk SH2 domain selected predominantly small residues such as Ala, Thr, Ser, and Abu (Figure 2b), consistent with previous reports from other laboratories.15, 16 At position pY+2, data from the LD library indicates that the Csk SH2 domain can accept a variety of amino acids but with some preference for small residues such as Abu and Thr. Like most other SH2 domains, it has no obvious selectivity at the pY-2 and -1 positions. When individually synthesized and tested for binding, peptide R(Abu)pYRSILN had only a modest affinity toward the Csk SH2 domain (KD = 4.1 μM), while the corresponding peptide containing an Ala at the pY+1 position [R(Abu)pYASILN] exhibited a 5-fold higher affinity (KD = 0.9 μM) (Table 2). We conclude that the positively charged residues (Arg and Lys) were selected from the HD library because they provide additional binding affinity by interacting with the negative charges on the SH2 domain surface (Figure 1, mechanism b). At the pY+3 position, a similar set of amino acids (Val, Ile, Pro, Abu, Nle, and Leu) were selected from both libraries, indicating that the Csk SH2 domain requires an aliphatic hydrophobic amino acid at this position. Presumably, substitution of Arg or Lys at this critical position would reduce the overall affinity to such an extent that it cannot be compensated for by the addition of charge-charge interactions. Note that there is an increased frequency of appearance for His at the pY-2 position among the peptides selected from the LD library. This is caused by some other bias of yet unknown origin and can be eliminated by the addition of 10 mM imidazole into the screening reaction (R. Hard and D.P., unpublished observations). Since it has been observed with other protein domains9, 11 and both HD and LD libraries, it is unlikely caused by the avidity effect discussed above.
Table 1.
Csk SH2 domain-Binding Sequences Selected from the LD librarya
| CCpYSQL | DEpYTTV | PN pYKLC |
| HV pYAKC | CLpYARC | HV pYTCP |
| EN pYSAI | HApYCLR | HK pYRVP |
| RT pYAQV | DLpYARI | HMpYTTV |
| AH pYACC | GN pYRCV | AT pYHCI |
| MC pYPPE | AE pYACC | HC pYART |
| HT pYTTL | TD pYAKV | DC pYASM |
| HT pYSQI | YR pYATS | PQ pYTEI |
| MC pYKYP | QD pYCRA | TP pYICS |
| KH pYAVC | HY pYKQM | PG pYACP |
| DS pYSCI | TNpYKMA | DY pYTAS |
| HQ pYAGI | PTpYCRI | HSpYAEC |
| QE pYAVL | HK pYTCL | EEpYCCM |
| HG pYACM | QEpYARV | HSpYTLV |
| KP pYKCV | KQpYTWI | YLpYITF |
| HG pYTTM | HK pYCRV | NL pYQTP |
| PI pYSSP | HQ pYACC |
C, (S)-2-aminobutyric acid; M, norleucine.
Table 2.
Dissociation Constants (KD, μM) of Selected Peptides against the Csk, Fyn, and SLAP SH2 domainsa
| Peptide sequence | Csk SH2 | Fyn SH2 | SLAP SH2 |
|---|---|---|---|
| Biotin-miniPEG-BBR(Abu)pYRSILN-NH2 | 4.1 ± 0.2b | ND | ND |
| Ac-R(Abu)pYASILNK(dPEG4-biotin)-NH2 | 0.9 ± 0.1 | ND | ND |
| Ac-AYpYAEILNBK(dPEG4-biotin)-NH2 | ND | 0.22 ± 0.04 | ND |
| Ac-RQpYFRRLNK(dPEG4-biotin)-NH2 | ND | >10 | ND |
| Ac-QApYEQVLNK(dPEG4-biotin)-NH2 | ND | ND | 2.2 ± 0.2 |
| Ac-QApYAQVLNK(dPEG4-biotin)-NH2 | ND | ND | 3.6 ± 0.1 |
| Ac-LTpYENDLNK(dPEG4-biotin)-NH2 | ND | ND | 0.48 ± 0.03 |
B, β-alanine; ND, not determined.
Data from ref. 9.
Sequence Specificity of Fyn and SLAP SH2 Domains
We have subsequently used the LD library to determine the sequence specificity of 9 other SH2 domains (Fyn, Fgr, Grb2, Lck, Lnk, SHP-1N, SLAP, SOCS2, and SOCS3). These domains were also screened against the HD library in parallel, to determine whether the improvement observed for the Csk SH2 domain is a general phenomenon. Out of the 9 SH2 domains, two (Fyn and Fgr) showed dramatic improvements. When screened against the HD library, the Fyn SH2 domain selected only false positive sequences, while the Fgr SH2 domain selected a mixture of bona fide, biased, and false positive sequences. Both domains selected almost exclusively bona fide ligands from the LD library. Three domains (SLAP, SOCS2 and 3) gave biased results when screened against the HD library, but the biases were absent with the LD library. The remaining four SH2 domains gave similar results with both libraries (all successful). The results of the Fyn and SLAP SH2 domains are described in detail below, as representatives of the first two categories. The data for the other seven SH2 domains will be reported elsewhere.
Fyn is a member of the Src family kinases and contains a single SH2 domain.18 The sequence specificity of the Fyn SH2 domain has previously been assessed by screening against oriented peptide libraries.15, 16 The earlier studies reported consensus sequences of pY(E/T)(E/D/Q)(I/V/M)15 and (N/P)(Y/F)pY(E/D/Y)(N/E/T/M)(I/L/V/P)(D/E),16 but did not give the individual binding sequences. Screening of the Fyn SH2 domain against the HD library produced peptides that are overwhelmingly positively charged and rich in hydrophobic, aromatic residues (Figure 3a and Table S1 in Supporting Information). One of the representative sequences, RQpYFRR, was resynthesized and found to have no detectable binding to the Fyn SH2 domain (Figure 4 and Table 2). Next, the Fyn SH2 domain was screened against 180 mg of the LD library (∼540,000 beads) under the identical conditions to obtain 97 binding sequences (Table S2). Inspection of these sequences revealed that they are in general agreement with the consensus sequences previously reported15, 16 and do not contain an overrepresentation of positively charged or aromatic residues (Figure 3b). The SH2 domain selected predominantly aliphatic hydrophobic residues at the pY+3 position including Ile, Nle, Val, Pro, and Leu. At position pY+1, it has a strong preference for acidic residues Glu and Asp and to a lesser extent other hydrophilic residues (e.g., His, Gln, and Arg). A small subset of the selected sequences also contained an Ala as the pY+1 residue. The Fyn SH2 domain has no significant selectivity at other positions. A representative sequence from the minor family (AYpYAEI) was resynthesized and shown to bind to the Fyn SH2 domain with high affinity (KD = 0.22 μM), providing an important confirmation of our screening results (Figure 4 and Table 2). Note that both of the earlier studies failed to reveal this minor family of sequences. This is due to the fact that the methods employed by the earlier studies are designed to select for both the affinity and abundance of a certain type of sequences in a library; as such, they cannot identify sequences that are tight binding but underrepresented in the library. In our method, an active compound is selected solely based on its affinity to the target molecule and therefore, any high-affinity ligand is selected regardless of its abundance.
Figure 3.
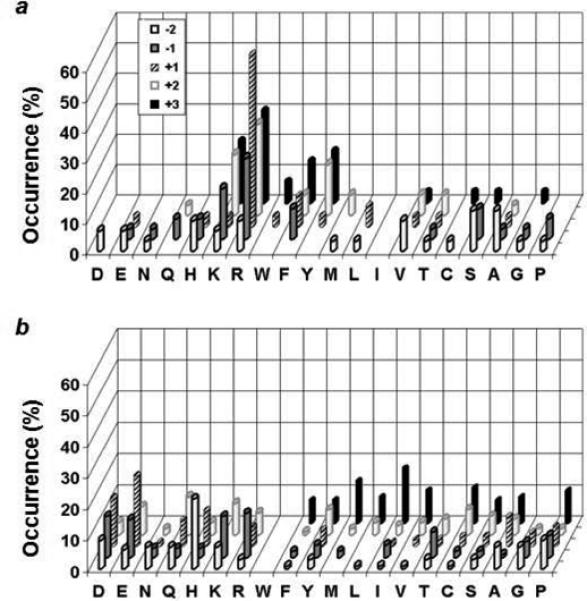
A comparison of the peptide sequences selected from the HD (a) vs LD library (b) against the Fyn SH2 domain. The histograms represent the amino acids identified at each position from pY-2 to pY+3. Number of occurrence on the z axis represents the percentage of selected sequences that contain a particular amino acid at a certain position. M, Nle; C, Abu.
Figure 4.
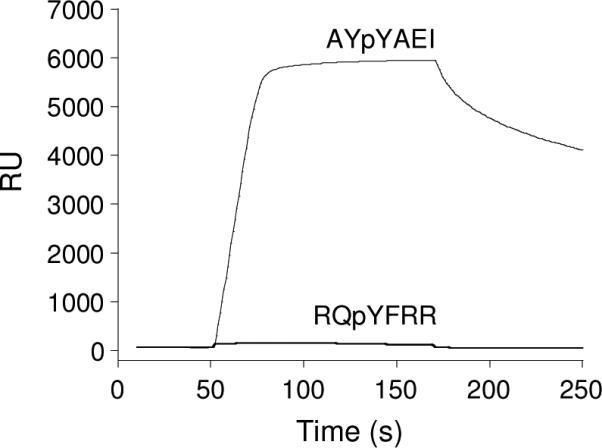
SPR sensograms showing the binding properties of the Fyn SH2 domain to peptides selected from HD (RQpYFRR) vs LD libraries (AYpYAEI). The peptides were biotinylated and immobilized onto a streptavidin-coated sensorchip and the Fyn SH2 domain (5 μM) was flowed over the chip surface. RU, response units.
SLAP (Src-like adaptor protein) belongs to a subfamily of adapter proteins that negatively regulate cellular signaling initiated by tyrosine kinases.19 SLAP has a myristylated N-terminus, followed by SH3 and SH2 domains with high homology to Src family tyrosine kinases and a unique C-terminal tail, which is important for c-Cbl binding.20 SLAP can compete with the Src family kinases for binding to pY proteins and works together with c-Cbl to direct some pY proteins to proteosome-mediated degradation. However, to our knowledge, there has not been any report on the sequence specificity of the SLAP SH2 domain. We initially expressed the SLAP SH2 domain by itself and found that the protein was nonfunctional. We therefore expressed the N-terminal fragment containing both the SH3 and SH2 domains of SLAP (amino acids 1−187) plus C-terminal ybbR21 (for specific biotinylation) and six-histidine tags (to facilitate protein purification). We screened the resulting SH2 domain against both HD and LD pY libraries (∼100 mg each) under identical conditions to obtain 62 and 94 complete sequences, respectively (Tables S3 and S4). Similar sequences were selected from both libraries, which can be classified into two different families. The SLAP SH2 domain has the most stringent selectivity at the pY+3 position, with both families requiring an aliphatic hydrophobic residue such as Val, Nle, Abu, Ile, and Pro (Figure 5). The difference between the two families is at the pY+1 position, where one family contains a small residue such as Ala, Abu, Ser, and Thr, whereas the other family prefers acidic residues Glu, Asp, and structurally similar Gln, and His. The SLAP SH2 domain has some preference for an Asn at the pY+2 position, although most amino acids are tolerated. There is no significant sequence selectivity on the N-terminal side of pY other than an overrepresentation of His at position pY-2. There are, however, subtle differences between the screening results from the two libraries. While the same two families of sequences were selected from both libraries, the HD library produced a higher percentage (71%) of the family one sequences (with a small residue at the pY+1 position). On the other hand, screening against the LD library gave a much higher percentage (44%) of the family two sequences (with Glu, Asp, or Gln at the pY+1 position). The HD library also selected more sequences with Arg and Lys at the less critical positions (e.g., positions pY-2, pY-1, and pY+2), whereas the LD library selected a few peptides with an acidic residue at the pY+3 position, which apparently form a distinct third family (LTpYEND, HWpYEND, RLpYINE, and GFpYVNE). Three representative sequences (one from each family) were individually synthesized and their affinities to the SLAP SH2 domain were determined by surface plasmon resonance (Table 2). Peptide QApYEQV, which was selected from the LD library, has a KD value of 2.2 μM, while peptide QApYAQV selected from the HD library has a 1.5-fold lower affinity (KD = 3.6 μM). This suggests that the family two sequences bind to the SLAP SH2 domain with slightly higher affinities than the family one sequences. The family three sequence (LTpYEND) had the highest affinity (KD = 0.48 μM). We attribute the lower occurrence of acidic residues (and the increased number of positively charged residues) from the HD library to the nonspecific charge-charge interactions between the SLAP protein and neighboring peptides. SLAP is negatively charged under the screening conditions (pI = 6.4) and is presumably repelled from those beads that carry a high density of negatively charged ligands but attracted to positively charged beads. This charge-charge repulsion/attraction should be greatly diminished in the LD library.
Figure 5.
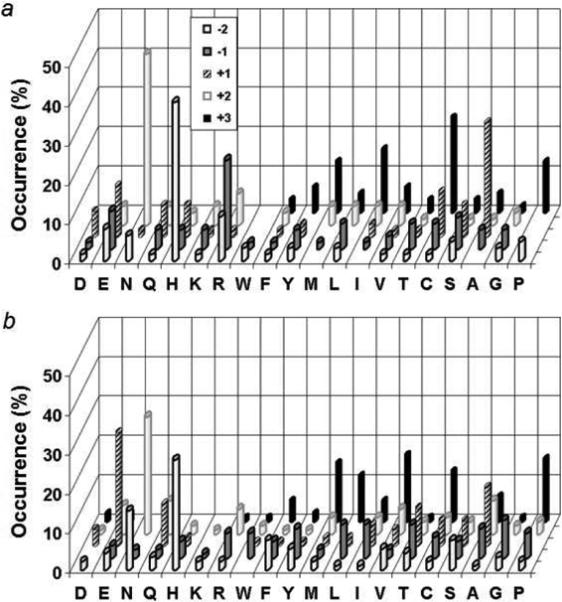
A comparison of the peptide sequences selected from the HD (a) vs LD library (b) against the SLAP SH2 domain. The histograms represent the amino acids identified at each position from pY-2 to pY+3. Occurrence on the z axis represents the percentage of selected sequences that contain a particular amino acid at a certain position. M, Nle; C, Abu.
Database Search of Potential Partner Proteins of SLAP
We searched the Phosphosite database (webpage: http://www.phosphosite.org/) using the sequence motif Y[E/D/A/S]X[P/V/L/I] (X denotes any amino acid) to identify the potential target proteins of SLAP. The search identified 346 human pY proteins which contain at least one pY motif that matches the consensus sequence of the SLAP SH2 domain. Among these pY proteins, 186 have already been assigned physiological functions. Since SLAP is known to function as a negative regulator during B- and T-cell signaling,22-24 we further restricted the candidate proteins to those that have previously been implicated in B- and/or T-cell signaling. This resulted in a total of 47 proteins (Table 3), which we consider as “highly probable” SLAP-binding proteins. Among these 47 proteins, seven [T-cell receptor ζ chain, protein tyrosine kinases Syk and ZAP-70, proto-oncogene product Vav, lymphocyte cytosolic protein SLP-76, linker for activation of T-cell (LAT), and B-cell receptor Ig-α) have previously been reported to physically interact with SLAP via its SH2 domain.20, 22-24 It should be pointed out that SLAP is ubiquitously expressed and therefore expected to also function in other cell types. Therefore, it is likely that additional SLAP-binding proteins will be identified from the other 141 proteins (Table S5 in Supporting Information).
Table 3.
Human Proteins Predicted to Bind to SLAP via the SH2 Domaina
| Protein | Predicted binding sites | Ref |
|---|---|---|
| Activated leukocyte cell adhesion molecule (ALCAM) | SVQyDDVPE | |
| Ankyrin repeat and SAM domain-containing protein 1A (ANKS1) | EHPyELLLT; DRPyEEPPQ | |
| Tyrosine-protein kinase Abl2 | VALyDFVAS | |
| B-cell linker protein (BLNK) | DSDyENPDE; DDSyEPPPV | |
| CRK-associated substrate-related protein (hEF1) | GYVyEYPSR; KDVyDIPPS | |
| E3 ubiquitin-protein ligase Cbl | SCTyEAMYN; DDGyDVPKP | |
| E3 ubiquitin-protein ligase Cbl-b | SEEyDVPPR; SQDyDQLPS | |
| B-lymphocyte antigen CD19 | GEGyEEPDS; GSGyENPED; SQSyEDMRG; ADSyENMDN |
|
| B-cell receptor CD22 | VGDyENVIP; GIHySELIQ | |
| Myeloid cell surface antigen CD33 | STEySEVRT | |
| T-cell surface glycoprotein CD3 epsilon chain (CD3ε) | NPDyEPIRK | |
| T-cell surface glycoprotein CD3 zeta chain (CD3ζ)b | REEyDVLDK; EGLyNELQK AEAySEIGM; KDTyDALHM |
20, 22, 23 |
| T-cell differentiation antigen CD6 | GEWyQNFQP; NDDyDDISA | |
| SLAM family member 5 (CD84) | SRIyDEILQ; NTVySEVQF | |
| Proto-oncogene C-crk | PNAyDKTAL | |
| Crk-like protein (CrkL) | RTLyDFPGN; AHAyAQPQT PCAyDKTAL |
|
| Catenin beta-1 (CTNNB1) | LINyQDDAE; TYTyEKLLW | |
| Disabled homolog 1 (DAB1) | EGVyDVPKS; ENIyQVPTS | |
| B lymphocyte adapter protein Bam32 (DAPP1) | PSIyESVRV | |
| Drebrin-like protein (DBNL) | EAVyEEPPE; ETFyEQPPL | |
| Erythropoietin receptor (EpoR) | DGPySNPyENS; LWLyQNDGC | |
| Tyrosine-protein kinase ZAP-70b | TSVyESPySDP | 20, 23 |
| Proto-oncogene vav (VAV1)b | EDLyDCVEN; DEIyEDLMR | 23 |
| Tyrosine-protein kinase Txk | KALyDFLPR | |
| Short transient receptor potential channel 5 (TRPC5) | SFRyEVLDL | |
| Tyrosine-protein kinase Tec | YTNyEVVTM | |
| Src-activating and signaling molecule protein (Srcasm) | SHAyDNFLE; EAIyEEIDA | |
| Switch-associated protein 70 (SWAP-70) | LEQyEEVKK | |
| Tyrosine-protein kinase Sykb | TEVyESPYA | 20 |
| Lymphocyte cytosolic protein 2 (SLP-76)b | EDDyESPND; DGDyESPNE | 23 |
| Suppressor of cytokine signaling 3 (SOCS3) | LDSyEKVTQ; LDQyDAPL | |
| SH2 domain-containing inositol-5'-phosphatase 1 (SHIP) | EKLyDFVKT | |
| Tyrosine-protein phosphatase non-receptor type 11 (SHP-2) | ARVyENVGL | |
| Src kinase-associated phosphoprotein 1 (SKAP55) | EETyDDIDG; EDIyEVLPD | |
| Signaling lymphocytic activation molecule (SLAM) | LTIyAQVQK | |
| FYN-binding protein (SLAP-130) | QEVyDDVAE; GCIyDND | |
| SH2 domain-containing adapter protein B (Shb) | ADEyDQPWE | |
| Microtubule-associated protein RP/EB family member 2 (RP1) | GKEyDPVEA | |
| Src kinase-associated phosphoprotein 2 (RA70) | GELyDDVDH; DEIyEELPE | |
| Beta-type platelet-derived growth factor receptor (PDGFRβ) | AELySNALP; MAPyDNYVP DEIyEIMQK |
|
| Linker for activation of T-cells family member 2 (LAB) | ANSyENVLI | |
| Leukocyte-associated immunoglobulin-like receptor 1 (LAIR1) | EVTyAQLDH | |
| Linker for activation of T-cells family member 1 (LAT)b | VASyENEGA; APDyENLQE | 20, 23 |
| B-cell antigen receptor complex-associated protein alpha-chain (Ig-α)b | ENLyEGLNL; CSMyEDISR QGTyQDVGS |
24 |
| Focal adhesion kinase 1 (FAK) | TDDyAEIID; DKVyENVTG | |
| Lck-interacting transmembrane adapter 1 (LIME1) | LEATySNVG | |
| Hematopoietic lineage cell-specific protein (HS1) | PENDyEDVEE; EPEGDyEEVLE |
y, phosphotyrosine.
Proteins the have previously been shown to bind SLAP via its SH2 domain.
Conclusion
A major problem with OBOC libraries has been the formation of false positives and biases during on-bead library screening. In this work, we have shown that the problem is primarily caused by the high ligand concentration on beads, which allows a macromolecular target to simultaneously interact with multiple ligands on a bead. The problem can be largely overcome by synthesizing OBOC libraries on spatially segregated beads that carry a reduced ligand loading on the bead surface. Reduction of ligand loading can also increase the screening stringency and facilitate the identification of the most potent ligands.25 Although only demonstrated with peptide libraries in this work, the general principle should be applicable to other types of OBOC libraries and any studies involving the interaction between a macromolecule and surface-immobilized ligands. Operationally, the LD libraries are easy to prepare, requiring only a minor modification of the conventional OBOC libraries. Thus, LD libraries of this type should become the method of choice for on-bead screening of immobilized ligands against macromolecular targets. In addition, this work has generated the first specificity profile for the SLAP SH2 domain and revealed a novel class of previously unrecognized high-affinity ligands of the Fyn SH2 domain.
Experimental Section
Materials
All DNA modifying enzymes were purchased from New England Biolabs (Ipswich, MA). All oligonucleotides were purchased from Integrated DNA Technologies (Coralville, IA). The prokaryotic vector for the addition of the ybbR tag (pET22-ybbR) and the Sfp-overproducing plasmid (pET29-Sfp) were generous gifts from Dr. C. T. Walsh (Harvard Medical School). BCIP, N-hydroxysuccinimido-biotin, Boc-Phe-OSu, and organic solvents were obtained from Sigma-Aldrich (St. Louis, MO). Fmoc-Met-OSu was from Chem-Impex International (Wood Dale, IL). NHS-dPEG4™-biotin was from Quanta BioDesign, Ltd. Talon resin was purchased from Clontech (Mountain View, CA). Reagents for peptide synthesis were from Advanced ChemTech (Louisville, KY), Peptides International (Louisville, KY), or NovaBiochem (La Jolla, CA). Protein concentrations were determined by the Bradford method using bovine serum albumin as the standard.
Expression, Purification, and Biotinylation of SH2 Domains
Human Csk SH2 domain (containing a C-terminal ybbR tag followed by a six-histidine tag) was expressed and purified as previously described.9 The DNA fragment coding for the SH3 and SH2 domains of SLAP (amino acids 1−187) was isolated by the polymerase chain reaction (PCR) from the human fetus Marathon-Ready cDNA library (Clontech) with the following primers: 5'-AGATATACATATGGGAAACAGCATGAAATCCACCCCTG-3' and 5'-CCGGAATTCGCGGCCCTCACTGCTGGGG -3'. The PCR product was digested with restriction endonucleases NdeI and EcoRI, and ligated into the corresponding sites in vector pET22b-ybbR-His. The DNA fragment coding for the Fyn SH2 domain (amino acids 144−251) was isolated by PCR from the same cDNA library with primers 5’-CGAATTCCATATGCAAGCTGAAGAGTGGTACTTTGGA-3’ and 5’-CATCGTGAAGCTTTCAGTCGACCCTTGGCATCCCTTTGTG-3’. The PCR product was digested with restriction endonucleases NdeI and SalI, and ligated into the above vector. The authenticity of the DNA constructs was confirmed by dideoxy sequencing of the entire coding regions. Expression, purification, and biotinylation (enzymatically at the ybbR tag) of the SLAP and Fyn SH2 domains were performed as previously described.9
Library Synthesis
The synthesis of the HD library has previously been described.9 The LD library was synthesized on 1.0 g of TentaGel S NH2 resin (90 μm, 0.26 mmol/g). All of the manipulations were performed at room temperature unless otherwise noted. To segregate the beads into outer and inner layers, the beads were soaked in water overnight, drained, and suspended in 30 mL of 55:45 (v/v) DCM/diethyl ether containing Fmoc-Met-OSu (0.06 mmol, 0.10 equiv) and Boc-Phe-OSu (0.24 mmol, 0.40 equiv). The mixture was incubated on a rotary shaker for 30 min. After washing with 55:45 DCM/diethyl ether (3 × 30 mL) and DMF (8 × 30 mL), the resin was treated with 4 equiv of Fmoc-Met-OH and HBTU/HOBt/NMM in DMF (90 min). The Boc group was removed by treatment with 50% (v/v) trifluoroacetic acid (TFA)/DCM for 30 min. After washing with DCM and 5% (v/v) DIPEA/DCM, the resin was treated with excess acetic anhydride and N,N-dimethylaminopyridine/NMM. The Fmoc group was removed by treatment twice with 20% piperidine in DMF (5 + 15 min) and the beads were exhaustively washed with DMF (6 × 30 mL). The linker sequence (LBBRM) was synthesized with 4 equiv of Fmoc-amino acids, using HBTU/HOBt/NMM as the coupling reagents. The coupling reaction was typically allowed to proceed for 1.5 h and the beads were washed with DMF (3 × 10 mL) and DCM (3 × 10 mL). For the addition of random residues, the resin was split into 20 equal portions and each portion (50 mg) was coupled twice, each with 5 equiv of a different Fmoc-amino acid for 1 h. To differentiate isobaric amino acids during MS sequencing, 5% (mol/mol) CD3CO2D was added to the coupling reactions of Leu and Lys, whereas 5% CH3CD2CO2D was added to norleucine reaction.5b Side-chain deprotection was carried out with a modified reagent K (6.5% phenol, 5% water, 5% thioanisole, 2.5% ethanedithiol, 1% anisole, and 1% triisopropylsilane in TFA) for 2 h. The resin was washed with TFA and DCM, and dried under vacuum before storage at −20 °C.
Library Screening
Library screening essentially followed the published procedures.9 The screening conditions including the SH2 protein concentration and staining time were adjusted by trial and error so that 0.01−0.05% of the library beads became positive. A typical screening experiment involved 30−50 mg of the library suspended in 1 mL of binding buffer containing 500 nM (for Csk and Fyn) or 50 nM SH2 protein (for SLAP). The positive beads (typically ∼30 beads) were sequenced by PED/MS as previously described.5b For each SH2 domain, the screening experiment was repeated at least three times to insure that the screening results were reproducible. Control experiments with biotinylated MBP produced no colored beads under identical conditions.
Synthesis and Binding Analysis of Selected Peptides
Each peptide was synthesized on 100 mg of CLEAR-amide resin (0.49 mmol/g) using the standard Fmoc/HBTU chemistry. After the addition of the last residue, the resin was acylated by treatment of acetic anhydride. Cleavage from the resin and side-chain deprotection were carried out using reagent K. The solvents were removed by evaporation under a N2 stream and trituration with cold diethyl ether. The crude peptide was dissolved in NaHCO3 buffer (pH 8) and biotinylated by treating with 1.5 equiv of commercially available NHS-dPEG4™-biotin. The biotinylated peptide was purified by reversed-phase HPLC on a C18 column (Vydac 300Ǻ, 4.6 × 250 mm) and their identity was confirmed by MALDI-TOF mass spectrometric analyses.
The dissociation constants were determined by surface plasmon resonance on a BIAcore 3000 instrument. The pY peptides were immobilized onto a streptavidin-coated sensorchip. All measurements were performed at room temperature as previously described.1e Increasing concentrations of an SH2 protein (typically 0−5 μM) in HBS-EP buffer (10 mM HEPES, pH 7.4, 150 mM NaCl, 3 mM EDTA, and 0.005% polysorbate 20) were passed over the sensorchip for 120 s at a flow rate of 15 μL/min. A blank flow cell (no immobilized pY peptide) was used as control to correct for any signal due to the solvent bulk and/or nonspecific binding interactions (no significant bulk effect or nonspecific binding was observed). In between two runs, the sensorchip surface was regenerated by flowing a strip solution (10 mM NaCl, 2 mM NaOH, and 0.025% SDS in H2O) for 5−10 s at a flow rate of 100 μL/min. The equilibrium response unit (RUeq) at a given SH2 protein concentration was obtained by subtracting the response of the blank flow cell from that of the sample flow cell. The dissociation constant (KD) was obtained by nonlinear regression fitting of the data to the equation
where RUeq is the measured response unit at a certain SH2 protein concentration and RUmax is the maximum response unit.
Supplementary Material
Acknowledgments
This work was supported by a grant from the NIH (GM062820).
Footnotes
Supporting Information Available. Sequences of selected peptides and additional proteins predicted to bind to SLAP via its SH2 domain. This information is available free of charge from the Internet at http://pubs.acs.org.
REFERENCES
- 1.For examples see: Chen JK, Lane WS, Brauer AW, Tanaka A, Schreiber SL. J. Am. Chem. Soc. 1993;115:12591–12592.Muller K, Gombert FO, Manning U, Grossmuller F, Graff P, Zaegel H, Zuber JF, Freuler F, Tschopp C, Baumann G. J. Biol. Chem. 1996;271:16500–16505.Smith HK, Bradley M. J. Comb. Chem. 1999;1:326–332. doi: 10.1021/cc990013c.Alluri PG, Reddy MM, Bachhawat-Sikder K, Olivos HJ, Kodadek T. J. Am. Chem. Soc. 2003;125:13995–14004. doi: 10.1021/ja036417x.Sweeney MC, Wavreille A-S, Park J, Butchar J, Tridandapani S, Pei D. Biochemistry. 2005;44:14932–14947. doi: 10.1021/bi051408h.Peng L, Liu RW, Marik J, Wang X-B, Takada Y, Lam KS. Nature Chem. Biol. 2006;2:381–389. doi: 10.1038/nchembio798.Zhang Y, Zhou S, Wavreille A-S, DeWille J, Pei D. J. Comb. Chem. 2008;10:247–255. doi: 10.1021/cc700185g.
- 2.For examples see: Wu J, Ma QN, Lam KS. Biochemistry. 1994;33:14825–14833. doi: 10.1021/bi00253a022.Meldal M, Svendsen I, Breddam K, Auzanneau F-I. Proc. Natl. Acad. Sci. U.S.A. 1994;91:3314–3318. doi: 10.1073/pnas.91.8.3314.Hu Y-J, Wei Y, Zhou Y, Rajagopalan PTR, Pei D. Biochemistry. 1999;38:643–650. doi: 10.1021/bi9820412.Rosse G, Kueng E, Page MGP, Schauer-Vukasinovic V, Giller T, Lahm H-W, Hunziker P, Schlatter D. J. Comb. Chem. 2000;2:461–466. doi: 10.1021/cc000019y.Garaud M, Pei D. J. Am. Chem. Soc. 2007;129:5366–5367. doi: 10.1021/ja071275i..
- 3.For reviews see: Hoveyda AH. Chem. Biol. 1998;5:R187–R191. doi: 10.1016/s1074-5521(98)90155-7.Revell JD, Wennerners H. Topics Curr. Chem. 2007;277:251–266..
- 4.a Lam KS, Salmon SE, Hersh EM, Hruby VJ, Kazmierski WM, Knapp RJ. Nature. 1991;354:82–84. doi: 10.1038/354082a0. [DOI] [PubMed] [Google Scholar]; b Houghten RA, Pinilla C, Blondelle SE, Appel JR, Dooley CT, Cuervo JH. Nature. 1991;354:84–86. doi: 10.1038/354084a0. [DOI] [PubMed] [Google Scholar]; c Furka A, Sebestyen F, Asgedom M, Dibo G. Int. J. Pept. Protein Res. 1991;37:487–493. doi: 10.1111/j.1399-3011.1991.tb00765.x. [DOI] [PubMed] [Google Scholar]
- 5.a Sweeney MC, Pei D. J. Comb. Chem. 2003;5:218–222. doi: 10.1021/cc020113+. [DOI] [PubMed] [Google Scholar]; b Thakkar A, Wavreille A-S, Pei D. Anal. Chem. 2006;78:5935–5939. doi: 10.1021/ac0607414. [DOI] [PubMed] [Google Scholar]; c Thakkar A, Cohen AS, Connolly MD, Zuckermann RN, Pei D. J. Comb. Chem. 2009;11:294–302. doi: 10.1021/cc8001734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.a Blom KF, Combs AP, Rockwell AL, Oldenburg KR, Zhang JH, Chen TM. Rapid Commun. Mass Spectrom. 1998;12:1192–1198. [Google Scholar]; b Biederman KJ, Lee H, Haney CA, Kaczmarek M, Buettner JA. J. Pept. Res. 1999;53:234–243. doi: 10.1034/j.1399-3011.1999.00047.x. [DOI] [PubMed] [Google Scholar]; c Redman JE, Wilcoxen KM, Ghadiri MR. J. Comb. Chem. 2003;5:33–40. doi: 10.1021/cc0200639. [DOI] [PubMed] [Google Scholar]; d Paulick MG, Hart KM, Brinner KM, Tjandra M, Charych DH, Zuckermann RN. J. Comb. Chem. 2006;8:417–426. doi: 10.1021/cc0501460. [DOI] [PubMed] [Google Scholar]; d Garske AL, Craciun G, Denu JM. Biochemistry. 2008;47:8094–8102. doi: 10.1021/bi800766k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.a Ohlmeyer MHJ, Swanson RN, Dillard LW, Reader JC, Asouline G, Kobayashi R, Wigler M, Still WC. Proc. Natl. Acad. Sci. U.S.A. 1993;90:10922–10926. doi: 10.1073/pnas.90.23.10922. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Youngquist RS, Fuentes GR, Lacey MP, Keough T. J. Am. Chem. Soc. 1995;117:3900–3906. [Google Scholar]; c Maclean D, Schillek JR, Murphy MM, Ni ZJ, Gordon EM, Gallop MA. Proc. Natl. Acad. Sci. U. S. A. 1997;94:2805–2810. doi: 10.1073/pnas.94.7.2805. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Song AM, Zhang JH, Labrilla CB, Lam KS. J. Am. Chem. Soc. 2003;125:6180–6188. doi: 10.1021/ja034539j. [DOI] [PubMed] [Google Scholar]
- 8.Beebe KD, Wang P, Arabaci G, Pei D. Biochemistry. 2000;39:13251–13260. doi: 10.1021/bi0014397. [DOI] [PubMed] [Google Scholar]
- 9.Wavreille A-S, Garaud M, Zhang Y, Pei D. Methods. 2007;42:207–219. doi: 10.1016/j.ymeth.2007.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Imhof D, Wavreille A-S, May A, Zacharias M, Tridandapani S, Pei D. J. Biol. Chem. 2006;281:20271–20282. doi: 10.1074/jbc.M601047200. [DOI] [PubMed] [Google Scholar]
- 11.Joo SH, Pei D. Biochemistry. 2008;47:3061–3072. doi: 10.1021/bi7023628. [DOI] [PubMed] [Google Scholar]
- 12.a Samson I, Rozenski J, Samyn B, Van Aerschot A, Van Beeumen J, Herdewijn P. J. Biol. Chem. 1997;272:11378–11383. doi: 10.1074/jbc.272.17.11378. [DOI] [PubMed] [Google Scholar]; b Aggarwal S, Harden JL, Denmeade SR. Bioconjugate Chem. 2006;17:335–340. doi: 10.1021/bc0502659. [DOI] [PubMed] [Google Scholar]
- 13.Waksman G, Kuriyan J. Cell. 2004:S116, S45–S48. doi: 10.1016/s0092-8674(04)00043-1. [DOI] [PubMed] [Google Scholar]
- 14.The linker sequence LNBBRM serves several purposes. The C-terminal Met permits peptide release by CNBr cleavage. The Arg provides a fixed positive charge to facilitate MS analysis in the positive ion mode and to improve the aqueous solubility. β-Ala provides additional flexibility to maximize interaction with a protein target, while LN increases the m/z values of peptides to >500 to avoid signal overlap with MALDI matrix materials. The N-terminal TA moves the positive charge associated with the free N-terminus away from the random region to minimize screening biases.
- 15.Songyang Z, Shoelson SE, McGlade J, Olivier P, Pawson T, Bustelo XR, Barbacid M, Sabe H, Hanafusa H, Yi T, et al. Mol. Cell Biol. 1994;14:2777–2785. doi: 10.1128/mcb.14.4.2777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Huang H, Li L, Wu C, Schibli D, Colwill K, Ma S, Li C, Roy P, Ho K, Songyang Z, Pawson T, Gao Y, Li S-C. Mol. Cell. Proteomics. 2008;7:768–784. doi: 10.1074/mcp.M700312-MCP200. [DOI] [PubMed] [Google Scholar]
- 17.Liu R, Marik J, Lam KS. J. Am. Chem. Soc. 2002;124:7678–7680. doi: 10.1021/ja026421t. [DOI] [PubMed] [Google Scholar]
- 18.Ulmer TS, Werner JM, Campbell ID. Structure. 2002;10:901–911. doi: 10.1016/s0969-2126(02)00781-5. [DOI] [PubMed] [Google Scholar]
- 19.a Roche S, Alonso G, Kazlauskas A, Dixit VM, Courtneidge SA, Pandey A. Curr. Biol. 1998;8:975–978. doi: 10.1016/s0960-9822(98)70400-2. [DOI] [PubMed] [Google Scholar]; b Manes G, Bello P, Roche S. Mol. Cell Biol. 2000;20:3396–3406. doi: 10.1128/mcb.20.10.3396-3406.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tang J, Sawasdikosol S, Chang JH, Burakoff SJ. Proc. Natl. Acad. Sci. USA. 1999;96:9775–9780. doi: 10.1073/pnas.96.17.9775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yin J, Straight PD, McLoughlin SM, Zhou Z, Lin AJ, Golan DE, Kelleher NL, Kolter R, Walsh CT. Proc. Natl. Acad. Sci. U. S. A. 2005;102:15815–15820. doi: 10.1073/pnas.0507705102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Myers MD, Sosinowski T, Dragone LL, White C, Band H, Gu H, Weiss A. Nat. Immunol. 2006;7:57–66. doi: 10.1038/ni1291. [DOI] [PubMed] [Google Scholar]
- 23.Sosinowski T, Pandey A, Dixit VM, Weiss A. J. Exp. Med. 2000;191:463–473. doi: 10.1084/jem.191.3.463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dragone LL, Myers MD, White C, Gadwai S, Sosinowski T, Gu H, Weiss A. Proc. Natl. Acad. Sci. U. S. A. 2006;103:18202–18207. doi: 10.1073/pnas.0608965103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.a Debenham SD, Snyder PW, Toone EJ. J. Org. Chem. 2003;68:5805–5811. doi: 10.1021/jo0207271. [DOI] [PubMed] [Google Scholar]; b Wang X, Peng L, Liu R, Xu B, Lam KS. J. Pep. Res. 2005;65:130–138. doi: 10.1111/j.1399-3011.2005.00192.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.