

Abstract
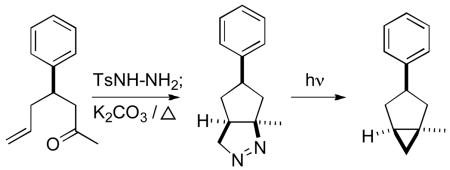
Heating the tosylhydrazone of an ω-alkenyl ketone or aldehyde to reflux in toluene in the presence of K2CO3 delivered the bicyclic diazene. Irradiation of the diazene converted it to the cyclopropane. This appears to be a generally useful method for the construction of substituted cyclopentanes and cyclohexanes.
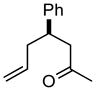
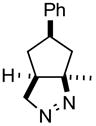
Carbocycle construction by intramolecular 1,3-dipolar cycloaddition has long been a workhorse of organic synthesis.1,2 Yet, one of the simplest of such ring-forming reactions, the generation and intramolecular cycloaddition of an ω-diazo alkene to form the cyclic diazene (Scheme 1, 1a → 2a), had not been developed as a generally useful synthetic method.3,4 We have found that simply heating the tosylhydrazone of a ketone such as 1a to reflux in toluene in the presence of K2CO3 delivered the bicyclic diazene 2a.5 Irradiation of 2a converted it to the cyclopropane 3a,6,7 while tautomerization3 of 2a equilibrated it to the more stable cyclic hydrazone 4. Hydrogenation of 2a followed by acylation led to the cyclic hydrazide 5, reduction8 of which with SmI2 generated the cyclopentane 6. This appears to be a generally useful method for the construction of substituted cyclopentanes and cyclohexanes.
Scheme 1.

There is precedent3,4 for the conversion of 1a to 2a. In 1980, Padwa3a reported that heating the sodium salt of an ω-alkenyl tosylhydrazone gave the tautomerized dihydropyrazole analogous to 4. Alternatively, when the diazo intermediate was generated by heating the ω-alkenyl ketone with an N-aminoaziridine, the cyclic diazene was isolated. Curiously, there had been very little further work3b–3e,4 on this approach to carbocyclic construction since that time.
We have briefly (Table 1; all yields in Table 1 and Table 2 are for pure isolated products) examined the scope of this cyclization. Both five-membered and six-membered ring formation proceeded efficiently. The geometry of the starting alkene appeared (entry 3) to be maintained in the product. While the cyclization to form the cyclopentane (Scheme 1, structure established by X-ray analysis) proceeded with significant diastereocontrol, cyclization to form the cyclohexane (entry 6) did not. As expected,3a ω-alkenyl aldehydes (entry 5) also cyclized efficiently, with useful diastereocontrol. It is also noteworthy (entry 7) that cyclic ketones participated smoothly. With the results reported here, the generation and intramolecular cycloaddition of a (presumed) intermediate diazo alkene to form the cyclic diazene appears to now be a generally applicable synthetic method.
Table 1.
Bicyclic and Tricylic Diazenes
| Entry | Ketone | Diazene | Yield (%)a |
|---|---|---|---|
| 1 |
 1a |
 2ab |
84 |
| 2 |
 1bc |
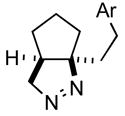 2b |
76 |
| 3 |
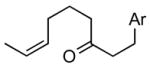 1cd |
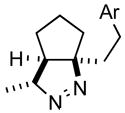 2ce |
70 |
| 4 |
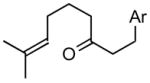 1d |
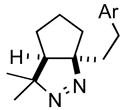 2d |
68 |
| 5 |
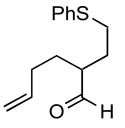 1e |
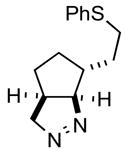 2ef |
91 |
| 6 |
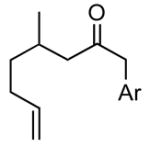 1f |
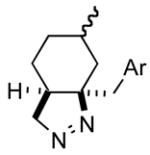 1fg |
72 |
| 7 |
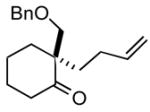 1g |
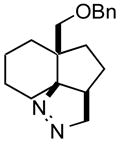 2g |
68 |
Yields are for pure isolated products.
The product was a ~ 5:1 mixture of diastereomers.
Ar = 4-methoxyphenyl.
The alkene was a ~ 5: 1 Z/E mixture.
The product was a ~ 5:1 mixture of diastereomers.
The product was a ~ 6:1 mixture of diastereomers. The major product had a 13C NMR methine at δ = 100.2. The minor diastereomer 13C NMR methine was at δ = 95.1
The product was a ~ 1:1 mixture of diastereomers.
Table 2.
Cyclopropanes from cyclic diazenes
| Entry | Diazene | Cyclopropane | Yield (%) |
|---|---|---|---|
| 1 |
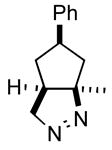 2a |
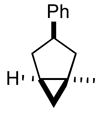 3a |
42 |
| 2 |
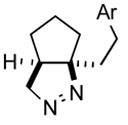 2b |
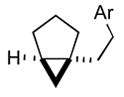 3b |
89 |
| 3 |
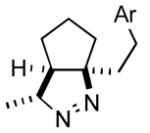 2c |
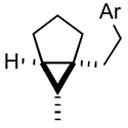 3c |
71 |
| 4 |
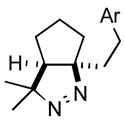 2d |
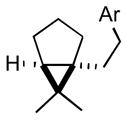 3d |
74 |
| 5 |
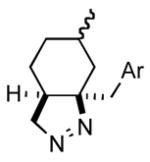 2f |
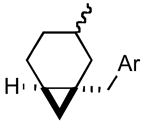 3f |
80 |
As outlined in Scheme 1, the product cyclic diazenes are versatile intermediates for further transformation. We were pleased to observe that the diazene 2 a was readily tautomerized to the dihydropyrazole 4, and was easily reduced to the tetrahydropyrazole, isolated as the bis benzamide 5. Such bicyclic dihydro and tetrahydro pyrazoles such as 4 and 5 should be useful scaffolds for pharmaceutical discovery. It was equally exciting that the benzamide 5 could be further reduced to the cyclopentane 6. Such aminated cyclopentanes, useful intermediates for alkaloid synthesis,9 are not readily prepared by other means.
The straightforward conversion of 1a to the cyclopropane 3 was particularly interesting. We have made a preliminary investigation (Table 2) of this reaction, which appears to be general. Intramolecular cyclopropanation is usually carried out with α-diazo ketones or esters.10 The net conversion of an ω-alkenyl ketone or aldehyde to the corresponding carbene with subsequent intramolecular cyclopropanation has been a long-standing goal. Although strategies have been developed for effecting this transformation,7 the protocol described here appears to be a practical alternative.
The approach delineated here makes cyclic diazenes such as 2a routinely available. We expect that this approach to carbocyclic construction by intramolecular dipolar cycloaddition will have many applications both in natural product synthesis and in medicinal chemistry.11,12
Experimental Section
Diazene 2a
Ketone 1a13 (85 mg, 0.45 mmol) and tosylhydrazine (1.07 equiv, 92 mg, 0.49 mmol) were stirred in MeOH (2 mL) at room temperature overnight. The MeOH was removed under reduced pressure, the crude hydrazone was redissolved in toluene (3 mL), K2CO3 (6 equiv, 385 mg, 2.8 mmol) was added and the reaction mixture was heated in a sealed vial at 120 °C (oil bath) for 18 h. After cooling to room temperature, the reaction mixture was partitioned between CH2Cl2 and, sequentially, water and brine. The combined organic extracts were dried (Na2SO4) and concentrated. The residue was chromatographed to yield diazene 2a (76 mg, 84% yield, 5:1 mixture of two isomers based on 1H NMR) as a pale yellow solid: mp 64 °C; TLC Rf (5% MTBE/CH2Cl2) = 0.45; IR (cm−1) 3027, 2958, 1442, 1270; 1H NMR δ 7.10–7.35 (m, 5H), 4.50 (dd, J = 2.2, 18.0 Hz, 1H), 4.30 (dd, J = 8.0, 18.0 Hz, 1H), 3.10 (m, 1H), 2.40 (m, 1H) 2.25 (m 1H), 2.05 (m, 2H), 1.40 (s 3H), 1.05 (m, 1H); 13C NMR (major isomer) δ u14 142.5, 100.8, 81.0, 44.2, 42.1; d 128.5, 126.9, 126.5, 42.3, 42.0; HRMS calcd for C13H17N2 (MH+) 201.1392, obsd 201.1397. The diazene 2a was recrystallized from hexane as a 20:1 mixture (1H NMR) of isomers, mp 69 °C.
(1 R*,3R*,5S*)-1-Methyl-3-phenylbicyclo[3.1.0]hexane (3a)
A solution of the recrystallized diazene 2a (76 mg, 0.38 mmol) in toluene was photolyzed for 24 h at room temperature in a Rayonet apparatus (350 nm). The reaction mixture was concentrated and chromatographed to yield cyclopropane 3a (28 mg, 42% yield) as a colorless oil: TLC Rf (2% MTBE/PE) = 0.54; IR (cm−1) 3011, 2932, 1449, 1021; 1H NMR δ 7.05–7.30 (m, 5H), 3.55 (m, 1H), 2.45 (m, 1H), 2.15 (m, 1H), 1.90 (dd, J = 6.4, 13.2 Hz, 1H), 1.80 (m, 1H), 1.20 (s, 3H), 1.05 (m, 1H), 0.45 (m, 1H), 0.35 (m, 1H); 13C NMR (major isomer) δ u 146.2, 42.5, 37.2, 26.3, 22.5; d 128.1, 127.2, 125.5, 47.2, 26.0, 22.6; HRMS calcd for C13H16 (MH+) 172.1252, obsd 172.1250.
Dihydropyrazole 4
To a stirred solution of the recrystallized diazene 2a (47 mg, 0.24 mmol) in MeOH (2 mL), was added K2CO3 (200 mg, 1.5 mmol) at rt. The reaction mixture was stirred in the dark at rt for 20 h, then concentrated. The residue was chromatographed to yield 4 (37 mg, 79% yield) as a colorless oil: TLC Rf (MTBE) = 0.42; IR (cm−1) 3309, 2956, 1649, 1451; 1H NMR (MeOD) δ 7.00–7.20 (m, 5H), 6.65 (s, 1H), 3.20 (s, 1H), 2.90–3.10 (m, 2H), 2.40 (m, 1H), 2.05 (m, 1H), 1.80 (t, J = 12 Hz,1H), 1.50 (q, 1H), 1.35 (s, 3H); 13C NMR (MeOD) δ u 144.6, 71.9, 50.0, 40.0; d 149.4, 129.5, 127.8, 127.3, 59.6, 45.4, 23.6; HRMS calcd for C13H16N2(M+) 200.1313, obsd 200.1314.
Bis-benzamide 5
A suspension of the recrystallized diazene 2a (85 mg, 0.43 mmol) and PtO2 (10 mg) in MeOH (3 mL) was stirred at rt under H2 atmosphere for 3 h. The reaction mixture was filtered and concentrated to give the crude diamine (84 mg).
To a stirred solution of the crude diamine and Et3N (182 mg, 1.8 mmol) in toluene (3 mL) was added benzoyl chloride (250 mg, 1.78 mmol) at 0 °C. After stirring at rt overnight, the reaction mixture was concentrated and chromatographed to yield pyrazolidine 5 (105 mg, 60% yield) as a pale yellow solid: mp 168 °C; TLC Rf (5% MTBE/CH2Cl2) = 0.62; IR (cm−1) 3054, 2955, 1667, 1396, 1234; 1H NMR δ 7.00–7.70 (m, 15H), 3.00–4.10 (m, 3H), 2.50–2.80 (m, 2H), 2.10–2.40 (m, 1H), 1.80 (s, 3H), 1.60–1.75 (m, 1H), 1.20 (m, 1H); 13C NMR δ u 172.3, 167.3, 142.5, 136.4, 133.7, 72.1, 54.0, 47.0, 40.2; d 131.6, 130.4, 128.7, 128.5, 128.2, 127.7, 127.6, 127.0, 126.5, 53.3, 45.2, 24.9; HRMS calcd for C27H27N2O2 (MH+) 411.2072, obsd 411.2067.
Bis-benzamide 6
To a stirred solution of pyrazolidine 5 (25 mg, 0.06 mmol) in MeOH (0.5 mL) was added SmI2 (3mL, 0.3 mmol, 0.1 M in THF) at rt. After stirring for 30 min at rt, the reaction mixture was concentrated and chromatographed to yield 6 (19 mg, 76% yield) as a pale yellow solid: mp 155 °C; TLC Rf (5% MTBE/PE) = 0.31; IR (cm−1) 3312, 2918, 1638, 1530, 1299; 1H NMR δ 7.00–7.90 (m, 15H), 6.30 (s, 1H), 3.75 (m, 1H), 3.20 (m, 2H), 2.10–2.40 (m, 3H), 1.60 (s, 3H), 1.70–1.80 (m, 1H), 1.10–1.30 (m, 2H); 13C NMR δ u 167.6, 166.0, 143.2, 133.9, 133.4, 61.8, 48.9, 40.2, 37.2; d 130.6, 130.2, 127.7, 127.6, 127.5, 126.0, 125.9, 125.8, 125.3, 48.8, 39.8, 26.0; HRMS calcd for C27H29N2O2 (MH+) 413.2229, obsd 413.2227.
Supplementary Material
Experimental procedures, details of the X-ray analysis, and 1H and 13C NMR spectra for all new compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
Acknowledgments
We thank Dr. John Dykins for mass spectrometric measurements, supported by the NSF (0541775), Dr. Glenn Yap for the X-ray analysis, and the NIH (GM60287) for financial support.
References
- 1.For a recent review of the use of intramolecular dipolar cycloaddition in target-directed organic synthesis, see Nair V, Suja TD. Tetrahedron. 2007;63:12247. It should be noted that the intramolecular cycloaddition of an ω-alkenyl diazo alkane to form the cyclic diazene is not mentioned in this thorough review.
- 2.For more recent examples of C-C ring construction by intramolecular dipolar cycloaddition, see Huang X, Zhang L. J Am Chem Soc. 2007;129:6398. doi: 10.1021/ja0717717.Belanger G, April M, Dauphin E, Roy S. J Org Chem. 2007;72:1104. doi: 10.1021/jo061556t.Coldham I, Burrell AJM, White LE, Adams H, Oram N. Angew Chem, Int Ed. 2007;46:6159. doi: 10.1002/anie.200701943.Pouilhes A, Amado AF, Vidal A, Langlois Y, Kouklovsky C. Org Biomol Chem. 2008;6:1502. doi: 10.1039/b800840j.
- 3.There have been isolated reports over the years of the cyclization of simple alicyclic ω-alkenyl ketones and aldehydes to the cyclic diazenes. For the most detailed account, see Padwa A, Ku H. J Org Chem. 1980;45:3756. For more recent references, see Brinker UH, Schrievers T, Xu L. J Am Chem Soc. 1990;112:8609.Ashby EC, Park B, Patil GS, Gadru K, Gurumurthy R. J Org Chem. 1993;58:424.Jung ME, Huang A. Org Lett. 2000;2:2659. doi: 10.1021/ol0001517. In each of these cases, ready tautomerization has commonly been observed. (e) For alternative methods for converting ketones to the corresponding diazo compounds, see Miller PC, Gaspar PP. J Org Chem. 1991;56:5101.
- 4.For the single instance of the use of intramolecular addition of a diazo alkane to an alkene in natural product synthesis, see Schultz AG, Puig S. J Org Chem. 1985;50:915. Note that in this case, conversion of the aldehyde to the intermediate diazo alkane by reaction with an N-aminoaziridine also led to the nitrile as a major byproduct.
- 5.The structure of 2a was established by X-ray analysis.
- 6.For an early report of the photolysis of cyclic diazenes to make cyclopropanes, see Rinehart KL, Jr, Van Auken TL. J Am Chem Soc. 1960;82:5251.Van Auken TL, Rinehart KL., Jr J Am Chem Soc. 1962;84:3736.
- 7.For leading references to efforts toward cyclopropane construction by the conversion of ketones into carbene equivalents, see Motherwell WB. J Organomet Chem. 2001;624:41.
- 8.For the reduction of hydrazine amides with SmI2, see Shirakawa S, Lombardi PJ, Leighton JL. J Am Chem Soc. 2005;127:9974. doi: 10.1021/ja052307+.
- 9.Kende AS, Liu K, Brands KMJ. J Am Chem Soc. 1995;117:10597. [Google Scholar]
- 10.For a representative cyclization of an ω-alkenyl diazo ketone, see Taber DF, Saleh SA, Korsmeyer RW. J Org Chem. 1980;45:4699.
- 11.de los Santos JM, López Y, Aparicio D, Palacios F. J Org Chem. 2008;73:550. doi: 10.1021/jo702050t. [DOI] [PubMed] [Google Scholar]
- 12.For a complementary carbacyclization of ω-alkenyl ketone tosylhydrazones via the derived alkyl radicals, see Taber DF, Wang Y, Stachel SJ. Tetrahedron Lett. 1993;34:6209.Taber DF, Wang Y, Pahutski TF., Jr J Org Chem. 2000;65:3861. doi: 10.1021/jo991866u.
- 13.Howarth J, James P, Dai J. J Mol Cat A: Chem. 2004;214:143. [Google Scholar]
- 14.13C multiplicities were determined with the aid of a JVERT pulse sequence, differentiating the signals for methyl and methine carbons as “d” and for methylene and quaternary carbons as “u”.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Experimental procedures, details of the X-ray analysis, and 1H and 13C NMR spectra for all new compounds. This material is available free of charge via the Internet at http://pubs.acs.org.