

Abstract
The intracellular juxtamembrane domain of the EGF receptor has been shown to be involved in the stimulation of the receptor’s tyrosine kinase activity. To further explore the function of this portion of the EGF receptor, a consensus site for protein palmitoylation was inserted at the beginning of the juxtamembrane domain of the receptor. The altered EGF receptor incorporated [3H]-palmitate demonstrating that it was palmitoylated. Compared to wild type EGF receptor, the palmitoylated EGF receptor was significantly impaired in EGF-stimulated receptor autophosphorylation as well as ligand-induced receptor internalization. While both the wild type and the palmitoylated EGF receptors showed a similar propensity to associate with lipid rafts, only the wild type receptor exited lipid rafts in response to EGF. Binding of 125I-EGF to the wild type EGF receptor showed a curvilinear Scatchard plot with both high and low affinity forms of the receptor. By contrast, the palmitoylated receptor exhibited only low affinity EGF binding. These data suggest that the cytoplasmic juxtamembrane domain is involved not only in the transmission of the proliferative signal generated by ligand binding but also in facilitating the adoption of the high affinity conformation by the extracellular ligand binding domain.
The EGF receptor is a classical receptor tyrosine kinase composed of an extracellular domain that recognizes and binds EGF, a single pass transmembrane segment, and an intracellular tyrosine kinase domain (1). The EGF receptor is thought to exist in the membrane as a monomer. However, upon ligand binding, the receptor forms a back- to-back dimer with a second EGF receptor monomer (2-4). This leads to the activation of the intracellular tyrosine kinase domain and phosphorylation of the receptor in trans (5).
Scatchard plots of the binding of 125I-EGF to its receptor show upward concavity indicating heterogeneity in the affinity of the receptor for EGF. We have recently shown that ligand binding in the EGF receptor system is best described by a model incorporating negative cooperativity in an aggregating system (6). In this model, monomers and dimers are present in a pre-existing equilibrium. Ligand can bind to receptor monomers, to the first site on a receptor dimer or to the second site on a singly-occupied receptor dimer. Binding to receptor monomers and the first site on receptor dimers is of high affinity (∼200 pM) whereas binding to the second site on a receptor dimer is of substantially lower affinity (∼3 nM). The difference in affinity between binding to the first site vs. the second site on an EGF receptor dimer is a classic example of negative cooperativity and gives rise to the curvilinear Scatchard plots.
Binding of EGF not only leads to dimerization of the extracellular domain of the EGF receptor, it also leads to the formation of an activating asymmetric dimer of the intracellular kinase domains (7). In this dimer, the N-terminal lobe of one kinase monomer interacts with the C-terminal lobe of a second kinase monomer, stimulating the kinase activity of the first monomer. The crystal structure of the asymmetric kinase dimer (7) shows that the interface on the C-terminal lobe is comprised of the loop between helices αG and αJ, the αH helix and the end of helix αI. The interface on the N-terminal lobe is formed by the C helix, the loop between strands ß4 and ß5 and the juxtamembrane extension of the kinase domain (residues 672-685). Recently, Thiel and Carpenter (8) demonstrated that the cytoplasmic juxtamembrane domain (roughly residues 645-672) is indispensable for kinase activation, even in the context of the purified soluble intracellular domain. Deletion of residues 645-662, a region not included in the asymmetric dimer interface, decreased autophosphorylation of the soluble kinase by 80%. This suggests that juxtamembrane sequences outside of the asymmetric dimer interface proper are important in kinase activation.
Exactly how ligand-induced dimerization of the extracellular domain of the EGF receptor leads to formation of asymmetric kinase dimers remains unclear. It has been reported that the transmembrane domain of the EGF receptor can form homodimers (9, 10) and that activation of the EGF receptor tyrosine kinase involves ligand-induced rotation of its transmembrane domain (11). Rotation of the transmembrane domains within an EGF receptor dimer could produce movement of the juxtamembrane domains that would bring the two intracellular kinase domains into close apposition. If so, mutation of the juxtamembrane domain might impair the ability of EGF to induce activation of the receptor kinase domain.
To test this hypothesis, we generated a consensus palmitoylation site in the EGF receptor by introducing two cysteine residues at the beginning of the cytoplasmic juxtamembrane domain. We reasoned that linking this region of the receptor to the membrane might restrict its mobility or limit its accessible conformations thereby impairing receptor activation. We report here that this mutant is palmitoylated and that it exhibits severely compromised activation properties. In addition, its interaction with lipid rafts is altered. Unexpectedly, this mutation also abrogated high affinity binding to the EGF receptor. These data suggest that the cytoplasmic juxtamembrane domain is involved not only in the transmission of the proliferative signal generated by ligand binding but also in facilitating the adoption of the high affinity conformation by the extracellular ligand binding domain.
Experimental Procedures
Materials
Methyl-ß-cyclodextrin was from Fluka. Recombinant mouse EGF was purchased from Biomedical Technologies. Na125I was from Perkin-Elmer. PfuTurbo DNA Polymerase was from Stratagene. The CHO-K1 Tet-On cell line and the pBI Tet vector were from Clontech. Lipofectamine 2000 and hygromycin were from Invitrogen. Anti-phosphotyrosine antibodies (PY20) were from BD Transduction Labs. Anti-pTyr-845, anti-pTyr-992, anti-pTyr-1045, anti-pTyr-1068 and anti-EGF receptor antibodies were from Cell Signaling. Anti-pTyr-1173 antibodies were from Upstate Biotechnology. Anti-Gq antibodies were from Santa Cruz. Anti-transferrin receptor antibodies were from Zymed Labs. Opti-Prep was from Greiner Bio-One. BS3 was from Pierce. All other chemicals were from Sigma.
Mutagenesis
The cDNA for wild type EGF receptor was ligated into pcDNA5/FRT between the Nhe1 and HindIII sites. The QuikChange Site-Directed Mutagenesis method was employed to generate the R647C/V650C (CC) and R647S/V650S (SS) mutants. Forward and reverse primers containing the desired mutations (underlined) were synthesized:
CC forward: 5′-CCTCTTCATGCGAAGGTGCCACATTTGCCGG-3′,
CC reverse: 5′-CCGGCAAATGTGGCACCTTCGCATGAAGAGG-3′,
SS forward: 5′-TCTTCATGCGAAGGAGCCACATCAGTCGGAAGCGCACGC-3′,
SS reverse: 5′-GCGTGCGCTTCCGACTGATGTGGCTCCTTCGCATGAAGA-3′].
PCR was carried out using pcDNA5/FRT-EGFR WT as template. The reaction mix was digested with DpnI and transformed directly into XL-1 Blue competent cells. Positive colonies were screened by PCR and the entire EGF receptor open reading frame sequenced. The EGF receptor fragment was then excised with NheI and EcoRV and the restriction fragment ligated into multiple cloning site 1 of the pBI Tet vector.
Cells and tissue culture
Chinese hamster ovary (CHO)-K1 tet-on cells were purchased from Clontech. Cells were co-transfected with pTK-Hyg and the pBI Tet vector engineered to express wild type or mutant EGF receptors from a single side, using Lipofectamine 2000 according to the manufacturer’s instructions. Stable clones were isolated by selection in 400 μg/ml hygromycin. Clonal lines were grown in DMEM containing 10% fetal bovine serum, 100 μg/ml hygromycin and 100 μg/ml G418. EGF receptor expression was induced by the addition of 1 μg/ml doxycycline for 48 hr prior to screening by Western blot for EGF receptor expression.
For experiments, cells were plated at 1 × 105 cells per 35 mm well into DMEM/10% fetal bovine serum containing the appropriate concentration of doxycycline. Concentrations of 140, 400 and 100 ng/ml were used to obtain equal levels of expression of receptors in wild type, CC and SS cells, respectively, after 48 hr.
Labeling of cells with [3H]-palmitate
Cells expressing wild type or CC-EGF receptors were grown to confluence in 60 mm diameter dishes. The media was removed and replaced with 1 ml Hams’ F12 medium containing 5% serum and 1 mCi/ml [3H]-palmitate. Cells were grown in labeling media for 4 hrs. At the end of the incubation, the medium was removed and the cells were washed with ice-cold Hepes-buffered saline. Monolayers were solubilized in 500 μl RIPA buffer containing protease inhibitors. After centrifugation, the supernatants were pre-cleared with Protein A-Sepharose and the EGF receptor immunoprecipitated by incubation with an anti-EGF receptor antibody pre-loaded onto Protein A-Sepharose. The beads were boiled in SDS sample buffer and the supernatants run on a 7% polyacrylamide gel. The gel was fixed and incubated in En3Hance before drying and exposure to x-ray film. Parallel samples were run on a gel and Western blotted for EGF receptor levels.
125I-EGF binding and internalization
125I-EGF was synthesized using the oxidative ICl method of Doran and Spar (12). Cells were plated into 6-well dishes and grown to confluence in the presence of the appropriate concentration of doxycycline for 48 hr. Cultures were washed twice in ice cold Hepes-buffered saline and then incubated overnight on ice in Hams F-12 medium containing 25 mM Hepes, pH 7.2, 3 mg/ml bovine serum albumin and 20 pM 125I-EGF with increasing concentrations of unlabelled EGF. Cultures were washed three times with 2 ml Hepes-buffered saline and the monolayers solubilized in 1 ml NaOH. The NaOH was transferred to tubes that were counted in a gamma counter. All points were done in triplicate. Data were analyzed using GraphPad Prism 4.0.
Internalization of 125I-EGF was measured in a similar fashion except that cells were incubated with 1 nM 125I-EGF for the indicated time at 37° C. To determine total cell-associated 125I-EGF, cultures were washed 3 times in 2 ml Hepes-buffered saline at 4° C. To determine internalized 125I-EGF, cultures were washed twice for 2 min with acid wash (50 mM glycine, pH 3.0, 100 mM NaCl). Non-specific binding was determining in replicate cultures containing 50 nM EGF.
EGF receptor autophosphorylation
Monolayers were treated with 30 μM phenylarsine oxide for one hour on ice in Hanks balanced salt solution to block phosphatase activity. Cells were washed twice with PBS at 37° C prior to stimulation with EGF for 2 min in DMEM containing 25 mM HEPES, pH 7.2 and 1 mg/ml bovine serum albumin. Cells were washed with cold PBS and scraped into RIPA buffer (10 mM Tris-HCl pH 7.2, 150 mM NaCl, 0.1% sodium dodecyl sulfate, 1% Triton X-100, 0.7% deoxycholate and 2.5mM EDTA) containing 20 mM p-nitrophenylphosphate, 100 μM sodium orthovanadate, and a protease inhibitor cocktail (Sigma). Protein concentrations were determined by BCA analysis. Equivalent amounts of protein were loaded onto 9% SDS-polyacrylamide gels. Gels were transferred onto PVDF and blotted with anti-phosphotyrosine or anti-EGF receptor antibodies.
For assays in which cells were depleted of cholesterol, monolayers were treated with 10 mM methyl-ß-cyclodextrin in DMEM containing 25mM HEPES, pH 7.2 and 1 mg/ml bovine serum albumin for 30 minutes at 37° C prior to stimulation by EGF.
Cross-linking of EGF receptors
CHO cells expressing wild type, SS- or CC- EGF receptors were incubated with 25 nM EGF for 3 min prior to the addition of BS3 to a final concentration of 3 mM in a reaction mixture buffered to pH 8.0. After 30 min, the cross-linking reactions were quenched by the addition of glycine to a final concentration of 1 M (pH 7.5). Cells were lysed as above, and equal amounts of protein were loaded onto a 4%-7.5% gradient SDS-polyacrylamide gel. After electrophoresis and transfer to PVDF, EGF receptors were visualized by Western blotting with anti-EGF receptor antibodies.
Isolation of lipid rafts
The protocol of Macdonald and Pike (13) for the production of detergent-free lipid rafts was followed. Briefly, three 150-mm diameter plates of cells were treated without or with 25 nM EGF for 3 minutes at 37° C. Cultures were washed in cold PBS, and scraped into lysis buffer (250 mM sucrose, 20 mM Tris pH 7.8, 100 μM calcium CaCl2, 100 μM MgCl2, 1 mM sodium orthovanadate, 20 mM p-nitrophenylphosphate and protease inhibitors). The lysate was passed through a 22 G needle 40 times, then centrifuged at 1000 x g, for 10 minutes. The post-nuclear supernatant was collected and mixed with an equal volume of 50% Opti-Prep in lysis buffer and placed in the bottom of a 12-ml centrifuge tube. The lysate was overlaid with a continuous gradient of 2-18% Opti-Prep, and centrifuged at 52,000 x g for 90 minutes. The gradient was fractionated into 666 μl fractions starting at the top of the gradient. Equal aliquots of each fraction were separated by polyacrylamide gel electrophoresis and analyzed by Western blotting for EGF receptors, Gq and the transferrin receptor.
Results
Generation of a palmitoylated EGF receptor
In transmembrane domain proteins, palmitoylation often occurs at cysteine residues adjacent to the membrane-cytoplasm interface (14, 15). Figure 1A shows the juxtamembrane domain sequence of the EGF receptor immediately C-terminal to the transmembrane domain. To generate a site for palmitoylation on the EGF receptor, we replaced Arg-647 and Val-650 with cysteines. We refer to this mutant as the CC-EGF receptor.
Figure 1. Palmitoylation of the CC-EGF receptor.
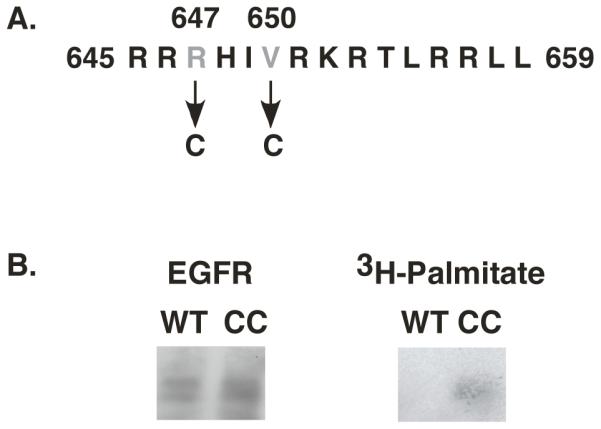
A) Sequence of the proximal juxtamembrane region of the EGF receptor showing the residues mutated to cysteines in the CC-EGF receptor. B) CHO cells expressing wild type or CC-EGF receptors were labeled with [3H]-palmitate and the EGF receptors immunoprecipitated as described under Experimental Procedures. Immunoprecipitates were run on SDS polyacrylamide gels and Western blotted for EGF receptors (left) or exposed to X-ray film to detect [3H] (right).
The wild type and the CC-EGF receptor were stably expressed in CHO cells that lack endogenous EGF receptors. To determine whether the CC-EGF receptor was palmitoylated, cells expressing either wild type or CC-EGF receptors were labeled with [3H]-palmitate as described in Experimental Procedures, immunoprecipitated with an anti-EGF receptor antibody and analyzed by SDS polyacrylamide gel electrophoresis. As shown in Figure 1B, although the levels of the wild type and CC-EGF receptors were comparable in the immunoprecipitates, only the CC-EGF receptor incorporated [3H]-palmitate during the labeling period. As the labeling period was short enough to preclude significant metabolism of the palmitate, these data indicate that the CC-EGF receptor is palmitoylated in vivo.
Because the CC-EGF receptor contains mutations in the juxtamembrane region that permitted receptor palmitoylation, it was necessary to distinguish the effects of palmitoylation on EGF receptor function from any effects that might be due simply to the mutation of Arg-647 and Val-650. To this end, a double serine mutation, the SS-EGF receptor, was constructed in which Arg-647 and Val-650 were replaced with serines. As serine residues cannot be palmitoylated, the SS-EGF receptor represents a control for phenotypic changes that might arise from mutation of residues 647 and 650 unrelated to receptor palmitoylation.
Effect of palmitoylation on EGF receptor function
Figure 2 shows Scatchard plots for the binding of 125I-EGF to wild type, CC- and SS-EGF receptors. As expected, the Scatchard plot for the wild type EGF receptor was upwardly concave, demonstrating the presence of high and low affinity forms of the EGF receptor that we have recently shown is the result of negative cooperativity in EGF binding (6). The Scatchard plot for the binding of 125I-EGF to the SS-EGF receptor was also curvilinear, indicating that mutation of Arg-647 and Val-650 to non-palmitoylatable serines does not significantly alter the ligand binding properties of the EGF receptor. By contrast, the Scatchard plot for the binding of 125I-EGF to the CC-EGF receptor was linear with a slope similar to that of the low affinity form of the wild type receptor. These data suggest that palmitoylation abolishes high affinity binding to the EGF receptor.
Figure 2. Scatchard analysis of 125I-EGF binding to EGF receptors.

CHO cells were stably transfected with wild type, CC- or SS-EGF receptors. Cells were subjected to 125EGF-binding and Scatchard analysis as described in Experimental Procedures. Values represent the mean +/- standard deviation of triplicate determinations.
Mutations of the EGF receptor that block receptor dimerization, such as Y246D (3, 4, 16), also lead to receptors that exhibit a single class of low affinity binding sites (6). To determine whether palmitoylation affected the ability of the CC-EGF receptor to oligomerize, a chemical crosslinking experiment was performed. Cells expressing wild type, CC- and SS-EGF receptors were incubated without or with EGF and the receptors were crosslinked with BS3. The lysates were separated on an SDS gel and blotted with an anti-EGF receptor antibody. The data in Figure 3 demonstrate that all three receptors showed a similar ability to be crosslinked into high molecular weight species, indicating that there was no obvious defect in oligomerization of the CC-EGF receptor.
Figure 3. Cross-linking of EGF receptors.
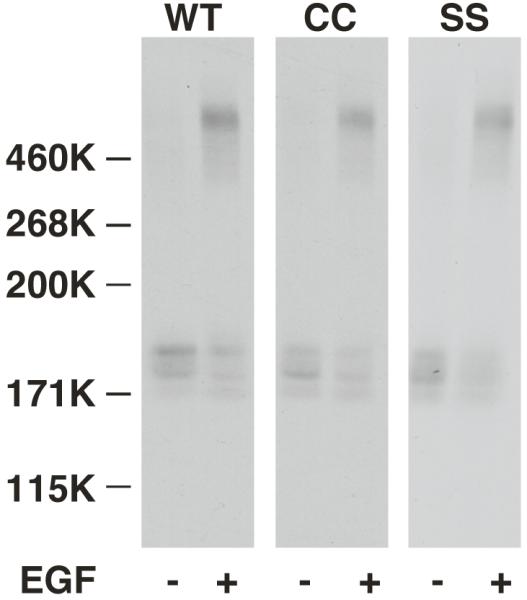
CHO cells expressing wild type, CC- or SS-EGF receptors were incubated in the absence or presence of 25 nM EGF for 3 min prior to the addition of BS3. Reaction mixtures were quenched, run on an SDS gel and Western blotted using anti-EGF receptor antibodies.
Although the CC-EGF receptor was able to bind ligand and to oligomerize, the ability of EGF to stimulate autophosphorylation was significantly reduced in the CC-EGF receptor as compared to the wild type and SS-EGF receptors. As shown in Figure 4A and quantitated in Figure 4B, the maximal level of phosphorylation of the CC-EGF receptor was only ∼20% of that seen for the wild type or SS-EGF receptors expressed at comparable levels (∼100,000 receptors/cell). Nevertheless, the EC50 for EGF was similar for all three receptors (2.6 nM, 2.0 nM, and 1.1 nM for wild type, CC- and SS-EGF receptors, respectively) suggesting that this was not the result of the differences in ligand binding affinity.
Figure 4. EGF-stimulated autophosphorylation of wild type and EGF receptor mutants.

CHO cells expressing similar levels of wild type, CC- and SS-EGF receptors were treated with increasing concentrations of EGF for 2 min at 37° C. Lysates were prepared and analyzed by SDS polyacrylamide gel electrophoresis. In panel C, cells were treated without or with 100 μM 2-bromopalmitate for 24 hr prior to assay. Panel A) Western blots of lysates with an anti-phosphotyrosine antibody (left) or an anti-EGF receptor antibody (right). Panel B) Quantitation of the Western blots in panel A. Panel C) Effect of 2-bromopalmitate on EGF receptor autophosphorylation.
If palmitoylation of the EGF receptor were responsible for the observed decrease in autophosphorylation of the CC-EGF receptor, then blocking palmitoylation of this receptor should reverse the decline in autophosphorylation observed in this mutant but should have no effect on the kinase activity of the wild type or SS-EGF receptors. As shown in Figure 4C, treatment of cells with 2-bromopalmitate, an inhibitor of protein palmitoylation (17), led to a marked increase in the kinase activity of the CC-EGF receptor compared to untreated controls. By contrast, treatment of cells expressing either wild type EGF receptors or SS-EGF receptors with 2-bromopalmitate failed to alter the autophosphorylation of these receptors. These data are consistent with the interpretation that palmitoylation of the CC-EGF receptor is responsible for its altered tyrosine kinase activity.
In addition to stimulating receptor phosphorylation, EGF also promotes the internalization of its receptor. To determine whether this function of the receptor was affected by palmitoylation, the internalization of wild type, CC- and SS-EGF receptors was compared by monitoring the internalization of 125I-EGF over time at 37° C. The data are presented as a plot of the ratio of internalized 125I-EGF to the surface-bound 125I-EGF (In/Sur) as a function of time. As shown in Figure 5, the rates of internalization of 125I-EGF by the wild type and SS-EGF receptors were similar but the palmitoylated CC-EGF receptor internalized ligand about 3-fold slower than either of these receptors. Thus, like receptor autophosphorylation, 125I-EGF internalization was significantly impaired in the palmitoylated EGF receptor.
Figure 5. Internalization of 125I-EGF by wild type and mutant EGF receptors.
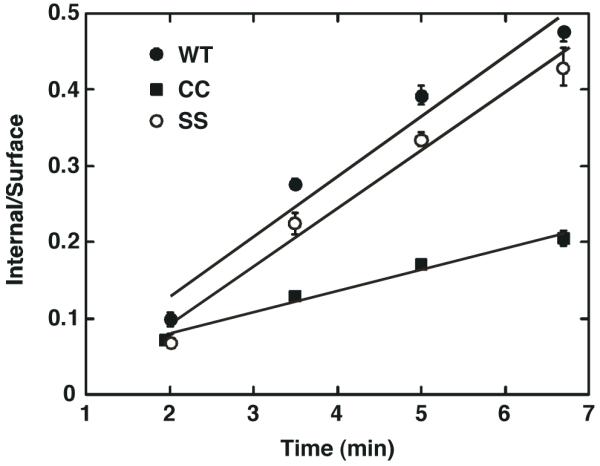
CHO cells expressing wild type, CC- or SS-EGF receptors were incubated with 1 nM 125I-EGF for the indicated times and internalized 125I-EGF measured as described in Experimental Procedures. Values represent the mean +/- standard deviation of duplicate determinations
Lipid raft localization of the palmitoylated EGF receptor
The EGF receptor is known to partition into lipid rafts (13, 18, 19). As palmitoylation can target proteins to lipid rafts (17, 20-22), it was possible that this modification could alter the interaction of the CC-EGF receptor with these cholesterol-rich domains. We therefore compared the ability of all three receptors to partition into lipid rafts. CHO cells expressing wild type, CC- or SS-EGF receptors were incubated without or with 25 nM EGF for 3 minutes and non-detergent lipid rafts were prepared by density gradient centrifugation (13). Equal aliquots of each fraction were run on SDS gels and blotted for EGF receptors, Gq and the transferrin receptor. Like the EGF receptor, Gq partitions into rafts and thus serves as an independent marker for these domains. The transferrin receptor is a non-raft plasma membrane protein used to determine the position of bulk plasma membrane proteins in the gradients. The data are shown in Figure 6.
Figure 6. Localization of EGF receptors to lipid rafts.

CHO cells expressing wild type, CC- or SS-EGF receptors were treated without or with 25 nM for 3 min then lysed and lipid rafts prepared by density gradient centrifugation as described in Experimental Procedures. Equal volumes of each fraction were analyzed by SDS polyacrylamide gel electrophoresis and Western blotting with the indicated antibodies.
As can be seen from the figure, in the absence of EGF, all three EGF receptors showed a similar propensity to partition into lipid rafts. Approximately half of each receptor was found in the low density, lipid raft fractions (fractions 1-5) of the gradient. A similar proportion of Gq was found in the low-density fractions, whereas the transferrin receptor, a non-raft plasma membrane protein, was found primarily in fractions of much higher density.
Addition of EGF did not change the distribution of either Gq or the transferrin receptor indicating that there was no general effect of the growth factor on membrane properties. However, treatment with EGF led to a significant shift of both the wild type and SS-EGF receptors out of the low-density fractions. By contrast, the distribution of the CC-EGF receptor changed very little in response to EGF. Thus, palmitoylation of the EGF receptor appeared to inhibit the ability of the EGF receptor to exit lipid rafts upon stimulation with growth factor.
This change in receptor distribution was reflected in the partitioning of autophosphorylated EGF receptors between raft and non-raft fractions. In the experiment shown in Figure 7, cells expressing wild type, CC- or SS-EGF receptors were stimulated with 25 EGF for 3 min and lipid rafts were prepared. The EGF receptor-containing raft and non-raft fractions were pooled separately and subjected to SDS polyacrylamide gel electrophoresis followed by Western blotting with a general anti-phosphotyrosine antibody (PY20) or with antibodies directed against specific sites of phosphorylation on the EGF receptor. The fractions were also blotted for EGF receptors. Receptor distribution between raft and non-raft fractions is quantitated for both control (Figure 7A) and EGF-stimulated cells (Figure 7B). However, since no receptor autophosphorylation was detected in the absence of EGF, the phosphorylation data are from EGF-treated cells only (Figure 7C through 7G).
Figure 7. Distribution of tyrosine phosphorylated EGF receptors between raft and non-raft compartments.

CHO cells expressing wild type, CC- or SS-EGF receptors were subjected to lysis and separation on density gradients to isolate lipid rafts. The low-density, lipid raft fractions containing the EGF receptor (fractions 1-5) were pooled as were the higher-density, non-raft fractions (either 8-12 or 9-13 depending on the gradient). Equal volumes of each fraction were separated by SDS polyacrylamide gel electrophoresis and subjected to Western blotting with anti-EGF receptor antibodies (panels A and B), anti-phosphotyrosine antibodies (panel C), or site-specific anti-phosphotyrosine antibodies (panels D-F). Blots were quantitated by densitometry.
As expected, addition of EGF stimulated the migration of the wild type and SS-EGF receptors out of lipid rafts (compare raft vs. non-raft fractions in Figure 7A vs. 7B). Western blots for total tyrosine phosphorylation of both the wild type and SS-EGF receptors (Figure 7C) showed that the vast majority of the phosphorylated receptor (∼90%) was present in the non-raft fraction. A similar distribution of phosphorylated receptors was apparent when individual sites of EGF receptor autophosphorylation were examined, including Tyr-992, Tyr-1045, Tyr-1068 and Tyr-1173. As the distribution of EGF receptor protein was ∼30% in rafts and ∼70% in the non-raft fraction in EGF-stimulated cells, the data suggest that phosphorylated EGF receptors are preferentially located in the non-raft fractions.
Like the wild type and SS-EGF receptors, the palmitoylated EGF receptor was evenly distributed between raft and non-raft fractions in control cells (Figure 7A). But unlike those receptors, the CC-EGF receptor did not shift into the non-raft fraction after EGF treatment (Figure 7B). This difference in receptor partitioning was associated with a more even distribution of the total tyrosine phosphorylated CC-EGF receptors between raft and non-raft fractions than was seen in the wild type or SS-EGF receptors (Figure 7C). Likewise, CC-EGF receptors phosphorylated at Tyr-992 and Tyr-1068 were relatively evenly distributed between raft and non-raft fractions (Figures 7D and 7F). However, CC-EGF receptors phosphorylated at Tyr-1045 and Tyr-1173 tended to partition preferentially into the non-raft fraction (Figures 7E and 7G).
Depletion of cholesterol from cells has been shown to disrupt lipid rafts and enhance EGF-stimulated receptor autophosphorylation (23-25). As shown in Figure 8, treatment of CHO cells expressing either wild type or SS-EGF receptors with methyl-ß-cyclodextrin did lead to an increase in EGF-stimulated receptor autophosphorylation. By contrast, depletion of cholesterol from cells expressing the palmitoylated-EGF receptor led to a decrease in the already compromised level of receptor autophosphorylation. These data indicate that the disruption of lipid rafts fails to reverse the impairment of receptor autophosphorylation seen in the palmitoylated receptor.
Figure 8. Effect of cholesterol depletion on EGF receptor autophosphorylation.
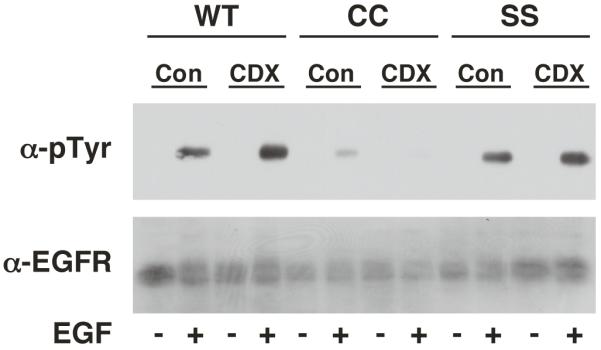
CHO cells expressing wild type, CC- or SS-EGF receptors were treated without or with 10 mM methyl-ß-cyclodextrin for 30 min to deplete cholesterol. Thereafter, cells were stimulated with 25 nM EGF for 2 min and lysates were prepared. Samples were analyzed by SDS polyacrylamide gel electrophoresis and Western blotting with an anti-phosphotyrosine antibody or an anti-EGF receptor antibody.
Discussion
A number of studies have pointed out the importance of the juxtamembrane domain of the EGF receptor in the process of EGF receptor kinase activation. Mutation or deletion of these residues has been shown to inhibit EGF-stimulated tyrosine kinase activity (26, 27). Similarly, Theil et al. (8) demonstrated that the allosteric activation of the EGF receptor kinase did not occur in receptors from which this sequence was deleted. Residues 645-657 have also been been implicated in the dimerization of the EGF receptor (8, 27, 28). This juxtamembrane segment contains a large number of basic amino acids, leading McLaughlin and colleagues to propose that this peptide lies along the membrane surface where it interacts with the negative charges of the phospholipid headgroups (29) and possibly calmodulin (29, 30). Alternatively, Aifa et al. (27, 28) have proposed that this positively-charged segment interacts with a negatively charged stretch of amino acids (residues 979-991) just C-terminal to the kinase domain. Though no consensus regarding the molecular role of this sequence has developed, it has consistently been found to be important in the activation of the EGF receptor kinase.
In these studies, we investigated the function of this sequence by linking it to the membrane via post-translational palmitoylation. We reasoned that if a specific conformation of the juxtamembrane domain were necessary for signal transduction, then linking this region to the membrane via addition of a palmitate group should restrict the mobility of this segment and should therefore impair activation of the EGF receptor. Consistent with this hypothesis, we found that maximal EGF-stimulated autophosphorylation of the CC-EGF receptor was only 20% of that seen in the wild type or SS-EGF receptors. Receptor internalization was also markedly diminished in the palmitoylated as compared to the wild type and SS-EGF receptors, further supporting the conclusion that palmitoylation of the juxtamembrane domain leads to a block in receptor-mediated signaling.
The inhibition of EGF-stimulated receptor kinase activity was not due to an inability of the palmitoylated receptor to bind ligand as this mutant bound EGF with nM affinity. Indeed, the EC50’s for receptor autophosphorylation were similar in the wild type, SS- and CC-EGF receptors, suggesting that ligand binding was not the limiting factor in the activation of the palmitoylated receptor. Nor did the block occur at the level of receptor oligomerization since the CC-EGF receptor showed a similar propensity to form high molecular weight oligomers as the wild type and SS-EGF receptors. As the kinase domain was wild type in all three receptors, these findings suggest that it is the transduction of the signal across the membrane that is impaired in the palmitoylated receptor.
Palmitoylation of the EGF receptor so close to the end of its transmembrane domain may interfere with helix rotation (31) or tilt (32) necessary for proper transmission of the signal through the membrane. Alternatively, the juxtamembrane extension has been shown to promote activation of the soluble intracellular domain of the EGF receptor kinase (8). This suggests that this segment functions via a mechanism that does not involve the membrane and may, in fact, require that it be freely accessible in the cytoplasm. It is therefore possible that tying the juxtamembrane domain to the plasma membrane via palmitoylation prevents it from accessing the cytosol where it is needed to promote proper formation of the activating asymmetric dimer.
Unexpectedly, we found that palmitoylation of the EGF receptor significantly changed the properties with which it bound EGF. A Scatchard plot of 125I-EGF binding to the wild type receptor exhibited the characteristic concave up shape demonstrating the existence of high and low affinity forms of the receptor and the presence of negative cooperativity (6). By contrast, the Scatchard plot for the binding of 125I-EGF to the palmitoylated CC-EGF receptor was linear, indicating the presence of only a low affinity form of the EGF receptor. The non-palmitoylatable SS-EGF receptor showed binding characteristics similar to those of the wild type EGF receptor, suggesting that mutation of Arg-647 and Val-650 by itself did not affect high affinity ligand binding. EGF receptors from which the entire intracellular domain has been deleted also exhibit a single class of low affinity binding sites (33, 34) suggesting that the cytoplasmic portion of the receptor contributes to the formation of the high affinity form of the EGF receptor. Since all other parts of the intracellular domain of the palmitoylated EGF receptor were wild type, our data suggest that it is the juxtamembrane region that is important for high affinity EGF binding.
Because palmitoylation has been shown to target proteins to lipid rafts (17, 20-22), we examined the effect of this modification on the association of the EGF receptor with these membrane domains. Under basal conditions, a similar fraction of wild type, CC- and SS-EGF receptors was associated with rafts. However, only the wild type and SS-EGF receptors moved out of rafts in response to EGF. As receptor tyrosine kinase activity has been shown to be required for movement of the EGF receptor out of rafts (35), it is possible that the failure of the palmitoylated receptor to exit rafts upon treatment with EGF is due to its relatively low level of autophosphorylation. However, it is also possible that the strength of its raft association has been altered by the addition of the palmitate group. Consistent with this interpretation is the finding that a significant amount of tyrosine phosphorylated CC-EGF receptor was found in the raft fractions. This indicates that even after phosphorylation, the palmitoylated receptors did not exit rafts. Thus, acylation appears to provide a raft localization signal that cannot be over-ridden by receptor activation/phosphorylation.
Previous reports have demonstrated that the kinase activity of the EGF receptor is suppressed when it is associated with lipid rafts (24, 36). It is therefore noteworthy that the palmitoylated receptor could undergo autophosphorylation while in the lipid raft compartment. Thus, while these cholesterol-enriched membrane domains may impair EGF receptor autophosphorylation, they do not completely inhibit it. Interestingly, for some sites of autophosphorylation (e.g. Tyr-992 and Tyr-1068), the fraction of phosphorylated receptors present in lipid rafts was similar to the fraction of receptor protein that was found in these domains. However, for other sites (e.g. Tyr-1045 and Tyr-1173), a higher proportion of the receptors phosphorylated on these sites was found in the non-raft fractions of the gradient. This suggests that phosphorylation on one or both of these latter two sites may be associated with movement of the receptor out of rafts.
Given the failure of the CC-EGF receptor to move out of rafts, the suppressive effect of these domains on the kinase activity of the EGF receptor could contribute to the observed decrease in autophosphorylation of the palmitoylated EGF receptor. However, the finding that disruption of rafts by cholesterol depletion did not reverse the autophosphorylation phenotype of the CC-EGF receptor suggests that raft localization is not the principal cause for the diminished kinase activity associated with receptor palmitoylation. We therefore favor the interpretation that a change in conformation or mobility of the juxtamembrane domain that occurs as a result of the palmitoylation is the proximal cause of the diminished capacity of this receptor to autophosphorylate in response to ligand binding.
In summary, we have found that palmitoylation of the EGF receptor at a position just beyond the transmembrane helix leads to significant changes in EGF receptor function. EGF-stimulated receptor autophosphorylation was impaired and high affinity ligand binding was abolished. The data are consistent with the hypothesis that the proximal intracellular juxtamembrane region of the receptor is required for efficient signal transduction as well as for the adoption of the high affinity conformation by the extracellular ligand binding domain.
Acknowledgements
The authors would like to thank Dr. Maurine Linder for many helpful discussions.
Abbreviations
- DMEM
Dulbecco’s modified Eagle’s medium
- EGF
epidermal growth factor
- SDS
sodium dodecyl sulfate
References
- 1.Ullrich A, Coussens L, Hayflick JS, Dull TJ, Gray A, Tam AW, Lee J, Yarden Y, Libermann TA, Schlessinger J, Downward J, Mayes ELV, Whittle N, Waterfield ND, Seeburg PH. Human Epidermal Growth Factor Receptor cDNA Sequence and Aberrant Expression of the Amplified Gene in A431 Epidermoid Carcinoma Cells. Nature. 1984;309:418–425. doi: 10.1038/309418a0. [DOI] [PubMed] [Google Scholar]
- 2.Ferguson KM, Berger MB, Mendrola JM, Cho H-S, Leahy DJ, Lemmon MA. EGF Activates Its Receptor by Removing Interactions that Autoinhibit Ectodomain Dimerization. Mol. Cell. 2003;11:507–517. doi: 10.1016/s1097-2765(03)00047-9. [DOI] [PubMed] [Google Scholar]
- 3.Garrett TPJ, McKern NM, Lou M, Elleman TC, Adams TE, Lovrecz GO, Zhu H-J, Walker F, Frenkel MJ, Hoyne PA, Jorissen RN, Nice EC, Burgess AW, Ward CW. Crystal Structure of a Truncated Epidermal Growth Factor Receptor Extracellular Domain Bound to Transforming Growth Factor α. Cell. 2002;110:763–773. doi: 10.1016/s0092-8674(02)00940-6. [DOI] [PubMed] [Google Scholar]
- 4.Ogiso H, Ishitani R, Nureki O, Fukai S, Yamanaka M, Kim J-H, Saito K, Sakamoto A, Inoue M, Shirouzu M, Yokoyama S. Crystal Structure of the Complex of Human Epidermal Growth Factor and Receptor Extracellular Domains. Cell. 2002;110:775–787. doi: 10.1016/s0092-8674(02)00963-7. [DOI] [PubMed] [Google Scholar]
- 5.Citri A, Yarden Y. EGF-ErbB Signalling: Towards the Systems Level. Nature Rev. Mol. Cell Biol. 2006;7:505–516. doi: 10.1038/nrm1962. [DOI] [PubMed] [Google Scholar]
- 6.Macdonald JL, Pike LJ. Heterogeneity in EGF Binding Affinities Arises From Negative Cooperativity in an Aggregating System. Proc. Natl. Acad. Sci. U.S.A. 2008;105:112–117. doi: 10.1073/pnas.0707080105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhang X, Gureasko J, Shen K, Cole PA, Kuriyan J. An Allosteric Mechanism for Activation of the Kinase Domain of Epidermal Growth Factor Receptor. Cell. 2006;125:1137–1149. doi: 10.1016/j.cell.2006.05.013. [DOI] [PubMed] [Google Scholar]
- 8.Thiel KW, Carpenter G. Epidermal Growth Factor Receptor Juxtamembrane Region Regulates Allosteric Tyrosine Kinase Activation. Proc. Natl. Acad. Sci. U.S.A. 2007;104:19238–19243. doi: 10.1073/pnas.0703854104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mendrola JM, Berger MB, King MC, Lemmon MA. The Single Transmembrane Domains of ErbB Receptors Self-associate in Cell Membranes. J. Biol. Chem. 2002;277:4704–4712. doi: 10.1074/jbc.M108681200. [DOI] [PubMed] [Google Scholar]
- 10.Duneau J-P, Vegh AP, Sturgis JN. A Dimerization Hierarchy in the Transmembrane Domains of the HER Receptor Family. Biochem. 2007;46:2010–2019. doi: 10.1021/bi061436f. [DOI] [PubMed] [Google Scholar]
- 11.Moriki T, Maruyama H, Maruyama IN. Activation of Preformed EGF Receptor Dimers by Ligand-induced Rotation of the Transmembrane Domain. J. Mol. Biol. 2001;311:1011–1026. doi: 10.1006/jmbi.2001.4923. [DOI] [PubMed] [Google Scholar]
- 12.Doran DM, Spar IL. Oxidative Iodine Monochloride Iodination Technique. J. Immunol. Methods. 1980;39:155–163. doi: 10.1016/0022-1759(80)90304-x. [DOI] [PubMed] [Google Scholar]
- 13.Macdonald JL, Pike LJ. A Simplified Method for the Preparation of Detergent-free Lipid Rafts. J. Lipid Res. 2005;46:1061–1067. doi: 10.1194/jlr.D400041-JLR200. [DOI] [PubMed] [Google Scholar]
- 14.Resh MD. Palmitoylation of Ligands, Receptors and Intracellular Signaling Molecules. Science’s stke. 2006;359:1–14. doi: 10.1126/stke.3592006re14. [DOI] [PubMed] [Google Scholar]
- 15.Smotrys JE, Linder ME. Palmitoylation of Intracellular Signaling Proteins. Regulation and Function. Ann. Rev. Biochem. 2004;73:559–587. doi: 10.1146/annurev.biochem.73.011303.073954. [DOI] [PubMed] [Google Scholar]
- 16.Walker F, Orchard SG, Jorissen RN, Hall NE, Zhang H-H, Hoyne PA, Adams TE, Johns TG, Ward CW, Nice EC, Burgess AW. CR1/CR2 Interactions Modulate the Functions of the Cell Surface Epidermal Growth Factor Receptor. J. Biol. Chem. 2004;279:22387–22398. doi: 10.1074/jbc.M401244200. [DOI] [PubMed] [Google Scholar]
- 17.Webb Y, Hermida-Matsumoto L, Resh MD. Inhibition of Protein Palmitoylation, Raft Localization, and T Cell Signaling by 2-Bromopalmitate and Polyunsaturated Fatty Acids. J. Biol. Chem. 2000;275:261–270. doi: 10.1074/jbc.275.1.261. [DOI] [PubMed] [Google Scholar]
- 18.Mineo C, James GL, Smart EJ, Anderson RGW. Localization of Epidermal Growth Factor-stimulated Ras/Raf-1 Interaction to Caveolae Membrane. J. Biol. Chem. 1996;271:11930–11935. doi: 10.1074/jbc.271.20.11930. [DOI] [PubMed] [Google Scholar]
- 19.Pike LJ, Miller JM. Cholesterol Depletion De-localizes PIP2 and Inhibits Hormone-Stimulated Phosphatidylinositol Turnover. J. Biol. Chem. 1998;273:22298–22304. doi: 10.1074/jbc.273.35.22298. [DOI] [PubMed] [Google Scholar]
- 20.Moffett S, Brown DA, Linder ME. Lipid-dependent Targeting of G Proteins into Rafts. J. Biol. Chem. 2000;275:2191–2198. doi: 10.1074/jbc.275.3.2191. [DOI] [PubMed] [Google Scholar]
- 21.Melkonian K. a., Ostermeyer AG, Chen JZ, Roth MG, Brown DA. Role of Lipid Modifications in Targeting Proteins to Detergent-resistant Membrane Rafts. J. Biol. Chem. 1999;274:3910–3917. doi: 10.1074/jbc.274.6.3910. [DOI] [PubMed] [Google Scholar]
- 22.Zacharias DA, Violin JD, Newton AC, Tsien RY. Partitioning of Lipid-Modified Monomeric GFPs into Membrane Microdomains of Live Cells. Science. 2002;296:913–916. doi: 10.1126/science.1068539. [DOI] [PubMed] [Google Scholar]
- 23.Roepstorff K, Thomsen P, Sandvig K, van Deurs B. Sequestration of EGF Receptors in Non-caveolar Lipid Rafts Inhibits Ligand Binding. J. Biol. Chem. 2002;277:18954–18960. doi: 10.1074/jbc.M201422200. [DOI] [PubMed] [Google Scholar]
- 24.Pike LJ, Casey L. Cholesterol Levels Modulate EGF Receptor-Mediated Signaling by Altering Receptor Function and Trafficking. Biochem. 2002;41:10315–10322. doi: 10.1021/bi025943i. [DOI] [PubMed] [Google Scholar]
- 25.Westover EJ, Covey DF, Brockman HL, Brown RE, Pike LJ. Cholesterol Depletion Results in Site-Specific Increases in EGF Receptor Phosphorylation Due to Membrane Level Effects: Studies with Cholesterol Enantiomers. J. Biol. Chem. 2003;278:51125–51133. doi: 10.1074/jbc.M304332200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yamane K, Toyoshima C, Nishimura S. Ligand-Induced Functions of the Epidermal Growth Factor Receptor Require the Positively Charged Region Asymmetrically Distributed Across the Plasma Membrane. Biochem. Biophys. Res. Commu. 1992;184:1301–1310. doi: 10.1016/s0006-291x(05)80024-5. [DOI] [PubMed] [Google Scholar]
- 27.Aifa S, Aydin J, Nordvall G, Lundstrom I, Svensson SPS, Hermanson O. A Basic Peptide Within the Juxtamembrane Region is Required for EGF Receptor Dimerization. Exp. Cell Res. 2005;302:108–114. doi: 10.1016/j.yexcr.2004.08.032. [DOI] [PubMed] [Google Scholar]
- 28.Aifa S, Miled N, Frikha F, Aniba MR, Svensson SPS, Rebai A. Electrostatic Interactions of Peptides Flanking the Tyrosine Kinase Domain in the Epidermal Growth Factor Receptor Provides a Model for Intracellular Dimerization and Autophosphorylation. Proteins. 2006;62:1036–1043. doi: 10.1002/prot.20780. [DOI] [PubMed] [Google Scholar]
- 29.McLaughlin S, Smith SO, Hayman MJ, Murray D. An Electrostatic Engine Model For Autoinhibition and Activation of the Epidermal Growth Factor Receptor (EGFR/ErbB) Family. J. Gen. Physiol. 2005;126:41–53. doi: 10.1085/jgp.200509274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li H, Ruano MJ, Villalobo A. Endogenous Calmodulin Interacts with the Epidermal Growth Factor Receptor in Living Cells. FEBS Lett. 2004;559:175–180. doi: 10.1016/S0014-5793(04)00067-5. [DOI] [PubMed] [Google Scholar]
- 31.Bell CA, Tynan JA, Hart KC, Meyer AN, Robertson SC, Donoghue DJ. Rotational Coupling of the Transmembrane and Kinase Domains of the Neu Receptor Tyrosine Kinase. Mol. Biol. Cell. 2000;11:3589–3599. doi: 10.1091/mbc.11.10.3589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Joseph M, Nagaraj R. Interactions of Peptides Corresponding t Fatty Acylation Sites in Proteins with Model Membranes. J. Biol. Chem. 1995;270:16749–16765. doi: 10.1074/jbc.270.28.16749. [DOI] [PubMed] [Google Scholar]
- 33.Prywes R, Livneh E, Ullrich A, Schlessinger J. Mutations in the Cytoplasmic Domain of EGF Receptor Affect EGF Binding and Receptor Internalization. EMBO J. 1986;5:2179–2190. doi: 10.1002/j.1460-2075.1986.tb04482.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kashles O, Yarden Y, Fischer R, Ullrich A, Schlessinger J. A Dominant Negative Mutation Suppresses the Function of Normal Epidermal Growth Factor Receptors by Heterodimerization. Mol. Cell. Biol. 1991;11:1454–1463. doi: 10.1128/mcb.11.3.1454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mineo C, Gill GN, Anderson RGW. Regulated Migration of Epidermal Growth Factor Receptor from Caveolae. J. Biol. Chem. 1999;274:30636–30643. doi: 10.1074/jbc.274.43.30636. [DOI] [PubMed] [Google Scholar]
- 36.Ringerike T, Glystad FD, Levy FO, Madshus IH, Stang E. Cholesterol is Important in Control of EGF Receptor Kinase Activity but EGF Receptors are Not Concentrated in Caveolae. J. Cell Sci. 2002;115:1331–1340. doi: 10.1242/jcs.115.6.1331. [DOI] [PubMed] [Google Scholar]