

Abstract
-The design and solution-phase synthesis of an α-helix mimetic library as an integral component of a small molecule library targeting protein-protein interactions are described. The iterative design, synthesis, and evaluation of the candidate α-helix mimetic was initiated from a precedented triaryl template and refined by screening the designs for inhibition of MDM2/p53 binding. Upon identifying a chemically and biologically satisfactory design and consistent with the screening capabilities of academic collaborators, the corresponding complete library was assembled as 400 mixtures of 20 compounds (20 × 20 × 20-mix), where the added subunits are designed to mimic all possible permutations of the naturally occurring i, i+4, i+7 amino acid side chains of an α-helix. The library (8000 compounds) was prepared using a solution-phase synthetic protocol enlisting acid/base liquid-liquid extractions for purification on a scale that insures its long term availability for screening campaigns. Screening of the library for inhibition of MDM2/p53 binding not only identified the lead α-helix mimetic upon which the library was based, but also suggests that a digestion of the initial screening results that accompany the use of such a comprehensive library can provide insights into the nature of the interaction (e.g., an α-helix mediated protein-protein interaction) and define the key residues and their characteristics responsible for recognition.
Introduction
Protein-protein interactions1 play pivotal roles in nearly all biological processes including cellular signaling. Selective modulation of specific protein-protein interactions by small molecules has emerged as an important approach to investigating biological processes, for validating new drug targets, and for the development of new therapeutics. However, the development of small molecule modulators of protein-protein interactions has been slow.2 The challenges associated with such targets are often attributed to the large surface area generally covered by the two interacting proteins (approximately 800 Å2 per protein on average), the relatively flat binding interface, and the noncontiguous binding regions within the binding proteins. Although systematic case studies of selected protein-protein interactions have shown that such generalizations represent an oversimplification and overstatement of the challenges, few generalized approaches to targeting protein-protein interactions have yet emerged.
Over the past decade, we have enlisted a simple solution-phase library synthesis protocol, complementary to more conventional solid-phase techniques, for generation of libraries capable of targeting protein-protein or protein-DNA interactions.3-12 The protocol features acid/base liquid-liquid or liquid-solid extractions for the purification of products (>95% pure irrespective of the reaction efficiency), and offers the advantages of a less limiting scale, expanded repertoire of chemical reactions (use of heterogeneous catalysts and reagents), direct production of soluble intermediates and final products for assay, and the lack of required linking, attachment and detachment, or capping steps. It is readily amenable to convergent synthetic strategies, the synthesis of mixture libraries, or use of dynamic libraries. Notably, a number of effective small molecule modulators of protein-protein or protein-DNA interactions have been identified from screening these libraries.3-13
As part of a program to expand our current 80,000 compound library2c,3 and to prepare a general small molecule library designed to selectively modulate protein-protein interactions, the three main recognition motifs mediating protein-protein interactions (α-helix, β-turn, and β-strand) have been targeted. Thus, three libraries built on templates mimicking each such secondary structure and diversified with groups representing the 20 natural amino acid side chains might be expected to cover nearly all targetable protein-protein interactions with as few as 24,000 compounds (3 × 20 × 20 × 20). In principle, the three libraries would contain a member capable of modulating any protein-protein interaction mediated by an α-helix, β-turn, or β-strand, and would provide a unique resource for interrogating such targets. Even if the key recognition motif is unknown (no structure) or unrecognized (not yet mapped), the library screening would be capable of providing a lead structure, provide insights into the nature of the interaction (α-helix, β-turn, or extended β-strand), and identify the key amino acids residues responsible for the protein-protein interaction. Herein, we report the design and synthesis of the first of these three libraries, an α-helix mimetic library targeting protein-protein interactions.
Results and Discussion
α-Helix Mimetic Design
The α-helix is the most common protein secondary structure, constituting more than 40% of the polypeptide structure in proteins. Recently, Hamilton and coworkers have developed an impressive class of small molecule scaffolds that can mimic the structural and recognition binding features of an α-helix (Figure 1).14-16 A functionalized terphenyl scaffold 1 was found to provide a rigid framework from which aryl o-substituents are projected to mimic the side chains at the i, i+4, and i+7 positions of an α-helix.17 This design was extended to other closely related structures including terpyridine 2,14d oligoamide 3,15 and terephthalamide 416 derivatives. These rationally designed compounds were shown to effectively inhibit protein-protein interactions featuring α-helix-mediated binding and recognition including Bcl-xL/Bak,14c,f,16a and p53/HDM2,14a,b thus validating the design. Although the Hamilton terphenyl motif could serve as a template for an α-helix mimetic library, the linear syntheses and poor solubilities of the terphenyl systems can pose technical problems for library synthesis and subsequent screening. We hoped to address these issues with modifications to the original Hamilton design.
Figure 1.

Hamilton's α-helix mimetic scaffolds.
We initiated our efforts with the triaryl amide scaffold 5 that closely resembles Hamilton's model 3 (Figure 2). Not only would the individual subunits now be joined by a simple amide coupling reaction subject to purification by acid/base extraction, but side chain diversification through o-alkoxy substituents could be achieved by a well established alcohol aromatic substitution reaction of 3-fluoro-4-nitrobenzoates in which the activating nitro substituent additionally serves as a “protected” aniline nitrogen for eventual coupling. In addition to the improved synthetic accessibility allowing its simple extension to libraries, the amide bonds connecting the aryl units provide an inherently greater flexibility, higher polarity, and improved aqueous solubility. As detailed below, 5 and several subsequent iterative designs that maintain these underlying simplifications were examined for their ability to function as α-helix mimetics.
Figure 2.
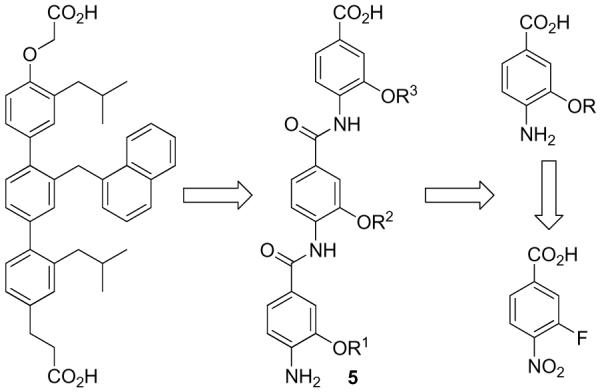
First-generation triaryl amide based scaffold.
Testing and Refinement of the α-Helix Mimetic Design against MDM2/p53
The structurally well-characterized MDM2/p53 protein-protein interaction was chosen as the target protein pair against which the α-helix design would be tested and refined. The MDM2/p53 (HDM2/p53) interaction has attracted considerable attention because of its therapeutic potential for the treatment of tumors with disregulated p53 resulting from overexpression of MDM2.18 The x-ray structure of a bound p53 peptide revealed a well-defined MDM2 hydrophobic binding pocket that is occupied by three key amino acid side chains (Phe19, Trp23, Leu26) on one face of a p53 α-helix.19 With this structural information in hand, the p53/MDM2 interaction has emerged as an important and prototypical target for the rational design and development of small molecule inhibitors of a protein-protein interaction.13a,14a-b,20-23 A prospective modeling study with template 5 suggested that the side chains overlap well with the three interacting amino acids of the p53 α-helix and might bind the MDM2 binding pocket (Figure 3).
Figure 3.
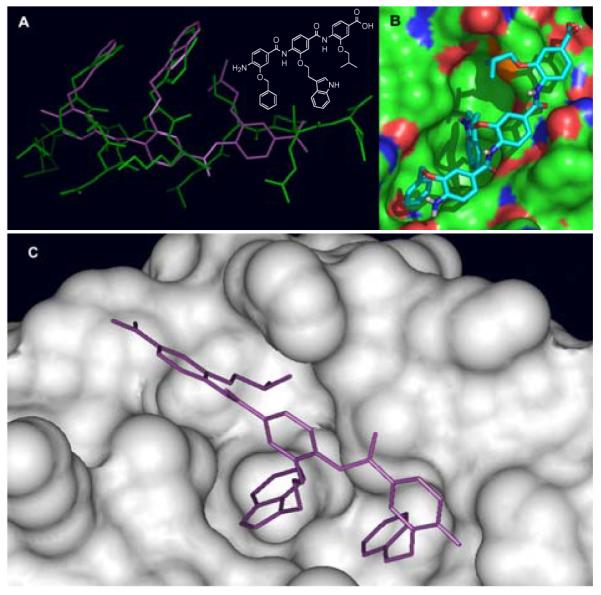
(A) An overlay of an α-helix mimetic compound (structure shown) with the p53 peptide. (B) Surface rendition of HDM2 with the α-helix mimetic located in the p53 binding pocket (calculated by Autodock3). (C) Further computational optimization of the α-helix compound bound at the HDM2/p53 binding pocket.
Ten different and iterative variants on the template 5 were prepared (80 compounds) and examined for inhibition of MDM2/p53 binding (Figure 4), in which the subunits and aryl substituents were chosen to mimic the side chains of Phe19, Trp23, and Leu26 of the p53 α-helix. In these efforts, the number of unnatural aryl subunits (1-3), the position of the aryl alkoxy substituent (3-alkoxy vs 2-alkoxy), the order of the side chain presentation (e.g., [Phe]-[Trp]-[Leu] vs. [Leu]-[Trp]-[Phe]), and the incorporation of a Nap versus Trp central side chain were examined (Figure 4). Activity was only observed with the C-terminus carboxylic acids (esters inactive) potentially reflecting an impact on compound solubility, while the N-terminus functional status had less or little impact on activity (NO2 ≈ NH2 ≈ NHBoc). The 3-alkoxyaryl derivatives exceeded the activity of the corresponding 2-alkoxyaryl derivatives, the [Trp] containing derivatives were typically or significantly more active than the [Nap] containing derivatives, and the projected subunit order represented by 5 (H2N-[Phe]-[Trp]-[Leu]-OH) proved more active than a presentation of the side chains in the reverse order (H2N-[Leu]-[Trp]-[Phe]-OH). Moreover, the partial two subunit structures often approached the activity of the three subunit derivatives, and the sequential incorporation of the more flexible natural amino acids at the termini maintained and often improved the activity. These latter observations suggested that the spatially more rigid side chain presentation embodied in the triaryl template permits two, but perhaps not three, effective side chain interactions and that this may be improved with the more flexible natural amino acids that may adjust the projected side chain distances. Additionally, the physical properties of the candidate compounds, especially the water solubility of the deprotected final derivatives, significantly improved as the number of aryl subunits was reduced as did their synthetic complexity. These latter features coupled with the activity of the modified design led to its selection for synthesis. Notably, its selection is not meant to suggest that this template represents the best α-helix mimetic design, but rather that it is sufficient for our screening objectives and chemically the most feasible to produce in a library format.
Figure 4.

Candidate MDM2/p53 inhibitors.
Synthesis of the α-Helix Mimetic Library
The plan for the construction of the library is outlined in Scheme 1. By introducing and utilizing 20 amino acid side chain variants at each of the positions of the trimer scaffold, all possible combinations produce 8,000 compounds representing all permutations on a naturally occurring α-helix. The final compounds can be obtained from the Boc/tert-buyl ester protected trimers, which are accessed from the aniline dimers. In turn, the aniline dimers are derived from the corresponding aryl nitro dimers that can be obtained from the aryl nitro subunits. The aryl nitro group serves both as an amine protecting group and also allows the introduction of the R2 diversity elements via a nucleophilic aromatic substitution reaction. Consistent with our objectives and the screening capabilities of academic collaborators,4-11 the library was to be assembled as 400 mixtures of 20 compounds (20 × 20 × 20-mix) by conducting the final coupling with a full mixture of the 20 amino acids. The identification of the individual compounds responsible for any mixture activity is conducted by resynthesis of the individual 20 compounds in the mixture from archived samples of the precursor dimers (1 step) and their individual rescreening. Facilitating the library synthesis, the isolation and purification of each intermediate is conducted by simple acid/base liquid-liquid extractions.3
Scheme 1.
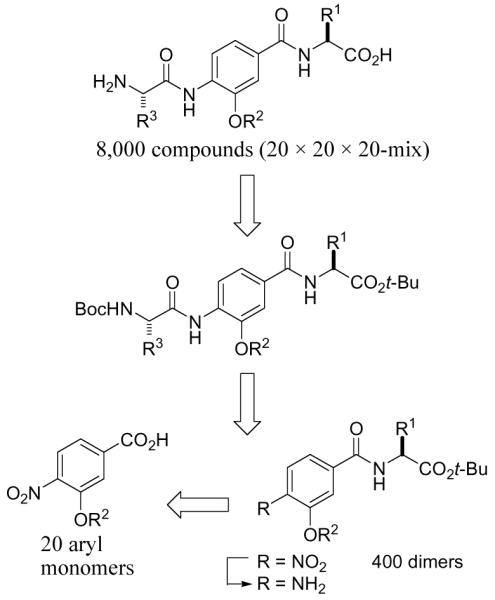
Plan for construction of the library.
Central to the selected template and the basis of the original design rested with access to and the subsequent coupling of 3-alkoxy-4-nitrobenzoates. These were anticipated to be accessed enlisting an aromatic nucleophilic substitution reaction of 3-fluoro-4-nitrobenzoates with the appropriate alkoxide representing an amino acid side chain. Given a perceived degeneracy of incorporating both aspartic acid (Asp) and glutamic acid (Glu) and both asparagine (Asn) and glutamine (Gln) side chains, only a single side chain (Asp and Asn) was used to represent each. The former (Asp and Asn) provide stable α-alkoxy linkages to the aryl core whereas the latter (Glu and Gln) would entail a more problematic and less stable β-alkoxy linkage. Similarly, an arginine side chain was not incorporated, but lysine (Lys) was. Finally, no attempt was made to incorporate a cysteine side chain anticipating that it would provide problematic storage and stability issues, and we found no direct manner to incorporate a proline. These five natural amino acid side chains were replaced with additional aromatic or hydrophobic side chains found to be effective against protein-protein interactions: O-methyl tyrosine [Tyr(Me)], ethyl representing an aminobutyric acid (Abu), 4-chlorophenylalanine [Phe(4-Cl)], naphthyl (Nap), and homophenylalanine (HoPhe) representing a one carbon extension of phenylalanine itself. The aryl subunits were accessed in good yields (Figure 5) by a room temperature nucleophilic aromatic substitution of 3-fluoro-4-nitrobenzoic acid (6). Sodium hydride served as the base for alkoxide formation as well as carboxylic acid deprotonation, with the exceptions of the formation of 7 and 8 where NaOMe and NaOEt were employed, respectively (entries 1 and 2). THF was found to be a suitable solvent and THF/DMF (6:1) was enlisted for the synthesis of 19 and 20 to overcome solubility issues (entries 13 and 14). Initial efforts to conduct the aromatic nucleophilic substitution reaction with the methyl and t-butyl esters of 3-fluoro-4-nitrobenzoic acid were not as satisfactory and both suffered competitive transesterification reactions (Me > t-Bu) with the reacting alkoxide that was especially prominent with the smaller nucleophiles. This competitive reaction is not possible enlisting 6, which additionally provides the free carboxylic acid directly thereby avoiding an intermediate deprotection step prior to coupling. What is remarkable and unappreciated at the time we initiated our efforts, the reaction can be conducted with 6 at room temperature for typically 1-3 h even though the substrate carboxylic acid is deprotonated.
Figure 5.

Synthesis of aryl subunits.
All attempts to access 26 by a direct aromatic substitution with the alkoxide derived from glycolamide were complicated by competitive amide versus alkoxide addition. Instead, 26 was prepared by nucleophilic substitution with the alkoxide derived from methyl glycolate to afford 25, and subsequent aminolysis of the methyl ester (Scheme 2). Finally, commercially available 4-nitrobenzoic acid was employed as an aryl subunit lacking an amino acid side chain (Gly).
Scheme 2.
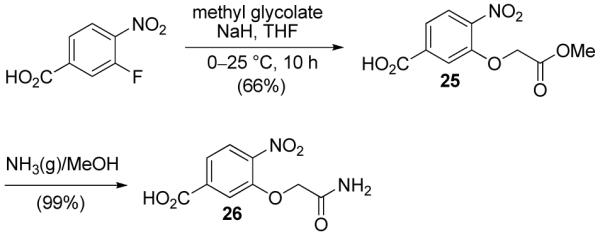
Synthesis of 26.
Four hundred individual dimers were subsequently synthesized employing the 20 aryl subunits and 20 t-butyl α-amino acids via EDCI/HOAt mediated peptide coupling enabling purification by simple acid/base extractions (equation 1).
 |
(1) |
All couplings proceeded in near quantitative yields affording the dimers in amounts ranging from 80-300 mg (Figure S1 in Supporting Information). The twenty diagonal matrix dimers featuring monomers with the same side chains were characterized and constitute a verification of the product derived from each coupling subunit (Figure 6).
Figure 6.

Characterized O2N-[XXX]-XXX-Ot-Bu.24
The next stage in the library synthesis involved reduction of the 400 aryl nitro dimers to the corresponding anilines. We developed reduction conditions involving the use of activated zinc nanopowder in combination with ammonium chloride in aqueous acetone, as they were found to tolerate the presence of benzyl ethers, aryl halides, and sulfur. Remarkably, the reductions proceeded in near quantitative yields at room temperature in a matter of seconds under optimized conditions (equation 2).
 |
(2) |
Although a number of reagents that we examined can provide the analogous reduction [Zn(Hg), Al(Hg), H2-Pd/C, Fe/AcOH, SnCl2/EtOH, Zn/EtOH], none do so at a rate competitive with the zinc nanopowder, which insures complete clean reduction independent of the substrate and reaction period adopted. Other methods often provided incomplete nitro reduction, competitive reductions (side chain hydrogenolysis), or competitive protecting group cleavage, and even ordinary activated zinc powder is much slower requiring elevated temperatures and providing the intermediate hydroxyamines as contaminant or major products in our hands. As such, we anticipate that this simple modification making use of activated zinc nanopowder will find widespread utility beyond the application detailed herein.
The final library was obtained by coupling each of the 400 aniline dimers with an equimolar mixture of 20 Boc-protected amino acids. Subsequent acid-mediated deprotection afforded 400 mixtures of 20 compounds (Scheme 3).
Scheme 3.
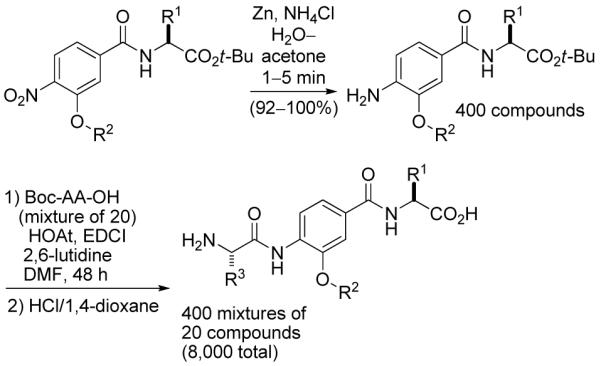
Final stage of library synthesis.
To ensure that each of the 20 Boc-protected amino acids fully and equally react with the aniline dimer, a slight excess of aniline was employed with respect to the mixture of 20 amino acids. The LCMS trace of the product mixture from a typical coupling of the aniline dimer 47 with an equimolar mixture of 20 amino acids exhibited the peaks corresponding to the 20 products and the remaining 47. The excess aniline in the product mixtures was anticipated to be removed by acidic aqueous extractions. However, the relatively nonbasic aniline dimers, especially the more nonpolar variants, did not effectively partition into an aqueous acidic layer. Consequently, the product mixtures were treated with sulfonyl chloride polystyrene resin and pyridine resulting in the effective removal of the anilines via their irreversible trapping as demonstrated for 47 (Figure 7).
Figure 7.

Removal of excess aniline from the product mixture. HPLC conditions: linear gradient 30-98% MeCN in H2O over 5 min, then 98% MeCN for 7 min with 0.75 mL/min flow rate.
The final global deprotection was achieved upon treatment with 4 M HCl in 1,4-dioxane providing each of the final mixtures in amounts ranging from 40-60 mg. Complete Boc, tert-butyl ester, TIPS, trityl, and tert-butyl ether removal was achieved after treatment under these conditions for 8-10 h. Stability control studies indicated that the benzyl phenyl ethers of the unnatural aromatic subunits 14, 16, and 17 remained intact during the final acid-mediated deprotections and all 20 final products can be readily detected by MS in a representative final mixture (Figure S3 in Supporting Information).
To further verify the quality of the construction of the library as 20 compound mixtures, a representative final library mixture was compared by LC to an authentic equimolar mixture of the 20 compounds prepared from the individual components (Figure 8). Although a separation of all 20 components is not possible on a single HPLC trace, the identical profile displayed by the two mixtures confirms that not only are all 20 compounds in the final mixture, but that they must be present in amounts that approach an equimolar mixture.
Figure 8.

Comparison of library (top) and authentic (bottom) equimolar mixture of final 20 compounds. HPLC conditions: linear gradient 30-98% MeCN in H2O over 5 min, then 98% MeCN for 7 min with 0.75 mL/min flow rate.
As a screening complement to the 8,000-member library (400 mixtures of 20 compounds), both the 400 protected (eg., O2N-[Ala]-Phe-OtBu) and 400 deprotected (eg., O2N-[Ala]-Phe-OH) dimers have been added to the screening library. Not only does this add an additional 800 single compounds to the evolving library, but it provides the opportunity to assess the screening leads (mixtures vs partial structure) upon completion of the initial screen.
Screening the Library Against MDM2/p53
The entire library composed of 400 mixtures of 20 compounds (400 wells) was screened for inhibition of MDM2/p53. Consistent with expectations and representative of the type of immediate informative results that will be available through screening the library, the most effective central subunit to emerge was [Trp] (Figure 9A). Thus, an examination of the data from the 400-well screening revealed that it was the central subunit versus C-terminus residue that dominated the MDM2/p53 inhibitory activity, so much so that even its representation as the summed average over the 20 mixtures constituting each of the entries in Figure 9A depicts the clear [Trp] preference. That is, each central residue representation in Figure 9A constitutes the average % inhibition of 20 mixtures (e.g., Trp is the summed average % inhibition of the 20 mixtures in Figure 9B). Although this representation of the data dampens the magnitude of the effect of uniquely active compounds or even that of a single mixture, it serves to illustrate the dominant effect of [Trp] and may illustrate that such treatments can statistically compensate for measurement errors made with single concentration assays conducted in duplicate. Further examination of the 20 defined C-terminus residues for [Trp] revealed that Leu is most preferred with significant activity observed for the closely related aliphatic residues Abu and Val as well as the bulky aromatic residues Nap and Phe(4Cl) (Figure 9B). Deconvolution by resynthesis of the individual 20 members of the [Trp]-Leu mixture (i.e., H2N-XXX- [Trp]-Leu-OH) and their individual assessment revealed that Phe is preferred at the N-terminus over the other 19 residues, followed by the closely related aromatic residues Phe(4Cl) and Nap (Figure 9C). Thus, the library screening and subsequent deconvolution led to the discovery of the lead inhibitor H2N-Phe-[Trp]-Leu-OH used to define the α-helix mimetic screening library template (see Figure 4). In contrast, the preparation and testing of the individual 20 compounds that make up the H2N-XXX-[Trp]-Phe(4Cl)-OH mixture that was comparable in the initial screening (Figure 9B) did not provide any effective inhibitors MDM2/p53 binding (IC50 > 50 μM). Although we did not conduct the related additional deconvolutions, the expectation would be that the H2N-XXX-[Trp]-Abu-OH and H2N-XXX-[Trp]-Val-OH may behave productively and analogous to H2N-XXX-[Trp]-Leu-OH, whereas H2N-XXX-[Trp]-Nap-OH would behave nonproductively as did H2N-XXX-[Trp]-Phe(4Cl)-OH. What is remarkable and most important in these examinations is that the library screening produced the expected lead structure from which information on the protein-protein interaction target may be confidently extrapolated and not an unexpected, more potent inhibitor of the MDM2/p53 interaction.
Figure 9.

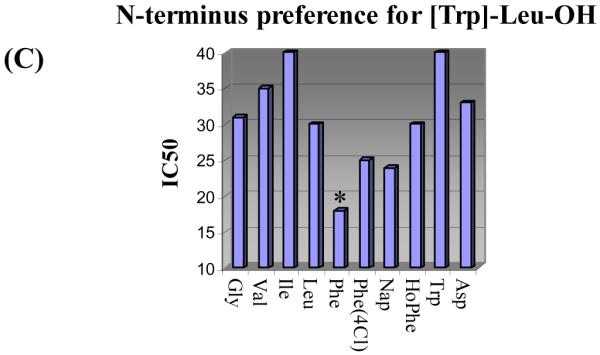
(A) Central subunit preferences, average % inhibition of MDM2/p53 binding for the 20 mixtures containing the defined central subunit (12.5 μM/compound; 250 μM mixture). (B) C-terminus preferences for [Trp], % inhibition of MDM2/p53 binding for the 20 compound mixtures H2N-mix-[Trp]-XXX-OH (12.5 μM/compound; 250 μM mixture). (C) N-terminus preference for [Trp]-Leu-OH, IC50 values for the inhibition of MDM2/p53 binding by individual compounds H2N-XXX-[Trp]-Leu-OH with IC50 < 40 μM. Assay conditions employed were those published by Searcey et al.25
Conclusions
The solution-phase synthesis of an α-helix mimetic library as the first component of a general library targeting protein-protein interactions is described. The initial designs were refined by screening against the MDM2/p53 protein-protein interaction resulting in the selection of a template that features a non-natural aryl monomer as the central unit and a natural amino acid at both ends. The library (8,000 compounds) consists of 400 mixtures of 20 compounds, where each added subunit is designed to mimic all possible permutations of the naturally occurring i, i+4, or i+7 amino acid side chains of an α-helix. Even if the recognition motif is unknown or unrecognized, the library screening should be capable of providing lead structures, provide insights into the nature of the interaction (α-helix), and identify the key amino acid residues responsible for the protein-protein interaction. Consistent with these expectations, the screening of the entire library for inhibition of MDM2/p53 binding provided immediate insights into the nature of the interaction, defining the key residues of a known α-helix mediated protein-protein interaction responsible for the recognition (H2N-XXX-[Trp]-Leu-OH). Testing the individual components of the identified active mixture revealed the key inhibitor (H2N-Phe-[Trp]-Leu-OH) used to design the library, providing an attractive lead structure available for further optimization. Notably, the library design is not meant to represent an optimal α-helix mimetic, but rather to represent a chemically tractable design that is a sufficiently good mimetic to be informative in screening against protein-protein interaction targets. Additional reports of such uses will be forthcoming.26
Supplementary Material
Figure 10.
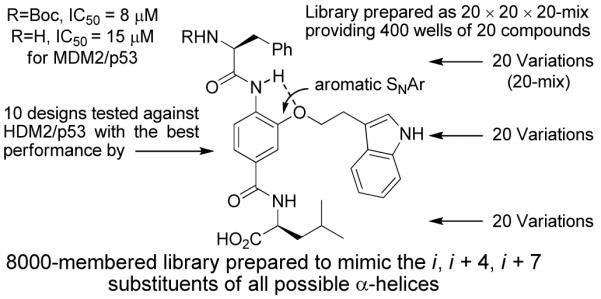
Summary figure of α-helix mimetic library design.
Acknowledgments
We gratefully acknowledge the financial support of the National Institutes of Health (CA78045) and the Skaggs Institute for Chemical Biology and wish to thank Wooyoung Hur for the synthesis and characterization of the reversed (H2N-[Leu]-[Trp]-[Phe]-OH) series of candidate MDM2/p53 inhibitors. LRW is a Skaggs Fellow.
Footnotes
Supporting Information Available: General procedures for the library synthesis and full experimental details for 7-25, 27-46, 51, and all compounds presented in Figure 4 are provided. Full author listing is provided for references 21c and 21f. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- (1).Stites WE. Chem. Rev. 1997;97:1233–1250. doi: 10.1021/cr960387h. [DOI] [PubMed] [Google Scholar]
- (2).Small molecule modulators of protein-protein interactions: Yin H, Hamilton AD. Angew. Chem. Int. Ed. 2005;44:4130–4163. doi: 10.1002/anie.200461786.. Arkin MR, Wells JA. Nature Rev. Drug Discovery. 2004;3:301–317. doi: 10.1038/nrd1343..Boger DL, Desharnais J, Capps K. Angew. Chem. Int. Ed. 2003;42:4138–4176. doi: 10.1002/anie.200300574.. Berg T. Angew. Chem. Int. Ed. 2003;42:2462–2481. doi: 10.1002/anie.200200558..Toogood PL. J. Med. Chem. 2002;45:1543–1558. doi: 10.1021/jm010468s.. Sharma SK, Ramsey TM, Bair KW. Curr. Med. Chem. Anti-Caner Agents. 2002;2:311–330. doi: 10.2174/1568011023354191.
- (3).Solution-phase preparation of libraries:Boger DL, Lee JK, Goldberg J, Jin Q. J. Org. Chem. 2000;65:1467–1474. doi: 10.1021/jo9916481..Boger DL, Goldberg J, Satoh S, Ambroise Y, Cohen SB, Vogt PK. Helv. Chim. Acta. 2000;83:1825–1845..Boger DL, Chai W, Jin Q. J. Am. Chem. Soc. 1998;120:7220–7225.. Boger DL, Chai W. Tetrahedron. 1998;54:3955–3970.. Boger DL, Ducray P, Chai W, Jiang W, Goldberg J. Bioorg. Med. Chem. Lett. 1998;8:2339–2344. doi: 10.1016/s0960-894x(98)00423-5.. Boger DL, Goldberg J, Jiang W, Chai W, Ducray P, Lee JK, Ozer RS, Andersson C-M. Bioorg. Med. Chem. 1998;6:1347–1378. doi: 10.1016/s0968-0896(98)00128-x.. Boger DL, Chai W, Ozer RS, Andersson C-M. Bioorg. Med. Chem. Lett. 1997;7:463–468.. Boger DL, Ozer RS, Andersson C-M. Bioorg. Med. Chem. Lett. 1997;7:1903–1908.. Cheng S, Comer DD, Williams JP, Boger DL. J. Am. Chem. Soc. 1996;118:2567–2573.. Cheng S, Tarby CM, Comer DD, Williams JP, Caporale LH, Boger DL. Bioorg. Med. Chem. 1996;4:727–737. doi: 10.1016/0968-0896(96)00069-7.. Boger DL, Tarby CM, Caporale LH. J. Am. Chem. Soc. 1996;118:2109–2110.. Boger DL, Fink BE, Hedrick MP. J. Am. Chem. Soc. 2000;122:6382–6394.. Boger DL, Schmitt H, Fink BE, Hedrick MP. J. Org. Chem. 2001;66:6654–6661. doi: 10.1021/jo010454u.. Chen Y, Bilban M, Foster CA, Boger DL. J. Am. Chem. Soc. 2002;124:5431–5440. doi: 10.1021/ja020166v.
- (4).(a) Boger DL, Goldberg J. Bioorg. Med. Chem. 2001;9:557–562. doi: 10.1016/s0968-0896(00)00276-5. [DOI] [PubMed] [Google Scholar]; (b) Boger DL. Bioorg. Med. Chem. 2003;11:1607–1613. doi: 10.1016/s0968-0896(03)00031-2. [DOI] [PubMed] [Google Scholar]
- (5).(a) Silletti S, Kessler J, Goldberg J, Boger DL, Cheresh DA. Proc. Natl. Acad. Sci.U.S.A. 2001;98:119–124. doi: 10.1073/pnas.011343298. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Boger DL, Goldberg J, Silletti S, Kessler T, Cheresh D. A. J. Am. Chem. Soc. 2001;123:1280–1288. doi: 10.1021/ja003579+. [DOI] [PubMed] [Google Scholar]
- (6).Goldberg J, Jin Q, Ambroise Satoh, S., Y., Desharnais J, Capps K, Boger DL. J. Am. Chem. Soc. 2002;124:544–555. doi: 10.1021/ja0118789. [DOI] [PubMed] [Google Scholar]
- (7).(a) Berg T, Cohen SB, Desharnais J, Sonderegger C, Maslyar DJ, Goldberg J, Boger DL, Vogt PK. Proc. Natl. Acad. Sci.U.S.A. 2002;99:3830–3835. doi: 10.1073/pnas.062036999. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Lu X, Vogt PK, Boger DL, Lunec J. Oncol. Rep. 2008;19:825–830. [PubMed] [Google Scholar]
- (8).Ambroise Y, Yuspan B, Ginsberg MH, Boger DL. Chem. Biol. 2002;9:1219–1226. doi: 10.1016/s1074-5521(02)00246-6. [DOI] [PubMed] [Google Scholar]
- (9).Capps KJ, Humiston J, Dominique R, Hwang I, Boger DL. Bioorg. Med. Chem. Lett. 2005;15:2840–2844. doi: 10.1016/j.bmcl.2005.03.094. [DOI] [PubMed] [Google Scholar]
- (10).Eubanks LM, Hixon MS, Jin W, Hong S, Clancy CM, Tepp WH, Malizio CJ, Goodnough MC, Barbieri JT, Johnson EA, Boger DL, Dickerson TJ, Janda KD. Proc. Natl. Acad. Sci. USA. 2007;104:2602–2607. doi: 10.1073/pnas.0611213104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Lee AM, Rojek JM, Spiropoulou CF, Gunderson A, Jin W, Shaginian A, York J, Nunberg JH, Boger DL, Oldstone MBA, Kunz S. J. Biol. Chem. 2008;283:18734–18742. doi: 10.1074/jbc.M802089200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Additional recent examples: Xu Y, Shi J, Yamamoto N, Moss JA, Vogt PK, Janda KD. Bioorg. Med. Chem. 2006;14:2660–2673. doi: 10.1016/j.bmc.2005.11.052.. Biros SM, Moisan L, Mann E, Carella A, Zhai D, Reed JC, Rebek J., Jr. Bioorg. Med. Chem. Lett. 2007;17:4641–4645. doi: 10.1016/j.bmcl.2007.05.075.. Volonterio A, Moisan L, Rebek J., Jr. Org. Lett. 2007;9:3733–3736. doi: 10.1021/ol701487g.. Moisan L, Odermatt S, Gombosuren N, Carella A, Rebek J., Jr. Eur. J. Org. Chem. 2008:1673–1676.. Restorp P, Rebek J., Jr. Bioorg. Med. Chem. Lett. 2008;18:5909–5911. doi: 10.1016/j.bmcl.2008.06.074.. Maity P, Konig B. Org. Lett. 2008;10:1473–1476. doi: 10.1021/ol8002749.
- (13).α-helix mimetic library: Lu F, Chi S-W, Kim D-H, Han K-H, Kuntz ID, Guy RK. J. Comb. Chem. 2006;8:315–325. doi: 10.1021/cc050142v.. Antuch W, Menon S, Chen Q-Z, Lu Y, Sakamuri S, Beck B, Schauer-Vukašinović V, Agarwal S, Hess S, Dömling A. Bioorg. Med. Chem. Lett. 2006;16:1740–1743. doi: 10.1016/j.bmcl.2005.11.102.. β-turn mimetic library: Souers AJ, Ellman JA. Tetrahedron. 2001;57:7431–7448.. β-strand or β-sheet mimetic library: Fuchi N, Doi T, Cao B, Kahn M, Takahashi T. Synlett. 2002:285–289.. Loughlin WA, Tyndall JDA, Glenn MP, Fairlie DP. Chem. Rev. 2004;104:6085–6117. doi: 10.1021/cr040648k..
- (14).Terphenyls and terpyridines Yin H, Lee G-I, Park HS, Payne GA, Rodriguez JM, Sebti SM, Hamilton AD. Angew. Chem. Int. Ed. 2005;44:2704–2707. doi: 10.1002/anie.200462316.. Chen L, Yin H, Farooqi B, Sebti S, Hamilton AD, Chen J. Mol. Cancer Ther. 2005;4:1019–1025. doi: 10.1158/1535-7163.MCT-04-0342.. Yin H, Lee G-I, Sedey KA, Kutzki O, Park HS, Orner BP, Ernst JT, Wang H-G, Sebti SM, Hamilton AD. J. Am. Chem. Soc. 2005;127:10191–10196. doi: 10.1021/ja050122x.. Davis JM, Truong A, Hamilton AD. Org. Lett. 2005;7:5405–5408. doi: 10.1021/ol0521228.. Ernst JT, Kutzki O, Debnath AK, Jiang S, Lu H, Hamilton AD. Angew. Chem. Int. Ed. 2002;41:278–281. doi: 10.1002/1521-3773(20020118)41:2<278::aid-anie278>3.0.co;2-a.. Kutzki O, Park HS, Ernst JT, Orner BP, Yin H, Hamilton AD. J. Am. Chem. Soc. 2002;124:11838–11839. doi: 10.1021/ja026861k.. Orner BP, Ernst JT, Hamilton AD. J. Am. Chem. Soc. 2001;123:5382–5383. doi: 10.1021/ja0025548.
- (15).Oligoamide, Ernst JT, Becerril J, Park HS, Yin H, Hamilton AD. Angew. Chem. Int. Ed. 2003;42:535–539. doi: 10.1002/anie.200390154. [DOI] [PubMed] [Google Scholar]
- (16).Terephthalamide: Yin H, Sedey KA, Rodriguez JM, Wang H-G, Sebti SM, Hamilton AD. J. Am. Chem. Soc. 2005;127:5463–5468. doi: 10.1021/ja0446404.. Yin H, Hamilton AD. Bioorg. Med. Chem. Lett. 2004;14:1375–1379. doi: 10.1016/j.bmcl.2003.09.096.
- (17).Fairlie DP, West ML, Wong AK. Curr. Med. Chem. 1998;5:29–62. [PubMed] [Google Scholar]
- (18).(a) Buolamwini JK, Addo J, Kamath S, Patil S, Mason D, Ores M. Curr. Cancer Drug Targets. 2005;5:57–68. doi: 10.2174/1568009053332672. [DOI] [PubMed] [Google Scholar]; (b) Chène P. Mol. Cancer, Res. 2004;2:20–28. [PubMed] [Google Scholar]; (c) Shir MD. Chem. Biol. 1997;4:791–794. doi: 10.1016/s1074-5521(97)90112-5. [DOI] [PubMed] [Google Scholar]
- (19).Kussie PH, Gorina S, Marechal V, Elenbaas B, Moreau J, Levine AJ, Pavletich NP. Science. 1996;274:948–953. doi: 10.1126/science.274.5289.948. [DOI] [PubMed] [Google Scholar]
- (20).(a) Vassilev LT. J. Med. Chem. 2005;48:4491–4499. doi: 10.1021/jm058174k. [DOI] [PubMed] [Google Scholar]; (b) Vassilev LT, Vu BT, Graves B, Carvajal D, Podlaski F, Filipovic Z, Kong N, Kammlott U, Lukacs C, Klein C, Fotouhi N, Liu EA. Science. 2004;303:844–848. doi: 10.1126/science.1092472. [DOI] [PubMed] [Google Scholar]; (c) Tovar C, Rosinski J, Filipovic Z, Higgins B, Kolinsky K, Hilton H, Zhao X, Vu BT, Qing W, Packman K, Myklebost O, Heimbrook DC, Vassilev LT. Proc. Natl. Acad. Sci. USA. 2006;103:1888–1893. doi: 10.1073/pnas.0507493103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).(a) Leonard K, Marugan JJ, Raboisson P, Calvo R, Gushue JM, Koblish HK, Lattanze J, Zhao S, Cummings MD, Player MR. Bioorg. Med. Chem. Lett. 2006;16:3463–3468. doi: 10.1016/j.bmcl.2006.04.009. [DOI] [PubMed] [Google Scholar]; (b) Parks DJ, LaFrance LV, Calvo RR, Milkiewicz KL, Marugán JJ, Raboisson P, Schubert C, Koblish HK, Zhao S, Franks CF. Bioorg. Med. Chem. Lett. 2006;16:3310–3314. doi: 10.1016/j.bmcl.2006.03.055. [DOI] [PubMed] [Google Scholar]; (c) Koblish HK, Zhao S, Franks CF, Donatelli RR, Tominovich RM, LaFrance LV, Leonard KA, Gushue JM, Parks DJ, Calvo RR, et al. Mol. Cancer Ther. 2006;5:160–169. doi: 10.1158/1535-7163.MCT-05-0199. [DOI] [PubMed] [Google Scholar]; (d) Raboisson P, Marugán JJ, Schubert C, Koblish HK, Lu T, Zhao S, Player MR, Maroney AC, Reed RL, Huebert ND. Bioorg. Med. Chem. Lett. 2005;15:1857–1861. doi: 10.1016/j.bmcl.2005.02.018. [DOI] [PubMed] [Google Scholar]; (e) Parks DJ, LaFrance LV, Calvo RR, Milkiewicz KL, Gupta V, Lattanze J, Ramachandren K, Carver TE, Petrella EC, Cummings MD. Bioorg. Med. Chem. Lett. 2005;15:765–770. doi: 10.1016/j.bmcl.2004.11.009. [DOI] [PubMed] [Google Scholar]; (f) Grasberger BL, Lu T, Schubert C, Parks DJ, Carver TE, Koblish HK, Cummings MD, LaFrance LV, Milkiewicz KL, Calvo RR, et al. J. Med. Chem. 2005;48:909–912. doi: 10.1021/jm049137g. [DOI] [PubMed] [Google Scholar]
- (22).(a) Tsukamoto S, Yoshida T, Hosono H, Ohta T, Yokosawa H. Bioorg. Med. Chem. Lett. 2006;16:69–71. doi: 10.1016/j.bmcl.2005.09.052. [DOI] [PubMed] [Google Scholar]; (b) Ding K, Lu Y, Nikolovska-Coleska Z, Wang G, Qui S, Shangary S, Gao W, Qin D, Stuckey J, Krajewski K, Roller PP, Wang S. J. Med. Chem. 2006;49:3432–2435. doi: 10.1021/jm051122a. [DOI] [PubMed] [Google Scholar]; (c) Ding K, Lu Y, Nikolovska-Coleska Z, Qiu S, Ding Y, Gao W, Stuckey J, Krajewski K, Roller PP, Tomita Y, Parrish DA, Deschamps JR, Wang S. J. Am. Chem. Soc. 2005;127:10130–10131. doi: 10.1021/ja051147z. [DOI] [PubMed] [Google Scholar]; (d) Galatin PS, Abraham DJ. J. Med. Chem. 2004;47:4163–4165. doi: 10.1021/jm034182u. [DOI] [PubMed] [Google Scholar]; (d) Kumar SK, Hager E, Pettit C, Gurulingappa H, Davidson NE, Khan SR. J. Med. Chem. 2003;46:2813–2815. doi: 10.1021/jm030213+. [DOI] [PubMed] [Google Scholar]; (e) Bowman AL, Nikolovska-Coleska Z, Zhong H, Wang S, Carlson HA. J. Am. Chem. Soc. 2007;129:12809–12814. doi: 10.1021/ja073687x. [DOI] [PubMed] [Google Scholar]
- (23).Peptide-based MDM2 inhibitors: Hara T, Durell SR, Myers MC, Appella DH. J. Am. Chem. Soc. 2006;128:1995–2004. doi: 10.1021/ja056344c.. Kritzer JA, Leudtke NW, Harker EA, Schepartz A. J. Am. Chem. Soc. 2005;127:14584–14585. doi: 10.1021/ja055050o.. Murray JK, Farooqi B, Sadowsky JD, Scalf M, Freund WA, Smith LA, Chen J, Gellman SH. J. Am. Chem. Soc. 2005;127:13271–13280. doi: 10.1021/ja052733v.. Sadowsky JD, Schmitt MA, Lee H-S, Umezawa N, Wang S, Tomita Y, Gellman SH. J. Am. Chem. Soc. 2005;127:11966–11968. doi: 10.1021/ja053678t.. Fasan R, Dias RLA, Moehle K, Zerbe O, Vrijbloed JW, Obrecht D, Robinson JA. Angew. Chem., Int. Ed. 2004;43:2109–2112. doi: 10.1002/anie.200353242.. Fasan R, Dias RLA, Moehle K, Zerbe O, Obrecht D, Mittl PRE, Grutter MG, Robinson JA. ChemBioChem. 2006;7:515–526. doi: 10.1002/cbic.200500452.. Kritzer JA, Lear JD, Hodsdon ME, Schepartz A. J. Am. Chem. Soc. 2004;126:9468–9469. doi: 10.1021/ja031625a.. Sakurai K, Chung HS, Kahne D. J. Am. Chem. Soc. 2004;126:16288–16289. doi: 10.1021/ja044883w.. Duncan SJ, Gruschow S, Williams DH, McNicholas C, Purewal R, Hajek M, Gerlitz M, Martin S, Wrigley SK, Moore M. J. Am. Chem. Soc. 2001;123:554–560. doi: 10.1021/ja002940p.. García-Echeverría C, Chène P, Blommers MJJ, Furet P. J. Med. Chem. 2000;43:3205–3208. doi: 10.1021/jm990966p.
- (24).A figure displaying a complete table of all coupling yields and product amounts (400 compounds) is provided in Supporting Information
- (25).Woon ECY, Arcieri M, Wilderspin AF, Malkinson JP, Searcey M. J. Org. Chem. 2007;72:5146–5151. doi: 10.1021/jo070450a. For details on the MDM2/p53 binding assay conditions see supporting information. [DOI] [PubMed] [Google Scholar]
- (26).The library is stored (-20 °C) as a sealed 50 mM solution in DMSO (master plate) that is dispensed and routinely stored (-20 °C) at a 10-fold dilution as a 5 mM solution in DMSO (typically 10 μL total volume, daughter plates). The further 100-fold dilution of the daughter plate solution into an assay (e.g.; 1 μL into a 100 μL assay volume, now 1% DMSO) provides a 50 μM solution of the tested compounds. The daughter plates (typically 3-6 prepared at a time) are accessed routinely as needed up to 5-10 times before being depleted. In this manner and for our uses, the master plate is accessed infrequently and even the daughter plates experience limited freeze/thaw access that can erode their quality (H2O absorption). At our present usage rate of the α-helix mimetic library, we anticipate only accessing the master plate approximately every 2 years.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.