

Abstract
The di-heme enzyme MauG catalyzes the biosynthesis of tryptophan tryptophylquinone (TTQ), the protein-derived cofactor of methylamine dehydrogenase (MADH). This process requires the six-electron oxidation of a 119 kDa MADH precursor protein with incompletely synthesized TTQ (PreMADH). The kinetic mechanism of the initial two-electron oxidation of this natural substrate by MauG was characterized. The relative reactivity of free MauG towards H2O2 and the O2 analog CO was essentially the same as that of MauG in the pre-formed enzyme-substrate complex. The addition of H2O2 to di-ferric MauG generated a di-heme bis-Fe(IV) species (i.e., Fe(IV)=O/Fe(IV)) which formed at a rate >300 s-1 and spontaneously returned to the di-ferric state at a rate of 2×10-4 s-1 in the absence of substrate. The reaction of bis-Fe(IV) MauG with PreMADH exhibited saturation behavior with a limiting first-order rate constant of 0.8 s-1 and a Kd ≤ 1.5 μM for the MauG-PreMADH complex. The results were the same whether bis-Fe(IV) MauG was mixed with PreMADH, or H2O2 was added to the pre-formed enzyme-substrate complex to generate bis-Fe(IV) MauG followed by reaction with PreMADH. Stopped-flow kinetic studies of the reaction of di-ferrous MauG with CO yielded a faster major transition with a bimolecular rate constant of 5.4 × 105 M-1s-1, and slower transition with a rate 16 s-1 which was independent of [CO]. The same rates were obtained for binding of CO to di-ferrous MauG in complex with PreMADH. This demonstration of a random kinetic mechanism for the first two-electron oxidation reaction of MauG-dependent TTQ biosynthesis, in which the order of addition of oxidizing equivalent and substrate does not matter, is atypical of those of heme-dependent oxygenases that are not generally reactive towards oxygen in the absence of substrate. This kinetic mechanism is also distinct from that of the homologous di-heme cytochrome c peroxidases that require a mixed valence state for activity.
Tryptophan tryptophylquinone (TTQ)1 (1) is the protein-derived cofactor (2) of methylamine dehydrogenase (MADH), a 119 kDa heterotetrameric α2β2 protein with a TTQ present on each β subunit (3, 4). TTQ biosynthesis requires incorporation of two oxygens into βTrp57 and cross-linking of the indole rings of βTrp57 and βTrp108. This is not a self-processing event but requires the action of at least one processing enzyme. Deletion of mauG, a gene in the methylamine utilization (mau) gene cluster (5), allowed isolation of a biosynthetic precursor of MADH (PreMADH) in which βTrp57 is mono-hydroxylated at C7 and the cross-link is absent (6, 7). MauG is a 42.3 kDa enzyme containing two c-type hemes (8). It exhibits homology to di-heme cytochrome c peroxidase (CCP) (9, 10) but exhibits significant differences in catalytic and redox behavior (11, 12). MauG catalyzes the six-electron oxidation of PreMADH to yield oxidized MADH with the mature protein-derived TTQ cofactor (Scheme 1). MauG-dependent TTQ-biosynthesis from PreMADH was achieved in vitro using either O2 plus electrons from an external donor, or H2O2, as oxidizing equivalents ([O] in Scheme 1) (12, 13).
Scheme 1.
We previously developed a steady-state in vitro assay for MauG-dependent TTQ biosynthesis in which TTQ formation was monitored by the appearance of the absorption spectrum characteristic of TTQ in oxidized MADH (14). This allowed determination of values of kcat and Km for the multistep biosynthetic reaction. Previous studies have shown that di-ferric MauG tightly binds PreMADH, as evidenced by co-elution of the MauG-PreMADH complex during size-exclusion chromatography (14). It was also shown that addition of PreMADH to a high-valent bis-Fe(IV) form of MauG (i.e., Fe(IV)=O/Fe(IV)), which was generated by addition of H2O2, caused return of MauG to the di-ferric state with concomitant production of a PreMADH-based radical (15). Analysis of this high-valent MauG species by EPR and Mössbauer spectroscopy revealed the presence of two distinct Fe(IV) species (15). One heme exhibits spectral properties consistent with an Fe(IV)=O (ferryl). The other is assigned to an Fe(IV) heme with two axial ligands from the protein. Thus, in this di-heme bis-Fe(IV) form of MauG, the second oxidizing equivalent is stored not as a porphyrin cation radical (i.e., Compound I), but as a second Fe(IV) heme.
Given the complexity of the overall reaction of MauG-dependent TTQ biosynthesis, it is impossible to determine the reaction mechanism solely from steady-state kinetic studies. As the first step towards kinetically isolating and describing the individual reaction steps in the overall biosynthetic reaction, this paper describes the kinetic mechanism for the first two-electron oxidation of PreMADH by MauG. In this study we characterize changes in the visible absorption spectrum of MauG which correlate with the formation and decay of the bis-Fe(IV) MauG state, and use stopped-flow spectroscopy to kinetically characterize the initial two-electron oxidation step in MauG-dependent TTQ biosynthesis. The reaction of di-ferrous MauG with the O2 analog CO was also examined. This has allowed us to determine rate and binding constants for the initial reaction steps in MauG-dependent TTQ biosynthesis and demonstrate that the order of addition of the oxidizing equivalent and substrate to MauG is not critical as it is for other heme-dependent monooxygenases (16, 17). The results indicate that the kinetic mechanism of this MauG-catalyzed reaction is also distinct from that of the di-heme CCP (10).
Experimental Procedures
The methods for homologous expression of MauG in Paracoccus denitrificans and its purification were as described previously (8). The biosynthetic precursor of MADH with incompletely synthesized TTQ, which contains monohydroxylated βTrp57 and no crosslink to βTrp108 (6), was heterologously expressed in Rhodobacter sphaeroides and purified as described previously (18).
Transient kinetic experiments were performed using an On-Line Instrument Systems (OLIS, Bogart GA) RSM stopped-flow spectrophotometer. Kinetic data collected in the rapid-scanning mode were reduced by factor analysis using the singular value decomposition algorithm and then globally fit using the fitting routines of the OLIS Global Fit software. All reactions were performed in 0.01 M potassium phosphate buffer, pH 7.5, at 25 °C.
The reactions of MauG with H2O2 and PreMADH were performed in two configurations. (i) One syringe contained bis-Fe(IV) MauG that was generated by stoichiometric addition of H2O2, and the other contained PreMADH. (ii) One syringe contained di-ferric MauG plus PreMADH, and the other contained H2O2. In each case the concentrations of MauG and H2O2 were fixed and the concentration of PreMADH was varied. Reactions were monitored between 360 and 440 nm. In all cases the observed rates were best fit to a single exponential. The limiting first-order rate constant for the reaction of MauG with PreMADH (k3) was determined from the concentration dependence of the observed rate using Eqs 1 and 2, where [S] is the concentration of PreMADH, E is bis-Fe(IV) MauG and E’ is di-ferric MauG.
| (1) |
| (2) |
For reaction with CO, di-ferrous MauG was prepared in deoxygenated 0.01 M potassium phosphate, pH 7.5, and stoichiometrically reduced by addition of sodium dithionite. Stock solutions of CO were prepared by bubbling O2-free N2 for 4 hr through the 0.01 M potassium phosphate, pH 7.5, and then flushing with CO for 45 min. The concentration of this saturated stock solution was taken to be 930 μM CO at 25 °C at atmospheric pressure (19, 20). Solutions of different concentrations of CO were prepared by diluting this stock solution with oxygen-free buffer. In the stopped-flow experiments one syringe contained di-ferrous MauG, plus or minus PreMADH, and the other syringe contained buffer with varying concentrations of CO. The final concentration of MauG after mixing was 2 μM. Reactions were monitored between 374 and 454 nm. In each reaction data were best fit a two-phase exponential transition.
Results
Conversion of di-ferric MauG to bis-Fe(IV) MauG by H2O2
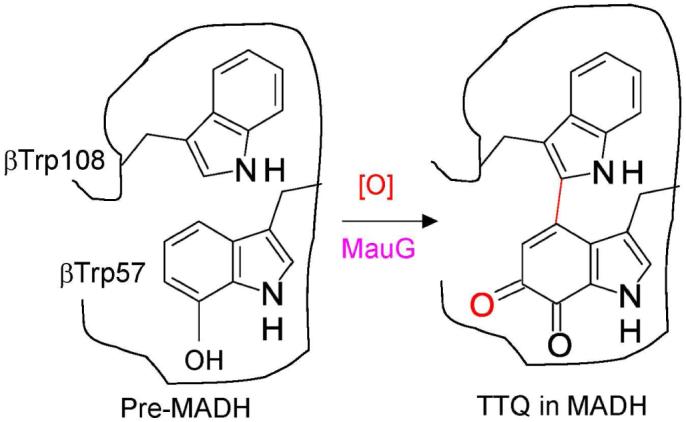
It was previously shown that addition of H2O2 to di-ferric MauG generated a di-heme bis-Fe(IV) species which is relatively stable in the absence of the PreMADH substrate, and which spontaneously decays back to the di-ferric state over a period of minutes (15). The time course of the reaction was previously monitored by stopping the reaction at various times by freezing and analyzing the samples by EPR spectroscopy. In order to facilitate kinetic analysis of the formation and reaction of bis-Fe(IV) MauG, we have characterized changes in the visible absorption spectrum of MauG which correlate with the formation of the bis-Fe(IV) MauG and decay back to the di-ferric state (Figure 1A). When MauG is mixed with H2O2 one observes a decrease in intensity of the Soret peak and a shift in its maximum from 405 to 407 nm. When monitored by stopped-flow spectroscopy (not shown) the rate of formation of bis-Fe(IV) species after addition of excess H2O2 occurs within the dead-time for mixing (∼ 2-3 ms). As such, that rate is estimated conservatively to be >300 s-1. In the absence of substrate, the absorption spectrum slowly returns to one similar to that of di-ferric MauG. The time course of the spontaneous decay of the bis-Fe(IV) species as judged by the increase in absorbance at 405 nm is shown in Figure 1B. The rate of increase in A405 with time was fit to a single exponential and displayed a rate constant of 2×10-4 s-1. The time course of these changes in absorbance parallels the time course for changes in the X-band EPR spectrum which was reported previously (15).
Figure 1.
Changes in the absorption spectrum of MauG on addition of H2O2.
A. Spectra were recorded in 0.01 M potassium phosphate, pH 7.5, of di-ferric MauG (black line), MauG immediately after addition of two equivalents of H2O2 (red line), and 20 min after addition of the two equivalents of H2O2 (blue line). The portion of the spectra of most interest is magnified in the Inset. B. Time course of the increase in absorbance at 405 nm after addition of H2O2 in the absence of substrate (i.e., the transition from red spectrum to blue spectrum in A).
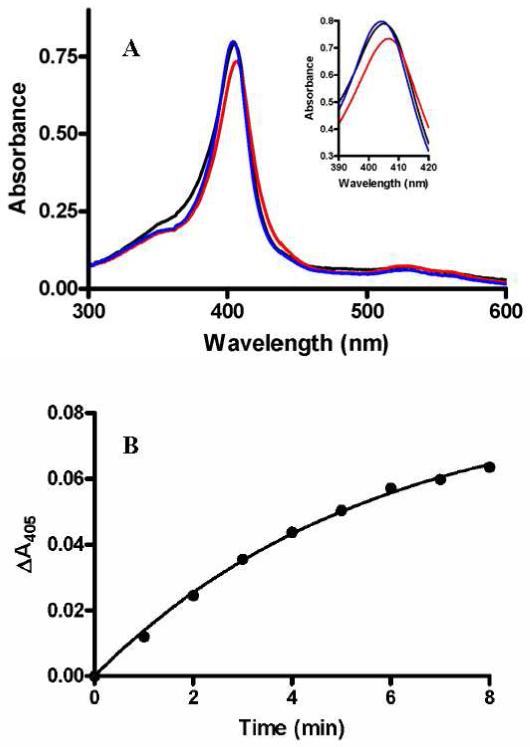
For many heme-dependent oxygenases, including cytochrome P450-dependent monooxygenases (16, 17), the enzyme is not reactive toward oxygen in the absence of substrate. The rapid reaction of MauG with H2O2 in the absence of substrate suggests that this is not true for MauG. To determine whether binding of PreMADH to MauG has any influence on its affinity for or reaction with H2O2, MauG was mixed with a sub-stoichiometric amount of H2O2 in the absence and presence of PreMADH. At these lower concentrations of H2O2 the rate of formation of the bis-Fe(IV) species is measurable by stopped-flow spectroscopy, and any changes in either binding affinity for H2O2 or the reaction rate constant will be reflected in the observed rate. When MauG alone is mixed with 0.25 equivalent of H2O2 the observed rate for the formation of the bis-Fe(IV) species as determined from a global fit of the total data at all wavelengths is 46±5 s-1. When the mixture of MauG and PreMADH is mixed with H2O2, the observed rate for the formation of the bis-Fe(IV) species is 47±5 s-1. The changes in A405 with time for each reaction are shown in Figure 2 for comparison. At the concentrations at which MauG and PreMADH were pre-incubated, it was previously shown that the two proteins co-elute as a complex during size-exclusion chromatography (14). On the basis of these results it may be concluded that the reactivity of MauG with H2O2 is the same for free MauG and MauG in the enzyme-substrate complex.
Figure 2.
Reaction of di-ferric MauG with sub-stoichiometric H2O2 in the absence and presence of its natural substrate.
A. MauG (2μM) was mixed with H2O2 (0.5 μM). B. MauG (2μM) plus PreMADH (2μM) was mixed with H2O2 (0.5 μM). The reactions were performed in 0.01 M potassium phosphate, pH7.5, at 25 °C. Each tick on the y-axis represents a ΔA405 of 0.01. The lines represent fits of the data to a single-phase exponential decay.
Reaction of bis-Fe(IV) MauG with its natural substrate
Because bis-Fe(IV) MauG is an unusually stable high-valent iron species it is possible to pre-form this high-valent state and then directly monitor its reaction with its natural substrate, PreMADH. The bis-Fe(IV) MauG intermediate was first generated by mixing with stoichiometric H2O2. Then it was mixed with PreMADH and the reaction was monitored by rapid scanning stopped-flow spectroscopy. The same spectral changes discussed above that are associated with the slow spontaneous conversion of the bis-Fe(IV) MauG species to the di-ferric state can be monitored during the faster reaction with substrate. That MauG has truly returned to the di-ferric state after the reaction was previously confirmed by EPR analysis of the product (15). These kinetic studies were performed with a fixed limiting concentration of bis-Fe(IV) MauG and varying concentrations of PreMADH, which were in sufficient excess of MauG to maintain pseudo-first-order reaction conditions. The dependence of the observed rate of reaction on substrate concentration is shown in Figure 3. Saturation behavior was observed with a limiting first-order rate constant for the reaction of 0.80±0.02 s-1. This reaction rate with the natural substrate is 4000-fold faster than the spontaneous rate of decay of 2×10-4 s-1 that was described earlier. The fit of the data to Eq 2 also yields an apparent Kd value of 1.3±0.2 μM for the MauG-PreMADH enzyme-substrate complex. Because of the relatively low Kd value it was not possible to fully define the lower portion of the curve in Figure 3 since use of lower concentrations of substrate would violate pseudo-first order conditions. This could affect the fitted value of Kd, however it is clear that the Kd is ≤ 1.5 μM.
Figure 3.
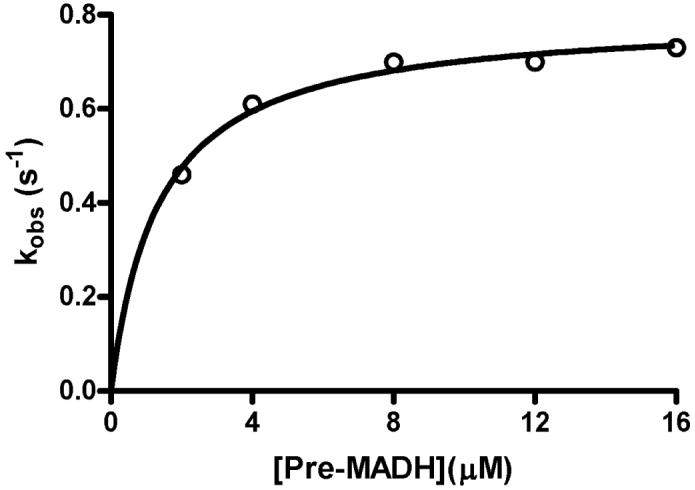
Concentration dependence of the rate of reaction of bis-Fe(IV) MauG with its natural substrate.
Prior to each reaction MauG (2μM) was mixed with a stoichiometric amount of H2O2 to generate the bis-Fe(IV) state. The reaction was then initiated by mixing this with PreMADH at varying concentrations. The line is a fit of the data to Eq. 2.
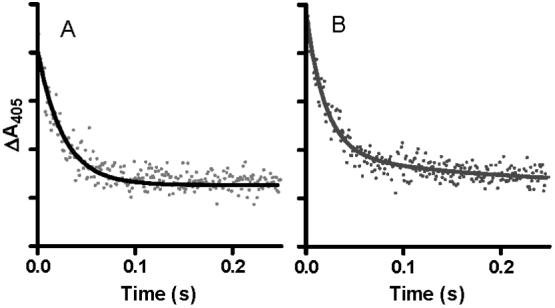
An important question is whether the MauG-dependent reaction is dependent on the order of addition of the substrates; in this case H2O2 and PreMADH. In the experiments described above, PreMADH was mixed with the pre-formed bis-Fe(IV) MauG. In parallel experiments H2O2 was mixed with the pre-formed enzyme-substrate complex of di-ferric MauG and PreMADH. Under saturating conditions the limiting first-order rate constant also was 0.8 s-1 (not shown). For a more definitive comparison of the effect of order of addition of H2O2 and PreMADH to MauG, the reactions were performed at a sub-saturating concentration of PreMADH. Under these conditions any changes in either binding affinity for the substrate or the reaction rate constant (Kd and k3 in Eqs 1 and 2) will be reflected in the observed rate. When PreMADH was mixed with the pre-formed bis-Fe(IV) MauG, the observed rate determined from a global fit of the data was 0.33±0.05 s-1. When H2O2 was mixed with the pre-formed MauG-PreMADH complex an initial rapid transition associated with the formation of the bis-Fe(IV) species was observed within the dead-time of the instrument (∼3 ms). This was followed by the slower transition associated with return to the di-ferric state which exhibited a rate of 0.42±0.03 s-1. The changes in A405 with time for each of these reactions are shown in Figure 4 for comparison. From these data it may be concluded that the rate of reaction of the bis-Fe(IV) MauG with substrate is essentially independent of whether or not MauG and PreMADH pre-exist as a complex.
Figure 4.
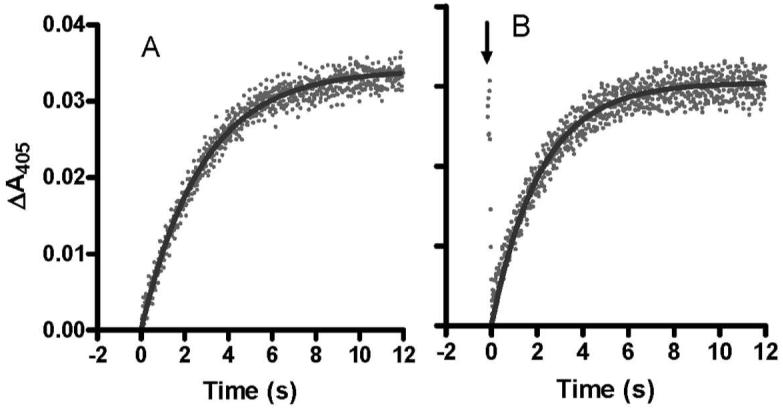
Reaction of bis-Fe(IV) MauG with the MADH biosynthetic precursor protein substrate.
A. Prior to the reaction MauG (2μM) was mixed with a stoichiometric amount of H2O2 to generate the bis-Fe(IV) state. The reaction was then initiated by mixing this with PreMADH (2μM). B. MauG (2μM) was pre-mixed with PreMADH (2μM) and the reaction was initiated by mixing with a stoichiometric amount of H2O2. The initial rapid decrease indicated by the arrow describes the rapid formation of the bis-Fe(IV) state which is followed by the slower substrate-dependent return to the initial state. The lines represent fits of the data to a single-phase exponential rise. The reactions were performed in 0.01 M potassium phosphate, pH 7.5, at 25 °C.
Reaction of di-ferrous MauG with CO
The reason for examining the reactivity of di-ferrous MauG towards CO in the absence and presence of PreMADH is that CO is a convenient and useful probe of the accessibility of the oxygen-binding heme to O2. This is of particular interest for MauG since its substrate is not a small molecule but a large protein. One might expect that binding the 119 kDa substrate could either induce a conformational change in MauG or block access to O2 or both. CO will bind to the ferrous heme as does O2 but will not allow reaction with the substrate. An easily discernable spectral change is also associated with binding of CO to ferrous heme. The Soret band shifts from 416 nm to 414 nm and exhibits an increase in extinction coefficient, and the α- and β-bands at 552 nm and 554 nm, respectively, broaden (8). As such it was possible to mix buffer containing varying concentrations of CO with di-ferrous MauG and monitor the kinetics of the reaction by stopped-flow spectroscopy.
The reaction of CO with di-ferrous MauG exhibited biphasic kinetics. The major component was the faster phase. The rate of this reaction exhibited a linear dependence on [CO] and a bimolecular rate constant of 5.4±0.8 × 105 M-1s-1. The minor slower phase was relatively independent of [CO] with a rate of 16±2.0 s-1 (Figure 5). The latter rate may represent some slow change in MauG conformation or redistribution of electrons subsequent to CO binding. The faster reaction which is dependent on [CO] is the reaction of interest as it represents rate of diffusion of CO into the heme site and binding to Fe(II). Essentially the same rates were obtained when the reaction was carried out in the presence of PreMADH. For comparison the change in A414 with time for a representative reaction of di-ferrous MauG with CO in the absence and presence of PreMADH is shown in Figure 6. These results indicate that the reactivity of MauG towards CO is not significantly stimulated or impeded by prior binding of PreMADH to MauG.
Figure 5.
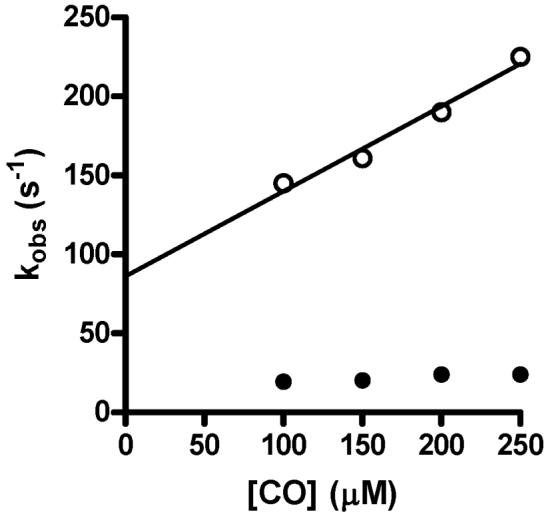
Concentration dependence of the rate of reaction of CO with di-ferrous MauG.
Di-ferrous MauG (final concentration 2 M) was mixed with buffer containing varying concentrations of CO to yield the final concentrations shown on this graph. Reactions were monitored between 374 and 454 nm and the data was globally fit. The solid line is a linear regression fit (R2=0.98) of the concentration dependency of the rates of the faster major phase (open circles). The rates of the slower minor phase (closed circles) were essentially independent of concentration. The reactions were performed in 0.01 M potassium phosphate, pH 7.5, at 25 °C.
Figure 6.
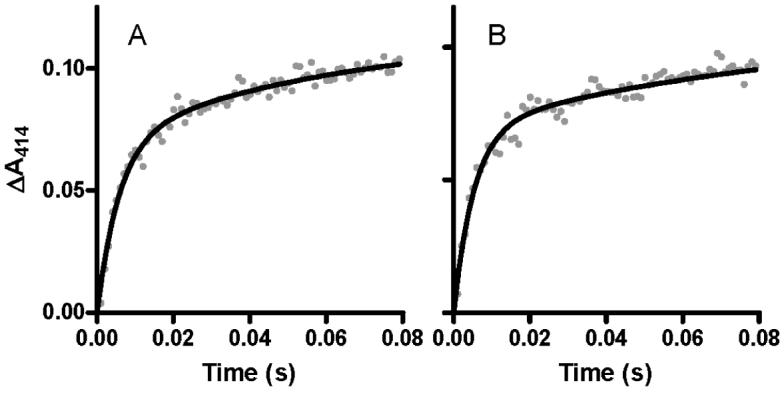
Reaction of CO with di-ferrous MauG in the absence and presence of its natural substrate.
Di-ferrous MauG was mixed with buffer containing CO in the absence (A) and presence (B) of PreMADH. Final concentrations were 2 μM MauG, 200 μM CO and 2 μM PreMADH when present. The reactions were performed in 0.01 M potassium phosphate, pH 7.5, at 25 °C. The lines represent fits of the data to a two-phase exponential transition.
Discussion
MauG catalyzes the six-electron oxidation of a 119-kDa protein precursor of MADH with mono-hydroxylated βTrp57 to yield oxidized MADH with the mature TTQ cofactor (Scheme 1). As such, three equivalents of H2O2 are required to complete MauG-dependent TTQ biosynthesis and MauG must complete three catalytic cycles, each of which presumably involves a different type of reaction. For each of these three diverse reactions that comprise the biosynthetic process it seems likely that the first step is the formation of a high-valent iron species, which we have shown to be a di-heme bis-Fe(IV) form of MauG (15).
The results presented in this study demonstrate that the order of addition of H2O2 and PreMADH to MauG does not matter for the first catalytic cycle (Scheme 2). When di-ferric MauG is first incubated with PreMADH and the reaction is initiated by addition of H2O2, the rates of formation of the bis-Fe(IV) species and subsequent reaction with PreMADH are essentially the same as when H2O2 is added first to pre-form the bis-Fe(IV) species (Figures 2 and 4). We had previously reported that di-ferrous MauG reacted with O2 in the absence of substrate (8). To better quantitate this phenomenon the reaction of di-ferrous MauG with the O2 analog CO was characterized. As observed with H2O2, the reactivity of MauG towards CO was neither stimulated nor impeded by prior complex formation with PreMADH. These results are in contrast to what is seen with cytochrome P450-dependent monooxygenases which are unreactive toward oxygen in the absence of substrate. In those enzymes, binding of substrate triggers a conformational change that allows the high-spin heme to bind and activate oxygen (16). There is also evidence that the presence or absence of substrate modulates the reactivity of cytochrome P450-dependent enzymes towards H2O2 (21) and CO (22, 23). This substrate-independence of the reactivity of MauG towards CO and H2O2 is also of interest since MauG must catalyze three sequential reactions to complete TTQ biosynthesis on PreMADH, which is not a small molecule but a 119 kDa protein substrate. One might intuitively expect the protein-protein interaction between the smaller enzyme and larger substrate to either induce a conformational change that would affect the reactivity of the active site to facilitate access to the smaller cosubstrate, O2 or H2O2, or to block access to the smaller cosubstrate because of its size. Neither is the case for MauG. The lack of an affect of substrate binding on the reaction with the oxidizing agent raises the possibility that the six-electron oxidation reaction is processive with the protein substrate remaining bound to MauG during the multiple reaction cycles of bis-Fe(IV) formation and reaction with substrate. These results indicate that multiple reactions of MauG with H2O2 while bound to pre-MADH and subsequent reaction intermediates is possible. That does not prove that the overall biosynthetic reaction is processive, but it does allow for that possibility.
Scheme 2.

MauG-dependent TTQ-biosynthesis from PreMADH was achieved in vitro using O2 plus electrons from an external donor as oxidizing equivalents ([O] in Scheme 1). The rate of reaction varied with the electron donor. Steady-state kinetic analysis of the MauG-dependent conversion of PreMADH to mature MADH with completely synthesized TTQ using the fastest electron donor yielded a value of kcat of 0.2 s-1 (14). The reaction using H2O2 as [O] is faster. Unfortunately it was not possible to obtain a kcat value for the steady-state reaction with H2O2 because the use of higher H2O2 concentrations caused damage to MauG and precluded determining the dependence of the rate of product formation on [H2O2] (14). The rate constant for the initial reaction of bis-Fe(IV) MauG with PreMADH is 0.8 s-1. This rate seems to be in the range of that of the rate-limiting step for the overall reaction. As the reaction of H2O2 with MauG is so much faster (>300 s-1), even within the MauG-PreMADH complex, it is likely that at least one of the catalytic reactions involving bis-Fe(IV) MauG, rather than formation of this high-valent species, is limiting the rate of TTQ biosynthesis.
MauG bears significant sequence homology to di-heme CCP (8), but exhibits significant differences in catalytic and redox behavior (11, 12). CCP is isolated in an inactive di-ferric state in which the H2O2-binding heme of CCP is six-coordinate with bis-axial ligation (10). Whereas the intrinsic oxidation-reduction potential values of the two hemes in MauG are similar (11), for the hemes of CCP they are separated by more than 600 mV (9). The reduction of the other six-coordinate heme in CCP generates a mixed-valence state that triggers a Ca2+-dependent conformational change in which the distal axial ligand of the H2O2-binding heme is replaced by water (10, 24). In contrast to CCP, and there is no evidence that MauG stabilizes a mixed-valence state or that a mixed-valence redox state plays any role in the reaction cycle of MauG. In fact the hemes of MauG exhibit redox cooperativity and are oxidized to Fe(III) and reduced to Fe(II) simultaneously (11). Interconversion of the two hemes between the Fe(III) and Fe(IV) states also occurs in concert (15).
MauG is the first Fe-dependent oxygenase to be shown to utilize c-type hemes to catalyze a monooxygenation reaction. Other heme-dependent oxygenases are not generally reactive toward oxygen in the absence of substrate. This may be beneficial in that it minimizes the chance of generation of reactive intermediates in the absence of substrate that could lead to deleterious autoxidation of the enzyme. In contrast, the reaction of di-ferric MauG with H2O2 in the absence of its natural substrate yields a bis-Fe(IV) intermediate that is stable for minutes, yet is catalytically competent. MauG is also mechanistically distinct from the di-heme CCPs which require a mixed valence state for reactivity. The kinetic results presented here indicate that MauG is atypical of heme-dependent oxygenases and peroxidases in that its activity is not strictly regulated by binding of substrate or redox state of the hemes. These distinctive properties of MauG may be necessitated by its unusual natural substrate, specific amino acid side-chains within a 119 kDa tetrameric precursor protein, and the fact that it catalyzes multiple reactions on this substrate. The unusual stability of the high-valent iron state and its relatively slow reaction with substrate also provide a means of preventing deleterious autooxidation which might otherwise occur during the steps of complex formation and possible rearrangements within the enzyme-substrate complex which are likely required to position the specific sites on the protein substrate for reaction. The fact that the bound protein substrate does not impede reaction with the oxidizing species also allows for the possibility of a processive reaction.
Acknowledgment
The authors thank Dr. Aimin Liu and Elizabeth Graichen for helpful advice and comments and Yu Tang for technical support.
Footnotes
- MADH
- methylamine dehydrogenase
- TTQ
- tryptophan tryptophylquinone
- PreMADH
- the biosynthetic precursor protein of MADH with incompletely synthesized TTQ
- CCP
- cytochrome c peroxidase
- bis-Fe(IV) MauG
- redox state of MauG with one heme as Fe(IV)=O and the other Fe(IV)
References
- 1.McIntire WS, Wemmer DE, Chistoserdov A, Lidstrom ME. A new cofactor in a prokaryotic enzyme: tryptophan tryptophylquinone as the redox prosthetic group in methylamine dehydrogenase. Science. 1991;252:817–824. doi: 10.1126/science.2028257. [DOI] [PubMed] [Google Scholar]
- 2.Davidson VL. Protein-derived cofactors. Expanding the scope of post-translational modifications. Biochemistry. 2007;46:5283–5292. doi: 10.1021/bi700468t. [DOI] [PubMed] [Google Scholar]
- 3.Chen L, Doi M, Durley RC, Chistoserdov AY, Lidstrom ME, Davidson VL, Mathews FS. Refined crystal structure of methylamine dehydrogenase from Paracoccus denitrificans at 1.75 A resolution. J. Mol. Biol. 1998;276:131–149. doi: 10.1006/jmbi.1997.1511. [DOI] [PubMed] [Google Scholar]
- 4.Davidson VL. Pyrroloquinoline quinone (PQQ) from methanol dehydrogenase and tryptophan tryptophylquinone (TTQ) from methylamine dehydrogenase. Adv. Protein Chem. 2001;58:95–140. doi: 10.1016/s0065-3233(01)58003-1. [DOI] [PubMed] [Google Scholar]
- 5.van der Palen CJ, Slotboom DJ, Jongejan L, Reijnders WN, Harms N, Duine JA, van Spanning RJ. Mutational analysis of mau genes involved in methylamine metabolism in Paracoccus denitrificans. Eur. J. Biochem. 1995;230:860–871. doi: 10.1111/j.1432-1033.1995.tb20629.x. [DOI] [PubMed] [Google Scholar]
- 6.Pearson AR, de la Mora-Rey T, Graichen ME, Wang Y, Jones LH, Marimanikkupam S, Aggar SA, Grimsrud PA, Davidson VL, Wilmot CW. Further insights into quinone cofactor biogenesis: Probing the role of MauG in methylamine dehydrogenase TTQ formation. Biochemistry. 2004;43:5494–5502. doi: 10.1021/bi049863l. [DOI] [PubMed] [Google Scholar]
- 7.Pearson AR, Marimanikkuppam S, Li X, Davidson VL, Wilmot CM. Isotope labeling studies reveal the order of oxygen incorporation into the tryptophan tryptophylquinone cofactor of methylamine dehydrogenase. J. Am. Chem. Soc. 2006;128:12416–12417. doi: 10.1021/ja064466e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang Y, Graichen ME, Liu A, Pearson AR, Wilmot CW, Davidson VL. MauG, a novel diheme protein required for tryptophan tryptophylquinone biogenesis. Biochemistry. 2003;42:7318–7325. doi: 10.1021/bi034243q. [DOI] [PubMed] [Google Scholar]
- 9.Fulop V, Watmouth NJ, Ferguson SJ. Structure and enyzmology of two bacterial diheme enzymes: cytochrome cd1 nitrite reductase and cytochrome c peroxidase. Adv. Inorg. Chem. 2001;51:163–204. [Google Scholar]
- 10.Pettigrew GW, Echalier A, Pauleta SR. Structure and mechanism in the bacterial dihaem cytochrome c peroxidases. J. Inorg. Biochem. 2006;100:551–567. doi: 10.1016/j.jinorgbio.2005.12.008. [DOI] [PubMed] [Google Scholar]
- 11.Li X, Feng M, Wang Y, Tachikawa H, Davidson VL. Evidence for redox cooperativity between c-type hemes of MauG which is likely coupled to oxygen activation during tryptophan tryptophylquinone biosynthesis. Biochemistry. 2006;45:821–828. doi: 10.1021/bi052000n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li X, Jones LH, Pearson AR, Wilmot CM, Davidson VL. Mechanistic possibilities in MauG-dependent tryptophan tryptophylquinone biosynthesis. Biochemistry. 2006;45:13276–13283. doi: 10.1021/bi061497d. [DOI] [PubMed] [Google Scholar]
- 13.Wang Y, Li X, Jones LH, Pearson AR, Wilmot CM, Davidson VL. MauG-dependent in vitro biosynthesis of tryptophan tryptophylquinone in methylamine dehydrogenase. J. Am. Chem. Soc. 2005;127:8258–8259. doi: 10.1021/ja051734k. [DOI] [PubMed] [Google Scholar]
- 14.Li X, Fu R, Liu A, Davidson VL. Kinetic and physical evidence that the di-heme enzyme MauG tightly binds to a biosynthetic precursor of methylamine dehydrogenase with incompletely formed tryptophan tryptophylquinone. Biochemistry. 2008;47:2908–2912. doi: 10.1021/bi702259w. [DOI] [PubMed] [Google Scholar]
- 15.Li X, Fu R, Lee S, Krebs C, Davidson VL, Liu A. A catalytic di-heme bis-Fe(IV)intermediate, alternative to an Fe(IV)=O porphyrin radical. Proc. Natl. Acad. Sci. U. S. A. 2008;105:8597–8600. doi: 10.1073/pnas.0801643105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Meunier B, de Visser SP, Shaik S. Mechanism of oxidation reactions catalyzed by cytochrome P450 enzymes. Chem. Rev. 2004;104:3947–3980. doi: 10.1021/cr020443g. [DOI] [PubMed] [Google Scholar]
- 17.Sono M, Roach MP, Coulter ED, Dawson JH. Heme-containing oxygenases. Chem. Rev. 1996;96:2841–2888. doi: 10.1021/cr9500500. [DOI] [PubMed] [Google Scholar]
- 18.Graichen ME, Jones LH, Sharma BV, van Spanning RJ, Hosler JP, Davidson VL. Heterologous expression of correctly assembled methylamine dehydrogenase in Rhodobacter sphaeroides. J. Bacteriol. 1999;181:4216–4222. doi: 10.1128/jb.181.14.4216-4222.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dean JA. Lange’s Handbook of Chemistry. 15th ed. McGraw-Hill; New York: 1998. [Google Scholar]
- 20.Lange R, Heiber-Langer I, Bonfils C, Fabre I, Negishi M, Balny C. Activation volume and energetic properties of the binding of CO to hemoproteins. Biophys. J. 1994;66:89–98. doi: 10.1016/S0006-3495(94)80772-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Prasad S, Mitra S. Substrate modulates compound I formation in peroxide shunt pathway of Pseudomonas putida cytochrome P450(cam) Biochem. Biophys. Res. Commun. 2004;314:610–614. doi: 10.1016/j.bbrc.2003.12.141. [DOI] [PubMed] [Google Scholar]
- 22.Koley AP, Robinson RC, Friedman FK. Cytochrome P450 conformation and substrate interactions as probed by CO binding kinetics. Biochimie. 1996;78:706–713. doi: 10.1016/s0300-9084(97)82528-x. [DOI] [PubMed] [Google Scholar]
- 23.Jung C, Bec N, Lange R. Substrates modulate the rate-determining step for CO binding in cytochrome P450cam (CYP101). A high-pressure stopped-flow study. Eur. J. Biochem. 2002;269:2989–2996. doi: 10.1046/j.1432-1033.2002.02980.x. [DOI] [PubMed] [Google Scholar]
- 24.Fulop V, Ridout CJ, Greenwood C, Hajdu J. Crystal structure of the dihaem cytochrome c peroxidase from Pseudomonas aeruginosa. Structure. 1995;3:1225–1233. doi: 10.1016/s0969-2126(01)00258-1. [DOI] [PubMed] [Google Scholar]