

Abstract
A method for the rapid sequence determination of peptoids [oligo(N-substituted glycines)] and peptide-peptoids hybrids selected from one-bead-one-compound combinatorial libraries has been developed. In this method, beads carrying unique peptoid (or peptide-peptoid) sequences were subjected to multiple cycles of partial Edman degradation (PED) by treatment with a 1:3 (mol/mol) mixture of phenyl isothiocyanate (PITC) and 9-fluorenylmethyl chloroformate (Fmoc-Cl) to generate a series of N-terminal truncation products for each resin-bound peptoid. After PED, the Fmoc group was removed from the N-terminus and any reacted side chains via piperidine treatment. The resulting mixture of the full-length peptoid and its truncation products was analyzed by matrix-assisted laser desorption ionization (MALDI) mass spectrometry, to reveal the sequence of the full-length peptoid. With a slight modification, the method was also effective in the sequence determination of peptide-peptoid hybrids. This rapid, high-throughput, sensitive, and inexpensive sequencing method should greatly expand the utility of combinatorial peptoid libraries in biomedical and materials research.
Keywords: Combinatorial chemistry, One-bead-one-compound library, MALDI-TOF MS, Partial Edman degradation, Peptoid Sequencing, High-throughput
Introduction
Oligomers of N-substituted glycines, or “peptoids”, are a new class of unnatural materials that are capable of mimicking the structure and function of peptides and proteins.1 A diverse array of biologically active peptoids have been discovered, including carriers for nucleic acid delivery,2 antimicrobials,3 lung surfactant mimetics,4 inhibitors of G-protein-coupled receptors,5 and ligands of both intracellular and cell surface proteins.6,7 Peptoids offer several advantages over peptides as therapeutic or diagnostic agents. They are resistant to proteases8 and more membrane permeable,9 due to the lack of amide -NH groups, thus overcoming two major problems associated with peptide drugs. Furthermore, peptoids containing a wide variety of side-chain functionalities can be readily prepared by using a two-step, submonomer method.10 These properties make peptoids attractive structures in drug discovery as well as materials engineering.
Combinatorial chemistry provides a powerful method for the rapid identification of high-affinity ligands against macromolecular targets (e.g., proteins). Most of the biologically active peptoids aforementioned were originally discovered by screening combinatorial peptoid libraries. Although insolution screening of peptoid libraries has been practiced,5 the more straightforward method involves one-bead screening of one-bead-one-compound (OBOC) libraries, during which peptoid ligands are displayed on the surface of individual beads and a soluble, properly labeled receptor (or intact cells) is allowed to bind to those beads that carry the high-affinity ligands.6,7 The advantage of on-bead screening is that OBOC libraries of very high sequence diversity can be readily prepared by the split-and-pool synthesis method11-13 and a large number of compounds can be rapidly screened in a short time (e.g., 107 beads/compounds can be screened in a few hours). However, on-bead screening of OBOC libraries necessitates post-screening identification of the selected “hit” compounds. Direct sequencing of resinbound peptoids by Edman degradation has been most commonly employed for this purpose.6,7,14 Unfortunately, Edman sequencing is time-consuming (hours to a day to sequence a short peptide/peptoid) and expensive (∼$300/peptide or peptoid). For peptoids and peptides containing unnatural amino acids, the Edman method is especially slow, since it requires the prior synthesis and analysis of phenylthiohydantoin standards for each unnatural residue. Liskamp and co-workers applied MS/MS techniques to determine the sequence of peptoids15-17 and found that peptoids in general fragment in a similar manner to peptides. However, the presence of at least two sets of fragment ions in the spectra (b and y ions) makes them inherently difficult to interpret. To alleviate this problem, we recently showed that spectral interpretation could be greatly simplified by the addition of an isotope tag to one end of the peptoids.18 In this method, the peptoids were attached to the solid phase via a cleavable linker. After a hit compound/bead was isolated from a library, the peptoid was released from the bead to form a C-terminal aldehyde, which was then derivatized with a bromine-containing amino-oxy compound that serves as an isotope tag. The tagged peptoid was subjected to MS/MS analysis, during which the y-ion fragments were readily identified by their doublet peaks (m/z M and M+2 due to 79Br and 81Br isotopes). This method works well with peptoids displayed on TentaGel macrobeads (which typically carry ≥1 nmol peptoids per bead), but becomes less reliable when the amount of peptoids is limited. It also requires chemical modification after the peptoid is released from the resin, resulting in sample loss during the manipulations. These features make the MS/MS method less convenient for the more popular TentaGel microbeads (which are ≤100 μm in diameter and carry ≤100 pmol of peptoids), especially when a large number of hits need to be sequenced. One of our laboratories has previously demonstrated that resin-bound peptides can be rapidly sequenced by partial Edman degradation/mass spectrometry (PED/MS).19-21 Briefly, a resin-bound peptide is converted into a series of progressively shorter fragments by multiple cycles of partial Edman degradation and the resulting peptide ladder is analyzed by MALDI MS. In this work, we have adapted the PED/MS concept to develop a reliable, sensitive, high-throughput, and inexpensive method to directly sequence resin-bound peptoids and peptide-peptoid hybrids derived from OBOC libraries.
Results and Discussion
PED/MS provides a powerful method for sequence determination of hit peptides identified from OBOC libraries.22-25 The key strategy of PED/MS is to convert a peptide to be sequenced into a family of progressively shorter fragments, followed by MALDI-TOF MS analysis. Thus, a support-bound peptide is treated with a mixture of phenyl isothiocyanate (PITC) and a small amount of capping agent (e.g., Fmoc-OSU21). PITC and the capping agent compete for reacting with the N-terminal amine of the peptide. Under properly controlled conditions, 90-95% of the peptide molecules react with PITC and subsequent treatment with trifluoroacetic acid (TFA) results in cleavage of the N-terminal residue (Edman degradation). The remaining 5-10% peptides react with the capping agent and become N-terminally blocked; TFA treatment does not cleave the N-terminal residue. Repetition of the PED reaction for n-1 cycles (where n is the number of residues to be sequenced) converts the original peptide into a series of sequence-related truncation products (or “a peptide ladder”). Subsequent analysis of the peptide mixture by MALDI-TOF MS reveals the amino acid sequence of the original peptide. Obviously, a proper capping agent is crucial for successful PED. The ideal capping agent should react readily with the N-terminal amine but not with any peptide side chain or solvent (e.g., pyridine and water). Alternatively, one can use a removable capping agent (traceless capping agent). The traceless capping agent may react with both the N-terminal amine and certain side-chain functionalities, but can be removed after PED is complete (but prior to MS analysis). For peptide sequencing, we have found that Fmoc-OSU is ideally suited for this purpose.21 It competes effectively with PITC for reacting with the N-terminal amines of peptides, is stable towards Edman degradation conditions (pyridine/water and TFA), and is readily removed by treatment with piperidine.
Synthesis and PED/MS Sequencing of Peptoid Libraries
Since peptoid oligomers have been sequenced by conventional Edman degradation,6,7,14 we reasoned that the PED/MS strategy previously developed for peptide sequencing should be applicable to peptoids. However, due to the very different nucleophilicity of peptide (primary amine) vs peptoid N-terminus (secondary amine), we anticipated that reagents and conditions for PED would have to be re-optimized for peptoids. To test this notion, we synthesized a peptoid library containing five random positions, XXXXX(Nmeg)4RM-resin (Library I, where X represents N-substituted glycines Nbsa, Nglu, Nleu, Nlys, Nphe, Npip, Nser, and Ntyr), on TentaGel S NH2 resin (90 μm, 0.28 mmol/g, 2.86 × 106 beads/g) (Figure 1). The C-terminal methionine allows cleavage of the peptoid from the resin by CNBr prior to MS analysis. The arginine residue improves the solubility of the peptoids in aqueous solution and provides a fixed positive charge to facilitate MS analysis. The four N-methoxyethylglycine (Nmeg) residues provide a hydrophilic linker and ensure that the peptoids and their PED fragments have masses greater than 500 amu, to avoid signal overlap with those of MALDI matrices (vide infra). Fmoc-OSU was initially tested as the capping agent for PED of library I peptoids, but failed to generate the desired peptoid ladder under all of the conditions tested (PITC/Fmoc-OSU ratios of 40:1 to 1:5). Most of the peptoids were completely degraded, indicating that Fmoc-OSU is not sufficiently reactive toward the sterically hindered secondary amines at the peptoid N-termini. We therefore examined several other electrophilic reagents including phenyl formate, N-formylbenzotriazole, and Fmoc-Cl as capping agents. The formyl ester and amide were chosen because their small acyl group (formyl) was expected to facilitate the nucleophilic attack by the bulky secondary amines. Fmoc-Cl is generally more reactive than Fmoc-OSU toward nucleophilic amines. Indeed, phenyl formate and N-formylbenzotriazole reacted readily with the N-terminal amines and, when added to PITC, generated the desired peptoid ladders (data not shown). However, they also formylated (albeit partially) some of the peptoid side chains, resulting in very complex MS spectra. Fmoc-Cl also had adequate reactivity with the peptoid N-termini; PED with 1:3 (mol/mol) PITC and Fmoc-Cl generated the desired percentage of chain termination after each cycle (5-20%). Fmoc-Cl undoubtedly also reacted with some of the nucleophilic side chains (e.g., the side-chain amine of Nlys), but any attached Fmoc group was quantitatively removed by subsequent piperidine treatment. We chose Fmoc-Cl as a traceless capping agent for further experimentation.
Figure 1.

The structures of peptoid library I and the building blocks used for its synthesis. The side-chain structure, the four-letter code, and the residue mass of 11 peptoid monomers are shown. Library I was constructed with 8 of the monomers (excluding Nasp, Ncyh, and Ncpl) and had a theoretical diversity 32,768. Library II contained 10 monomers (excluding Nasp) and a peptide linker, LNBBRM, with a theoretical diversity of 100,000. Library III was constructed with 7 N-substituted glycines (Nasp, Nbsa, Nleu, Nlys, Nphe, Npip, and Nser) and 3 amino acids (glycine, sarcosine, and L-proline), with the peptide linker LNBBRM and a theoretical diversity of 100,000.
To demonstrate the effectiveness of Fmoc-Cl as a capping agent for peptoid sequencing, we randomly selected 120 beads from library I and subjected them to five cycles of PED at a PITC/Fmoc-Cl ratio of 1:3 (Figure 2a). Twenty of these treated beads were randomly selected and their peptoids were released from the beads by treatment with CNBr and individually analyzed by MALDI-TOF MS. The 20 MS spectra are provided in Figure S1 under Supporting Information to show the overall quality of the spectra. Nineteen of the spectra had sufficient quality for unambiguous sequence assignment and their sequences are listed in Table 1. The remaining bead gave no peptoid signal, possibly due to failed peptoid synthesis or poor sample preparation. Figure 2b shows a representative MS spectrum obtained from one of the 20 beads. The spectrum contains peaks at m/z 1581.70, 1390.60, 1262.50, 1071.40, 958.37, and 718.28, derived from the full-length peptoid and its five N-terminal truncation products, respectively. The peptoid sequence is readily interpreted as Npip-Nlys-Npip-Nleu-Nbsa-(Nmeg)4-Arg-Met-resin by examining the mass differences between adjacent peaks. For example, the mass difference of 191.10 amu between the peak with the highest m/z value (m/z 1581.70, which corresponds to the full-length peptoid) and that of the second highest m/z value (m/z 1390.60, which corresponds to the peptoid missing the N-terminal residue) indicates that the N-terminal residue is Npip. The MS spectra were in general quite “clean”, containing relatively few extra peaks. Among some frequently observed extra signals was a peak at m/z 900.37, whose identity or origin has not yet been revealed. Other extra signals included a set of peaks that are always 85 amu heavier relative to the peptoid ladder (e.g., the peak at m/z 1347.50 in Figure 2b). These signals appear to be originated from peptoid library synthesis, most likely due to modification of the (Nmeg)4RM linker by a yet unknown functional group. However, none of the extra signals adversely affect the unambiguous sequence determination of the full-length peptoid.
Figure 2.

(a) Scheme showing the reactions involved in partial Edman degradation. Reagents and conditions: (a) 1:3-5 PITC:Fmoc-Cl; (b) TFA; (c) Repeat a and b four times; (d) 20% piperidine in DMF; and (e) CNBr in 70% TFA. M*, homoserine lactone. (b) Representative MS spectrum of a peptoid and its PED products derived from a 90-μm TentaGel bead carrying the sequence Npip-Nlys-Npip-Nleu-Nbsa-(Nmeg)4-Arg-Met-resin. The peaks labeled with “a” (at m/z 900.37) and “b” (at m/z 1347.50) are impurity peaks derived from library synthesis.
Table 1.
Sequences of 20 Randomly Selected Peptoids from Library I
| Bead No. | Peptoid sequence |
|---|---|
| 1 | Nlys-Nleu-Nglu-Nser-Ntyr |
| 2 | Nbsa-Nser-Nleu-Npip-Nglu |
| 3 | Nphe-Nglu-Nleu-Npip-Nbsa |
| 4 | Nglu-Nphe-Nglu-Ntyr-Nleu |
| 5 | Nglu-Nphe-Nser-Nleu-Nglu |
| 6 | Npip-Nphe-Nlys-Npip-Nser |
| 7 | Nser-Nlys-Nleu-Nphe-Nphe |
| 8 | Nleu-Npip-Nleu-Nphe-Nphe |
| 9 | Nglu-Nser-Nbsa-Nphe-Ntyr |
| 10 | Nser-Nglu-Nphe-Nlys-Nlys |
| 11 | Npip-Nphe-Ntyr-Nglu-Nphe |
| 12 | No ionization |
| 13 | Ntyr-Nphe-Nbsa-Nlys-Nleu |
| 14 | Nbsa-Nleu-Npip-Npip-Npip |
| 15 | Nphe-Npip-Nlys-Nphe-Ntyr |
| 16 | Ntyr-Npip-Nbsa-Nbsa-Nphe |
| 17 | Nleu-Nglu-Nbsa-Nphe-Ntyr |
| 18 | Npip-Nglu-Nphe-Nglu-Nlys |
| 19 | Ntyr-Nleu-Nleu-Nleu-Nglu |
| 20 | Nphe-Nlys-Nleu-Ntyr-Npip |
Due to the difference in reactivity between peptides (primary amines) and peptoids (secondary amines), we performed PED/MS under a variety of conditions in order to optimize the PED and MS analysis conditions. We found that a PITC/Fmoc-Cl ratio of 1:3-5 generally gave the proper percentage of chain termination during each PED cycle (5-20%). Compared to peptides, longer reaction times were necessary for both the carbamoylation step (20 vs 6 min) and the TFA-mediated cyclization reaction (40 vs 12 min). Variations in temperature showed that while higher temperature resulted in faster reactions, room temperature was operationally simpler and gave more consistent results. Another important modification involved sample preparation prior to MALDI MS analysis. Dissolution of PED products in 0.1% TFA in water produced poor or often no signal for larger peptoid fragments. This is likely due to the greater hydrophobicity and poorer aqueous solubility of peptoids as compared to peptides. This problem is largely resolved by dissolving the PED products in a 50:50:0.05 (v/v) mixture of acetonitrile, water, and TFA.
To determine the success rate and reproducibility of peptoid sequencing by PED/MS, we randomly selected two additional batches of beads from library I (70 and 50 beads) and repeated the PED procedure. Twenty and 14 of the degraded beads were subjected to MS analysis, resulting in 19 (95%) and 13 complete sequences (93%), respectively (Table 2, trials 2 and 3). We also synthesized a second peptoid library containing five random residues and a peptide linker, XXXXXLNBBRM-resin (library II, where X represents 10 N-substituted glycines Nbsa, Ncpl, Ncyh, Nglu, Nleu, Nlys, Nphe, Npip, Nser, and Ntyr and B is β-alanine) (Figure 1). Three batches of beads from library II were sequenced by PED/MS to give 16/16, 14/14, and 19/20 complete sequences (Table 2, trials 4-6). In addition, we individually synthesized 2 known peptoid sequences on 90-μm TentaGel beads and a few beads from each peptoid sequence were subjected to the PED/MS procedure (see Figure S2 for an example). In each case, the peptoid sequence was correctly determined (Table 2, trials 7 and 8). All together, we have used the PED/MS method to sequence 116 peptoid-containing beads and obtained 112 unambiguous sequences, with an overall success rate of 97% (Table 2).
Table 2.
Success Rates for Sequencing Support-Bound Peptoids by PED/MS
| Trial No. | Peptoid | PITC: Fmoc-Cl | No. of beads in PED | No. of beads analyzed by MS | No. of complete sequences obtained (%) |
|---|---|---|---|---|---|
| 1 | XXXXX(Nmeg)4RM-resin | 1:3 | 120 | 20 | 19 (95) |
| 2 | XXXXX(Nmeg)4RM-resin | 1:3 | 70 | 20 | 19 (95) |
| 3 | XXXXX(Nmeg)4RM-resin | 1:3 | 50 | 14 | 13 (93) |
| 4 | XXXXXLNBBRM-resin | 1:5 | 100 | 16 | 16 (100) |
| 5 | XXXXXLNBBRM-resin | 1:3 | 50 | 14 | 14 (100) |
| 6 | XXXXXLNBBRM-resin | 1:3 | 80 | 20 | 19 (95) |
| 7 | Ncpl-Npip-Nbsa-Nlys-Ncyh-LNBBRM-resin | 1:5 | 50 | 3 | 3 (100) |
| 8 | Nphe-Nlys-Nglu-Nser-Nleu-LNBBRM-resin | 1:5 | 50 | 9 | 9 (100) |
| Total | 570 | 116 | 112 (97) |
PED/MS Sequencing of Peptide-Peptoid Hybrids
While peptoids have improved protease resistance and membrane permeability, they lack the backbone -NH, which often serves as a critical element in both inter- and intramolecular interactions. There has been growing interest in the preparation of peptide-peptoid hybrids as molecular probes and therapeutics. We thus tested whether the above PED/MS approach can be adapted to sequence peptide-peptoid hybrids. We synthesized a third library containing both N-substituted glycines and α-amino acids at the random positions, XXXXXLNBBRM-resin (library III, where X represents N-substituted glycines Nasp, Nbsa, Nleu, Nlys, Nphe, Npip, and Nser and amino acids glycine, sarcosine, and L-proline and B represents β-alanine) (Figure 1). Our initial attempts to sequence the hybrid sequences under the conditions that had been optimized for peptoids (1:3-5 FITC/Fmoc-Cl) all failed; the peptoid/peptide ladder always stopped when PED reached the first amino acid in sequence (data not shown). We reasoned that the excess Fmoc-Cl probably dominated the Edman degradation reaction when the N-terminal residue was an amino acid, resulting in N-terminal blocking of all resin-bound molecules (instead of the desired 5-20%). To overcome this problem, we devised a two-stage PED protocol. The beads were first treated with 30:1 (mol/mol) PITC/Fmoc-OSU (i.e., the optimal condition for peptide sequencing) for 1 min; during this stage, 90-95% of the N-terminal amino acids (if any) would react with PITC and the remaining 5-10% would become N-blocked by Fmoc group, while the N-substituted glycines (which are less reactive) would have little reaction. The beads were quickly washed with pyridine and treated with 1:5 PITC/Fmoc-Cl for 19 min to partition the sequences that contained N-terminal N-substituted glycines. Five batches of the library III beads were subjected to the two-stage PED reaction and MS analysis. Out of a total of 75 beads analyzed by MS, we obtained 51 unambiguous, full-length sequences (68% success rate) (Table 3). Figure 3 shows a representative MS spectrum derived from one of the 75 beads, carrying the sequence Nleu-Pro-Gly-Nleu-Gly-Leu-Asn-βAla-βAla-Arg-Met-resin. The lower success rate as compared with peptide or peptoid sequencing is at least in part due to the poorer quality of the hybrid library. Synthesis of peptide-peptoid hybrid libraries is not yet well established. Our hybrid library also contained N-carboxymethylglycine (Nasp), which for yet unknown reasons interferes with PED/MS sequencing (AT and DP, unpublished observations).
Table 3.
Success Rates for Sequencing Peptide-Peptoid Hybrids by PED/MS
| Trial No. | PITC:Fmoc-OSU; PITC:Fmoc-Cl | No. of beads in PED | No. of beads analyzed by MS | No. of complete sequences obtained (%) |
|---|---|---|---|---|
| 1 | 30:1; 1:5 | 100 | 20 | 13 (65) |
| 2 | 30:1; 1:5 | 100 | 20 | 15 (75) |
| 3 | 30:1; 1:5 | 80 | 15 | 8 (53) |
| 4 | 20:1; 1:5 | 120 | 10 | 7 (70) |
| 5 | 30:1; 1:5 | 120 | 10 | 8 (80) |
| Total | 520 | 75 | 51 (68) |
Figure 3.
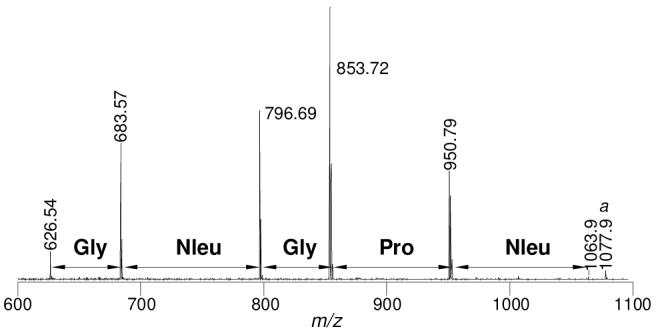
Representative MS spectrum of a peptide-peptoid hybrid and its PED products derived from a 90-μm TentaGel bead carrying the sequence Nleu-Pro-Gly-Nleu-Gly-Leu-Asn-βAla-βAla-Arg-Met-resin. The peak labeled with “a” (at m/z 1077.9) is the methylated peptide-peptoid full-length chain.
Identification of K-Ras Ligands from Peptoid Library
To demonstrate the compatibility of the PED/MS method with peptoid library screening, we screened peptoid library II (Figure 1) against G-protein K-Ras. K-Ras is involved in the G-protein signal transduction pathway, modulating cellular proliferation and differentiation; inhibitors against K-Ras may act as potential anticancer agents.26 Screening of 30 mg of library II (∼90,000 beads) against 500 nM K-Ras resulted in 34 positive beads, which were sequenced by the PED/MS method to give 28 unambiguous, complete sequences (82% success rate) (Table 4). Among the remaining 6 beads, two gave partial sequences while the other four did not produce interpretable mass spectra. Inspection of the selected sequences indicates that K-Ras has an overall preference for aromatic (Nphe, Ntyr, Npip, and Nbsa) and positively charged residues (Nlys). In contrast, the negatively charged Nglu was rarely selected. Individual synthesis and subsequent binding analysis of the selected peptoids are currently underway in one of our laboratories.
Table 4.
Sequences of K-Ras Ligands Selected from Library II
| Bead No. | Peptoid sequence |
|---|---|
| 1 | Nlys-Nlys-Nphe-Nlys-Ncyh |
| 2 | Nlys-Nser-Nbsa-Nlys-Ntyr |
| 3 | Npip-Nser-Ncyh-Nlys-Nlys |
| 4 | Ncpl-Nphe-Nleu-Nphe-Ntyr |
| 5 | Nglu-Nleu-Npip-Nphe-Npip |
| 6 | Ncyh-Ncpl-Nlys-Ntyr-Nlys |
| 7 | Ncpl-Ncyh-Ncyh-Nlys-Npip |
| 8 | Ncyh-Nlys-Nbsa-Nbsa-Nlys |
| 9 | Nlys-Nbsa-Nser-Nlys-Npip |
| 10 | Npip-Nphe-Nlys-Npip-Nlys |
| 11 | Nlys-Ncpl-Nlys-Nlys-Nbsa |
| 12 | Nphe-Nbsa-Ncpl-Nlys-Nlys |
| 13 | Nphe-Nlys-Nlys-Nbsa-Ncpl |
| 14 | Nlys-Nbsa-Npip-Nlys-Nphe |
| 15 | Nser-Nbsa-Ntyr-Nphe-Nphe |
| 16 | Nglu-Ncpl-Nbsa-Nlys-Nlys |
| 17 | Nbsa-Nphe-Nlys-Npip-Nser |
| 18 | Nphe-Ncyh-Nser-Ncyh-Ncyh |
| 19 | Nbsa-Nphe-Nlys-Nlys-Ncyh |
| 20 | Ncyh-Nbsa-Nlys-Nphe-Nlys |
| 21 | Npip-Ntyr-Nlys-Nlys-Nbsa |
| 22 | Nphe-Nleu-Nlys-Nlys-Nlys |
| 23 | Nser-Npip-Nphe-Nlys-Nlys |
| 24 | Ncyh-Nlys-Nlys-Nlys-Nphe |
| 25 | Npip-Nlys-Nlys-Ncyh-Nphe |
| 26 | Nphe-Nser-Nleu-Npip-Ntyr |
| 27 | Nphe-Nlys-Nser-Ntyr-Nlys |
| 28 | Nlys-Nglu-Nglu-Nbsa-Nphe |
Comparison of PED/MS and MS/MS Methods
The PED/MS method is advantageous over the conventional MS/MS methods in several aspects. First, while MS/MS generates at least two sets of fragment ions (y and b ions), PED/MS gives a single set of peaks. Additionally, in PED/MS, peptide/peptoid fragments are generated through well-defined chemical reactions and the relative abundance of fragment ions can be controlled by varying the ratio of PITC/capping agent. In contrast, collision- or ion-induced fragmentation in MS/MS is not as easy to control. Consequently, PED/MS gives much simpler spectra of more uniform peak intensities, making spectral interpretation simple and unambiguous. Second, with the addition of a few percent of capping agents during library synthesis, the PED/MS method can differentiate mass-degenerate residues,21 which is not possible by MS/MS. Third, our method is capable of sequencing peptoids derived from a single 90-μm TentaGel bead (∼0.1 nmol peptoid/bead). Although not yet tested, the PED/MS method should be compatible with still smaller beads (e.g., 30-μm TentaGel beads). The MS/MS methods, on the other hand, have only been demonstrated on macrobeads (300-500 μm), which typically contain 4-100 nmol peptoids per bead. With the more popular 90-μm microbeads, the MS/MS methods remain problematic. The use of smaller beads permits the construction of libraries of greater structural diversity (e.g., 1 g of 90-μm TentaGel resin contains 2.86 × 106 beads, whereas 1 g of 300-μm TentaGel resin has only 65,500 beads). Fourth, PED/MS has much higher throughput than the MS/MS methods. Because all of the selected beads/peptoids are pooled and degraded simultaneously and MALDI-TOF MS is run in an automated format, the PED/MS method can readily sequence 500 beads in a single day (on a single MS instrument). In fact, by using a Bruker Reflex III MALDI-TOF instrument in a core facility, one of our laboratories (The Ohio State University) has been sequencing ∼10,000 peptides per year over the past few years.22-25 Sequencing of peptoids and peptide-peptoid hybrids by PED/MS has a similar throughput (despite the longer PED cycle for peptoids), because the overall throughput is currently limited by the speed of MS analysis. Finally, the PED/MS method is inexpensive; the cost for sequencing a peptide/peptoid (reagents and instrument time) is $∼1. The method is also straightforward to carry out, does not require dedicated instrumentation or personnel, and can therefore be performed in any chemical or biochemical laboratory. The only drawback of the PED/MS method is that it is currently limited to resin-bound peptides/peptoids and cannot sequence peptides/peptoids that are free in solution.
Conclusion
We have modified the PED/MS method to effectively sequence peptoids and peptide-peptoid hybrids derived from combinatorial libraries. The PED/MS method is high-throughput, reliable, rapid, and inexpensive and is ideally suited for decoding “hit” peptoids identified from OBOC libraries. It should expand the utility of combinatorial peptoid (or peptide-peptoid hybrid) libraries in drug discovery and materials research.
Experimental Section
Materials
TentaGel-S NH2 resin (90 μm, 0.28 mmol/g loading, 2.86 × 106 beads/g) was purchased from Peptides International Inc. (Louisville, KY). Amino acids were purchased from NovaBiochem (Gibbstown, NJ). N,N’-Diisopropylcarbodiimide (DIC), N-hydroxybenzotriazole (HOBt), and 2-(1H-benzotriazole-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate (HBTU) were from Advanced ChemTech (Louisville, KY). 4-Methylpiperidine and isobutylamine were purchased from Alfa Aesar (Ward Hill, MA). O-TIPS-2-aminoethanol was synthesized according to a previously published procedure.27 4-Methoxybenzylamine, cyclohexylamine, 4-(2-aminoethyl)benzenesulfonamide, piperonylamine, 4-methylmorpholine, bromoacetic acid, 2-propanol, piperidine, and benzylamine were purchased from Sigma Aldrich (St. Louis, MO). Cyclopropylamine was purchased from Fluka (St. Louis, MO). Boc-1,4-diaminobutane and 9-fluorenylmethyl chloroformate were purchased from VWR (West Chester, PA). O-t-Butyl-β-alanine was purchased as the HCl salt from Bachem (Torrance, CA), and was free-based prior to use as described below. N,N’-Dimethylformamide (DMF) was purchased from Fisher Scientific (Pittsburgh, PA) and 1,2-dichloroethane was purchased from Mallinckrodt Chemicals (Hazelwood, MO). 2-(1H-7-Azabenzotriazol-1-yl)-1,1,3,3-tetramethyl uronium hexafluorophosphate (HATU) and 1-hydroxy-7-azabenzotriazole (HOAt) were purchased from GenScript Corporation (Piscataway, NJ). Bio-Rad columns were purchased from Bio-Rad Laboratories (Hercules, CA).
Free Base Procedure for O-t-Butyl-β-Alanine HCl
O-t-Butyl-β-alanine HCl (25 g, 138 mmol) was dissolved in dichloromethane (150 mL). One equivalent of NaOH was dissolved in water (40 mL) and added to the amine solution. The mixture was shaken in a separatory funnel and the organic layer was collected. The aqueous layer was extracted with dichloromethane (3 × 150 mL). The combined dichloromethane layers were washed with saturated NaCl (3 × 50 mL), dried with sodium sulfate, and the solvent removed under reduced pressure.
Solid-Phase Peptide and Peptoid Synthesis
Individual peptoid sequences were synthesized on TentaGel S NH2 resin at 35 °C using a custom automated synthesizer. All reactions and washings were performed in glass vessels equipped with a coarse frit. Resins were washed by the addition of either DMF or transfer solvent, with agitation so that a uniform slurry was obtained (1 min), followed by draining of the solvent by vacuum filtration through the fritted bottom of the reaction vessel. Agitation of the resin slurry was accomplished by bubbling nitrogen through the bottom of the fritted vessel. Prior to the start of the synthesis, the resin was swelled in DMF (1.1 mL/100 mg resin) for 5 min.
Amino acid monomer addition cycle
TentaGel S-NH2 resin (1.2 g, 0.34 mmol) was suspended in 1 mL of anhydrous DMF. Proper Fmoc-amino acid (0.67 mmol), HBTU (0.67 mmol), and HOBt (0.67 mmol) were dissolved in DMF (5 mL) in a plastic tube. 4-Methylmorpholine (1.3 mmol) was added to the tube and the resulting solution was briefly agitated (30 s) and added to the reaction vessel containing the TentaGel resin. The final reaction mixture contained 0.11 M Fmoc-amino acid, 0.11 M HBTU, 0.11 M HOBt, and 0.22 M 4-methylmorpholine. The reaction was allowed to proceed for 2 h on a rotary shaker and terminated by draining and washing with DMF (2 × 5 mL), DCM (2 × 5 mL), DMF (1 × 5 mL), and 20% piperidine in DMF (1 × 5 mL). The resin was treated twice with 5 mL of 20% piperidine in DMF at room temperature (5 min each time). The resin was washed with DMF (3 × 5 mL) and ready for the addition of the next monomer.
Peptoid submonomer addition cycle
To the resin-bound amine (0.028 mmol) was added bromoacetic acid in DMF (0.48 mL, 1.2 M, 0.57 mmol) followed by DIC (0.080 mL, 0.52 mmol). The resin was mixed for 20 min, drained, and washed with DMF (2 × 1 mL) followed by 20% isopropanol in 1,2-dichloroethane (3 × 1 mL). To the resin-bound bromide was added the appropriate amine (0.48 mL of a 1.0 or 2.0 M solution). O-TIPS-2-aminoethanol was used at 2.0 M in 1:1 DMF/chlorobenzene (0.95 mmol) and Boc-1,4-diaminobutane was used at 2.0 M in DMF (0.95 mmol). All other amines were used at 1.0 M in DMF (0.48 mmol). The amine displacement reaction was allowed to proceed for 120 min (with mixing), drained, and washed with DMF (2 × 1 mL) and 20% isopropanol in 1,2-dichloroethane (3 × 1 mL).
Synthesis of Peptoid Library
Peptoid library was synthesized on TentaGel S NH2 resin (800 mg, 0.224 mmol, 2.3 × 106 beads) using the procedure described above. For the addition of random residues, the resin was split into the desired number of reaction vessels by the isopycnic slurry method22 using 20% isopropanol in 1,2-dichloroethane as the resin transfer solvent. For the synthesis of library I & II, transfer solvent was added to the resin to bring the resin slurry to a final volume of 157 mL and the mixture was agitated briefly. Next, 18-mL aliquots of the slurry (2 × 9 mL) were transferred into each of the eight reaction vessels, with brief agitation of the slurry in between each transfer. The solvent was drained from all vessels and the procedure was repeated twice to ensure complete transfer of the resin. After the coupling reactions were complete, 14 mL of the transfer solvent was added to each reaction vessel and the resulting slurry from each vessel was agitated and transferred back into a large mixing vessel. Again, the transfer procedure was repeated twice to ensure complete transfer of the resin. The peptoid libraries were stored at -20 °C in the fully protected form with side chains deprotected prior to use. Typically, 2 mg of the resin (∼6000 beads) was placed in a Bio-Rad Micro-spin column (catalog No. 732-6204), washed with dichloromethane (3 × 1.5 mL), and treated with 800 μL of modified reagent B (94.5:2.5:2.5:0.5 TFA/water/TIPS/thioanisole) for 1 h at room temperature. The resin was washed with dichloromethane (5 × 1 mL).
Synthesis of Peptide-Peptoid Hybrid Library
The hybrid peptide-peptoid library (library III) was synthesized on TentaGel S NH2 resin using the peptoid synthesis procedure described above, but with the following modifications. The peptide linker (LNBBRM) was synthesized by standard Fmoc/HBTU chemistry. For the addition of random residues, 1 g of resin was split into four reaction vessels. Three vessels each contained 100 mg resin and were coupled to a different amino acid. The coupling reactions contained the properly protected Fmoc-amino acid (4 equiv), HATU (4 equiv), HOAt (4 equiv), DIPEA (8 equiv), and DMF (1 mL) and were allowed to proceed for 2 h at room temperature on a rotary shaker. The resin was then treated twice with 20% piperidine in DMF (1 mL and 10 min each time) and ready for the next coupling reaction. The 4th vessel contained the remaining resin (700 mg) and was washed with DMF (2 × 5 mL). A bromoacetic acid solution (516 mg in 3.1 mL of DMF) was added to the resin, followed by the addition of 521 μL of diisopropylcarbodiimide. The final concentrations of bromoacetic acid and DIC in DMF were 1.03 M and 0.93 M, respectively. The vessel was capped and shaken at 37 °C for 30 min. The resin was drained, washed with DMF (3 × 5 mL) and dichloromethane (3 × 5 mL), and dried for 4 h under vacuum suction. The resin was equally split into seven reaction vessels (100 mg each) and each aliquot was treated with 900 μL of a 2.0 M solution of different primary amine (for ammonium chloride salts, 1.1 equiv of diisopropylethylamine was also added). The reaction vessels were capped and placed on a rotary shaker for 2 h at 37 °C. The resin was drained and washed with DMF (3 × 1 mL) and dichloromethane (3 × 1 mL). Next, the resins from all ten vessels were pooled and mixed and were ready for the addition of the next random residue. During the library synthesis, whenever possible, the presence of primary or secondary amine was monitored by Kaiser28 and chloranil tests29, respectively.
Partial Edman Degradation
All reactions were performed in a homemade glass vessel (12-mm i.d.; 20-mm height) fitted with a ground glass Luer tip (1-mm i.d.) at one end and a fine porosity glass frit (10-20-μm pore size) just above the Luer tip. Typically, 10-120 beads were placed in the vessel, washed with water, pyridine, and a 2:1 (v/v) pyridine/water solution containing 0.1% triethylamine, and suspended in 160 μL of the latter solution. A 280-470 mM stock solution of Fmoc-Cl in pyridine (160 μL) was mixed with 1.8 μL (0.015 mmol) of PITC to obtain a final degradation reagent mixture containing 93 mM PITC and 280-470 mM Fmoc-Cl (PITC/Fmoc-Cl ratio of 1:3-5). An equal volume of this reagent mixture (160 μL) was added to the suspended beads and the reaction was allowed to proceed at room temperature for 20 min (with gentle shaking on a rotary mixer). The beads were drained, washed with pyridine (1.5 mL) and dichloromethane (3 × 1.5 mL), dried by suction, and treated twice with 1 mL of anhydrous TFA at room temperature (20 min each time). The beads were then washed with dichloromethane (3 × 1.5 mL) and pyridine (1.5 mL) and subjected to the next round of PED. After the final round of PED, the beads were treated twice with 1 mL of 20% piperidine in DMF solution at room temperature (10 min). The beads were suspended in 1 mL of anhydrous TFA containing 20 μL (0.27 mmol) of dimethyl sulfide and 25 mg (0.18 mmol) ammonium iodide and incubated on ice for 20 min to reduce any oxidized methionine. The beads were washed exhaustively with water and transferred to a Petri dish, where they were picked under a microscope and placed into separate microcentrifuge tubes.
For PED of peptide-peptoid hybrid sequences, the beads suspended in 160 μL of 2:1 (v/v) pyridine/water solution containing 0.1% triethylamine were first treated with a 30:1 (mol/mol) mixture of PITC/Fmoc-OSU solution. A 3.13 mM Fmoc-OSU stock solution in pyridine (160 μL) was mixed with 1.8 μL (0.015 mmol) of PITC to give a peptide degradation mixture containing 93 mM PITC and 3.1 mM Fmoc-OSU (PITC/Fmoc-OSU ratio of 30:1). An equal volume of this PITC/Fmoc-OSU mixture (160 μL) was added to the suspended beads and the degradation reaction was allowed to proceed at room temperature for 1 min with gentle mixing. The solution was drained, washed with pyridine (2 × 1 mL), and the beads were suspended in 160 μL of pyridine/water and treated with 160 μL of a peptoid degradation mixture containing 93 mM PITC and 464 mM Fmoc-Cl (PITC/Fmoc-Cl ratio of 1:5) at room temperature for 19 min. Following this step, the procedure is identical for that described for peptoid-only containing sequences.
MS Analysis
Microcentrifuge tubes containing individual beads were spun under vacuum in a SpeedVac concentrator to evaporate any residual water. Each bead was then treated with 20 μL of 40 mg/mL CNBr in 70% TFA in water for 14-18 h in the dark. The excess CNBr and TFA were removed under vacuum in a SpeedVac concentrator. The released peptoids were dissolved in 6 μL of 50:50:0.05 (v/v) acetonitrile/water/TFA. For MS analysis, 1 μL of the peptoid solution was mixed with 2 μL of a saturated solution of α-cyano-4-hydroxycinnamic acid in 50:50:0.05 (v/v) acetonitrile/water/TFA; 1 μL of the resulting mixture was applied to a 96- or 384-well sample plate. After drying, the samples were analyzed on a Bruker Reflex III MALDI-TOF mass spectrometer in the positive ion mode and automated format. Sequence assignment was made manually by examining the mass difference between two adjacent MS peaks.
Peptoid Library Screening
Library screening followed a previously reported procedure,22 but with the following modifications. In a Micro Bio-Spin column (1.0 mL, Bio-Rad), 30 mg of peptoid library II was swollen in dichloromethane and washed extensively with DMF and water. The resin was incubated in a blocking buffer (30 mM Hepes, pH 7.4, 150 mM NaCl, 0.05% Tween 20, and 0.1% gelatin) for 6 h with gentle mixing at 4 °C. The resin was drained and re-suspended in 800 μL of the blocking buffer containing 500 nM biotinylated K-Ras protein (as a glutathione-S-transferase fusion) and incubated overnight with gentle mixing at 4 °C. The resin was drained, re-suspended in the blocking buffer containing 1 μg/mL streptavidin-alkaline phosphatase (SA-AP), and incubated for 10 min at 4 °C with gentle mixing. The resin was again drained and washed with the blocking buffer (2 × 1 mL) and an SA-AP reaction buffer (30 mM Tris-HCl, pH 8.5, 100 mM NaCl, 5 mM MgCl2, 20 μM ZnCl2, and 0.05% Tween 20) (2 × 1 mL). The resin was transferred to a petri dish (60 × 15 mm) by using 2.7 mL (3 × 900 μL) of the SA-AP reaction buffer. Upon the addition of 300 μL of 5 mg/mL 5-bromo-4-chloro-3-indolyl phosphate, a turquoise color developed on positive beads in ∼40 min, when the staining reaction was terminated by the addition of 500 μL of 1 M HCl. The positive beads were picked manually with a pipet under a dissecting microscope.
Supplementary Material
Acknowledgment
This work was supported by the National Institutes of Health (GM062820 to D.P.) and portions of this work were performed at the Molecular Foundry, Lawrence Berkeley National Laboratory, which is supported by the Office of Science, Office of Basic Energy Sciences, U.S. Department of Energy, under Contract No. DE-AC02-05CH11231. A.T. was supported by an NIH Chemistry/Biology Interface training grant (T32 GM08512) and A.S.C. was supported by the Office of Naval Research (Grant No. 11398-23845-44-EKMAJ).
References and Notes
- 1).Patch JA, Kirshenbaum K, Seurynck SL, Zuckermann RN, Barron AE. Versatile Oligo(N-Substituted) Glycines: The Many Roles of Peptoids in Drug Discovery. In: Neilsen PE, editor. Pseudo-Peptides in Drug Discovery. Wiley-VCH Verlag; Weinheim, Germany: 2004. pp. 1–31. [Google Scholar]
- 2).Utku Y, Dehan E, Ouerfelli O, Piano F, Zuckermann RN, Pagano M, Kirshenbaum KA. Mol. BioSyst. 2006;2:312–317. doi: 10.1039/b603229j. [DOI] [PubMed] [Google Scholar]
- 3).Chongsiriwatana NP, Patch JA, Czyzewski AM, Dohm MT, Ivankin A, Gidalevitz D, Zuckermann RN, Barron AE. Proc. Natl. Acad. Sci. U. S. A. 2008;105:2794–2799. doi: 10.1073/pnas.0708254105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4).Brown NJ, Wu CW, Seurynck-Servoss SL, Barron AE. Biochemistry. 2008;47:1808–1818. doi: 10.1021/bi7021975. [DOI] [PubMed] [Google Scholar]
- 5).Zuckermann RN, Martin EJ, Spellmeyer DC, Stauber GB, Shoemaker KR, Kerr JM, Figliozzi GM, Goff DA, Siani MA, Simon RJ, Banville SC, Brown EG, Wang L, Richter LS, Moos WH. J. Med. Chem. 1994;37:2678–2685. doi: 10.1021/jm00043a007. [DOI] [PubMed] [Google Scholar]
- 6).Alluri PG, Reddy MM, Bachhawat-Sikder K, Olivos HJ, Kodadek T. J. Am. Chem. Soc. 2003;125:13995–14004. doi: 10.1021/ja036417x. [DOI] [PubMed] [Google Scholar]
- 7).Udugamasooriya DG, Dineen SP, Brekken RA, Kodadek T. J. Am. Chem. Soc. 2008;130:5744–5752. doi: 10.1021/ja711193x. [DOI] [PubMed] [Google Scholar]
- 8).Miller SM, Simon RJ, Ng S, Zuckermann RN, Kerr JM, Moos WH. Drug Dev. Res. 1995;35:20–32. [Google Scholar]
- 9).Tan NC, Yu P, Kwon Y-U, Kodadek T. Bioorg. Med. Chem. 2008;16:5853–5861. doi: 10.1016/j.bmc.2008.04.074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10).Zuckermann RN, Kerr JM, Kent SBH, Moos WH. J. Am. Chem. Soc. 1992;114:10646–10647. [Google Scholar]
- 11).Lam KS, Salmon SE, Hersh EM, Hruby VJ, Kazmierski WM, Knapp RJ. Nature. 1991;354:82–84. doi: 10.1038/354082a0. [DOI] [PubMed] [Google Scholar]
- 12).Houghten RA, Pinilla C, Blondelle SE, Appel JR, Dooley CT, Cuervo JH. Nature. 1991;354:84–86. doi: 10.1038/354084a0. [DOI] [PubMed] [Google Scholar]
- 13).Furka A, Sebestyen F, Asgedom M, Dibo G. Int. J. Pept. Protein Res. 1991;37:487–493. doi: 10.1111/j.1399-3011.1991.tb00765.x. [DOI] [PubMed] [Google Scholar]
- 14).Boeijen A, Liskamp RMJ. Tetrahedron Lett. 1998;39:3589–3592. [Google Scholar]
- 15).Heerma W, Versluis C, de Koster CG, Kruijtzer JAW, Zigrovic I, Liskamp RMJ. Rapid Commun. Mass Spectrom. 1996;10:459–464. doi: 10.1002/(SICI)1097-0231(19960315)10:4<459::AID-RCM501>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- 16).Heerma W, Boon J-PJL, Versluis C, Kruijtzer JAW, Hofmeyer LJF, Liskamp RMJ. J. Mass Spectrom. 1997;32:697–704. [Google Scholar]
- 17).Ruijtenbeek R, Versluis C, Heck AJR, Redegeld FAM, Nijkamp FP, Liskamp RMJ. J. Mass Spectrom. 2002;37:47–55. doi: 10.1002/jms.245. [DOI] [PubMed] [Google Scholar]
- 18).Paulick MG, Hart KM, Brinner KM, Tjandra M, Charych DH, Zuckermann RN. J. Comb. Chem. 2006;8:417–426. doi: 10.1021/cc0501460. [DOI] [PubMed] [Google Scholar]
- 19).Wang P, Arabaci G, Pei D. J. Comb. Chem. 2001;3:251–254. doi: 10.1021/cc000102l. [DOI] [PubMed] [Google Scholar]
- 20).Sweeney MC, Pei D. J. Comb. Chem. 2003;5:218–222. doi: 10.1021/cc020113+. [DOI] [PubMed] [Google Scholar]
- 21).Thakkar A, Wavreille A-S, Pei D. Anal. Chem. 2006;78:5935–5939. doi: 10.1021/ac0607414. [DOI] [PubMed] [Google Scholar]
- 22).Sweeney MC, Wavreille A-S, Park J, Butchar J, Tridandapani S, Pei D. Biochemistry. 2005;44:14932–14947. doi: 10.1021/bi051408h. [DOI] [PubMed] [Google Scholar]
- 23).Joo SH, Xiao Q, Ling Y, Gopishetty B, Pei D. J. Am. Chem. Soc. 2006;128:13000–13009. doi: 10.1021/ja063722k. [DOI] [PubMed] [Google Scholar]
- 24).Garaud M, Pei D. J. Am. Chem. Soc. 2007;129:5366–5367. doi: 10.1021/ja071275i. [DOI] [PubMed] [Google Scholar]
- 25).Wavreille A-S, Garaud M, Zhang Y, Pei D. Methods. 2007;42:207–219. doi: 10.1016/j.ymeth.2007.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26).Alvarado Y, Giles FJ. Expert Opin. Emerg. Drugs. 2007;12:271–284. doi: 10.1517/14728214.12.2.271. [DOI] [PubMed] [Google Scholar]
- 27).Zuckermann RN, Kerr JM, Siani MA, Banville SC. Int. J. Peptide Protein Res. 1992;40:497–506. doi: 10.1111/j.1399-3011.1992.tb00433.x. [DOI] [PubMed] [Google Scholar]
- 28).Kaiser E, Colescott RL, Bossinger CD, Cook PI. Anal. Biochem. 1970;34:595–598. doi: 10.1016/0003-2697(70)90146-6. [DOI] [PubMed] [Google Scholar]
- 29).Vojkovsky T. Pept. Res. 1995;8:236–237. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.