

Abstract
The Ala1—Gly28 glycopeptide fragment (28) of EPO was prepared by chemical synthesis as a single glycoform. Key steps in the synthesis include attachment of a complex dodecasaccharide (7) to a seven amino acid peptide via Lansbury aspartylation, native chemical ligation to join peptide 19 with the glycopeptide domain 18, and a selective desulfurization at the ligation site to reveal the natural Ala19. This glycopeptide fragment (28) contains both the requisite N-linked dodecasaccharide and a C-terminal αthioester handle, the latter feature permitting direct coupling with a glycopeptide fragment bearing N-terminal Cys29 without further functionalization.
Introduction
As a part of our continuing effort to bring chemical synthesis to the realm of “biologics”, we are pursuing the preparation of homogeneous glycoproteins.1,2 In contrast to biochemical methods that do not allow for homogenous expression of glycoproteins, de novo chemical synthesis offers precise structural control for the preparation of homogeneous products while potentially deconvoluting key structure—function relationships of such complex structures. Central to our efforts to apply chemical synthesis to the preparation of “biologics” is our proposed total synthesis of erythropoietin.
Erythropoietin (EPO, 1, Figure 1) is a glycoprotein hormone used to treat anemia associated with renal failure and cancer chemotherapy.3 Extensive efforts have been made to study and understand the structure and function of EPO, including the role of glycosylation. Specifically, Higuchi et al. demonstrated that erythropoietin’s (rhuEPO) three N-linked glycosyl groups were not required for in vitro activity but were required for in vivo activity, while the single O-linked glycosyl group did not bear any biological role.4 Furthermore, the in vivo activity of EPO was shown to be directly related to the sialic acid content.5 Interestingly, Kent and coworkers have synthesized a polymer-modified EPO that demonstrated superior in vivo activity compared to the glycosylated rhuEPO.6 Despite the impressive biological and chemical studies that have been made to understand the structure and function of EPO, efforts to determine the exact biological role of defined EPO glycoforms have been hindered by difficulties associated with isolating significant quantities of homogeneous glycoforms.7 In fact, this has not been accomplished by any biologically enabled means. As discussed in the preceeding papers,1 a potentially powerful solution to this problem is chemical synthesis, which would potentially enable access to homogeneous EPO glycoforms and related analogues.
Figure 1.

Retrosynthetic analysis and ribbon diagram of Erythropoietin (EPO, 1).
Chemical synthesis of glycoproteins or glycopeptides bearing complex carbohydrates has only recently been realized. This is in sharp contrast to the plethora of non-glycosylated proteins that have been prepared, aided by the power of native chemical ligation8,9 in polypeptide synthesis. Using native chemical ligation (NCL), Kajihara and Dawson reported the first synthesis of a complex glycoprotein, i.e., a single glycoform of monocyte chemotactic protein-3 containing human complex sialyloligosaccharide.10 Erythropoietin glycopeptide fragments containing complex sialyloligosaccharides have been prepared by our group1,2d using either NCL or direct condensation methods.2f,11 Recently, an asialo erythropoietin glycopeptide fragment was reported by Kajihara,12 in which a serine ligation was effectively employed. Clearly, the chemical synthesis of complex glycoproteins and glycopeptides remains a significant and daunting challenge.
Our program directed to reaching EPO by total synthesis has already yielded valuable contributions to the rapidly growing field of (glyco)peptide ligation tools and methods. Several different C-terminal functional groups have been employed in our glycoprotein synthesis studies, including cyanophenolic esters1b and nitrophenolic esters.2h Our ortho-disulfide phenolic ester2i was developed to serve as a stable, latent thioester. These C-terminal groups have enabled orthogonal ligations such that multiple polypeptide and glycopeptide couplings can be accomplished from the N→C terminus.1b,2f Our investigations into cysteine-free ligation methods have included the development of a thiol auxiliary that permits coupling of complex glycopeptide fragments,2c,13 the discovery of a two-component isonitrile/carboxylic acid coupling method to construct amide bonds,14 and alanine,2g,15 valine,16,17 and homocysteine18,19 ligations, which utilize a mild and selective desulfurization method that is also compatible with complex carbohydrates.20 The synthesis of homogeneous glycosylated EPO(1-28) (2, Figure 2), described below, serves to illustrate the value of “alanine” ligations.
Figure 2.

Structure of EPO 1-28 (2).
Results and Discussion
Our vision for the assembly of erythropoietin emphasizes maximum convergency. It projects the synthesis of three glycopeptide fragments that will subsequently be merged. Indeed the syntheses of EPO(78-166) and EPO(29-77) have been described earlier in this series.1 In each case we were able to overcome potentially serious complications inherent in those domains. The EPO(1-28) segment is the shortest of the three peptide fragments, containing less than 20% of the EPO sequence. As the smallest fragment and (potentially) the last fragment in the synthesis of EPO (assuming a linear synthesis from the C→N-terminus), it may appear to be the simplest of the segments. However, the inherent challenge of the EPO(1–28) segment lies in the absence of any functional cysteine and glycine/proline residues upon which to base a retrosynthetic analysis. This peculiarity precluded the use of either NCL or direct condensation methods21 for its assembly. This is critical because during our initial studies, and as reported in the preceeding paper,1a we found that the direct attachment of dodecasaccharide 722 to Asp24 of the EPO(1–28) peptide sequence 323 via Lansbury aspartylation24 was unsuccessful, providing only aspartimide by-products. In contrast, the joining of disaccharide 5 and hexasaccharide 6 to the 28-residue peptide 3 by Lansbury aspartylation proceeded in 70% and 30% yields, respectively. Such unacceptably poor reactivity must surely be the consequence of the increased steric bulk presented by dodecasaccharide 7, rendering aspartimide formation not only kinetically competitive (as with 6), but dominant.
Recognizing the need to employ a smaller peptide to permit functional aspartylations of larger glycosylamines, we revised our plan for the synthesis of the EPO(1–28) fragment to include a strategic dipeptide scission between Ala19 and Lys20. We first favored this disconnection with the goal of applying our TCEP-assisted phenolic ester-directed amide coupling method1f to assemble the fragment, despite concerns about the potential for epimerization at Ala19. Under this direction, the protected Lys20-Gly28 sequence (9) was prepared and effectively joined with dodecasaccharide 7 by Lansbury aspartylation to provide, after Fmoc removal, our target fragment 11 (35%). Efforts to ligate the FmocHN-Ala1-Ala19-CO2Ph(o-SSEt) fragment (13) with glycopeptide 11 using our direct condensation method proceeded successfully to give 12, but the stereo-integrity of the alanine ligation site (see asterisk) could not be verified.
One way to determine whether the condensation product (12) suffered from epimerization during the key peptide coupling is to compare the retention times of 12 and the corresponding D-Ala19 diastereomer. Of course, we recognized the possibility that the two diastereomers might co-elute. Following the same sequence of steps as shown in Scheme 2, and using the D-Ala19 peptide 29, the diasteroemer 30 was prepared (Scheme 3). Co-injection of the two glycopeptides yielded a single peak, offering no resolution. Increasing the concentration of the D-Ala19 diastereomer did not change the peak shape. Despite these efforts, the stereo-integrity of the alanine ligation site remained uncertain.25
Scheme 2.

a Synthesis of 12 via TCEP-assisted phenolic ester-directed amide coupling
a Reagents and conditions: (a) ethyl thiopropionate, EDCI, HOBt, DMF, 91%; (b) 88% TFA/CH2Cl2(1:2), 5% H2O, 5% phenol, 2% iPr3SiH, 23%; (c) HATU, iPr2NEt, DMSO, glycan 7; (d) piperidine, 35% over 2 steps; (e) 13, HCl·P(CH2CH2CO2H)3, HOOBt, 2,6-di-tBu-DMAP, DMSO.
Scheme 3.

a Synthesis of D-Ala19 diastereomer of EPO(1-28)
Given the knowledge that the attachment of dodecasaccharide 7 to a shorter sequence was successful, we turned to an alternative approach to address the issue of stereo-integrity. As stated earlier, the absence of any useful cysteine and glycine/proline residues precluded the use of either NCL or direct condensation methods. However, it was noted that several alanine residues are present in close proximity to Asp24, the site for glycosyl attachment. An established extension of NCL has been the conversion of cysteine to alanine via desulfurization,12a,b which enables ligation at alanine sites with the benefits of NCL (e.g., avoidance of epimerization, chemoselectivity). Recently, our laboratory had developed a desulfurization method that is compatible with oligosaccharides and is both extremely mild and selective.1g We anticipated that the versatility of this method would readily accommodate the different functional groups present within EPO(1–28), thus enabling the use of NCL to address our earlier problems in the synthesis of the EPO(1–28) fragment.
The EPO(1-28) fragment contains two alanine residues (Ala19 and Ala22) in close proximity to the N-glycan, either of which could serve as the ligation site. We elected to implement a ligation between Glu21 and Ala22 to obtain a shorter (glyco)peptide segment that would be more suitable for the essential Lansbury aspartylation as it is (1) smaller in size and (2) free of Lys20 and Glu21, the side chains of which would necessarily be protected during the aspartylation reaction. This disconnection also yields the longer 21-amino acid peptide terminating at Glu21. It should be noted that Botti has demonstrated that NCL at C-terminal glutamates and aspartates requires the side chains to be protected to avoid formation of the unnatural γ-amide bond during ligation.26 While inconvenient, a glutamate protecting group would be necessary regardless of which disconnection was selected.
The requisite glycopeptide segment 18 was prepared, as shown in Scheme 4. Starting from peptide 14 (95% yield via solid phase peptide synthesis), condensation with ethyl thiopropionate followed by removal of the t-butyl groups provided thioester 15. Attachment of dodecasaccharide 7 to peptide 15 via Lansbury aspartylation followed by in situ Fmoc cleavage proceeded in moderate yield to afford glycopeptide 17 in 65% yield over the two-step sequence. Finally, deallylation with Pd(PPh3)4 and PhSiH3 afforded the NCL partner 18 in 90% yield.
Scheme 4.

a Synthesis of glycopeptide 18
a Reagents and conditions: (a) ethyl thiopropionate, EDCI, HOBt, DMF; (b) 88% TFA, 5% H2O, 5% phenol, 2% iPr3SiH, 45% over two steps; (c) HATU, iPr2NEt, DMSO, glycan 7; (d) piperidine, 65% over two steps; (e) Pd(PPh3)4, PhSiH3, DMSO, 90%.
Under NCL conditions, glycopeptide 18 was coupled with the longer peptide segment 1927 to yield the desired ligation product 20, as well as the corresponding thiolactone, 21 (Scheme 5). Allowing the reaction to stir longer resulted in what was thought to be exclusive formation of the thiolactone (vide infra), which was isolated in 20% yield. Following ligation, the allyl protecting group on Glu21 was removed to avoid complications during the critical desulfurization.
Scheme 5.

a Native chemical ligation between 18 and 19
a Reagents and conditions: (a) 6M Guanidine·HCl, Na2HPO4, TCEP·HCl, TCEP, thiopropionic acid, 20%; (b) Pd(PPh3)4, PhSiH3, DMSO, 90%.
Before desulfurization could be attempted, it was necessary to free the cysteine sidechain from the thiolactone. Treatment of 22 with thiopropionic acid effectively opened the thiolactone to afford the acyclic 23 (Scheme 6A); however, a by-product (24), identified as EPO(1-21)COS(CH2)2CO2H, was also formed during the reaction. There are several possible explanations for the formation of this product. One possibility is that during the NCL reaction between 19 and 18, a competitive intramolecular NCL within glycopeptide 18 had led to lactam 26,28 which subsequently underwent a transthioesterification with 19 to afford thioester 27 (Scheme 6B). Following removal of the allyl side chain group, the αthioester bond was readily cleaved in the presence of thiopropionic acid to give the observed by-product. Alternatively, it is possible, though less likely, that under the mildly acidic conditions used to open the thiolactone, the Glu21-Cys22 amide bond underwent an N→S acyl tranfer to generate the corresponding αthioester, which was subsequently cleaved to give the observed αthiopropionate ester.
Scheme 6.

A: Thiolactone opening; B: Possible origin of observed by-product from thiolactone opening
Despite the loss of material, we were ready to test glycopeptide 23 in the key desulfurization reaction (Scheme 7). Treatment of 23 with VA-044 (a water soluble radical initiator), TCEP, and thiopropionic acid in buffered conditions at 37 °C cleanly afforded the reduced product 28 (67% yield). The use of thiopropionic acid as the radical propagator also served to open any thiolactone that formed during the reaction. The final product, 28, features both the biantennary glycan and a C-terminal αthioester, two critical features necessary for the convergent preparation of synthetic homogeneous EPO.
Scheme 7.
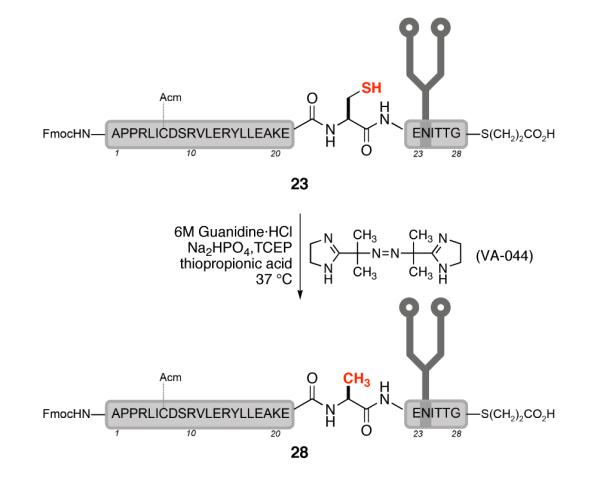
Key desulfurization step.
Conclusion
As described above, we have prepared the Ala1—Gly28 glycopeptide fragment (28) of EPO by chemical synthesis. Key steps in the synthesis included attachment of a complex dodecasaccharide (7) to a seven amino acid peptide via Lansbury aspartylation, native chemical ligation to join peptide 19 with the glycopeptide domain 18, and a selective desulfurization at the ligation site to expose the natural Ala19. This fragment presents both the requisite N-linked dodecasaccharide and a C-terminal αthioester handle, the latter feature permitting direct coupling with a glycopeptide fragment bearing N-terminal Cys29 without further functionalization.
In summary the preparation of this Ala1-Gly28, in the context of the accompanying reports, describing the syntheses of EPO(29-77), featuring the N-linked glycan, and EPO(78-166), presenting both the N-linked glycan and the O-linked glycophorin, suggests that the realization of our ultimate goal, i.e., biologically active homogeneous synthetic erythropoietin, is within reach. The assembly of these three fragments, corresponding to a full length homogenous erythropoietin, represents the closest and most convergent approach to the chemical synthesis of a single EPO glycoform. The primary drawback in our synthetic efforts is the limited availability of the complex dodecasaccharide 7. We note that much of the difficulty arises from the lack of commercial availability of the key building blocks. This situation could well change as the role of oligosaccharide chemistry in the synthesis of biologics becomes better understood.
We also think that the new amide bond forming chemistry delineated in this project, and summarized in the background section, could well allow for the combination of these fragments, recognizing that, with all complex target oriented total synthesis, intervention of the unexpected is predictable. That being said, we are hopeful that the convergent nature of our synthesis and its flexibility will enable its adaptation to reach our goals.
Supplementary Material
Scheme 1.

a Initial Lansbury aspartylation studies
a Reagents and conditions: (a) HATU, iPr2NEt, DMSO, with glycan 5, 70%; with glycan 6, 30%; with glycan 7, 0%.
Acknowledgment
This work was supported by the National Institutes of Health (Grant CA28824). Postdoctoral fellowship support is gratefully acknowledged by C.K. (Grant Number T32 CA062948 from the National Cancer Institute), by B.W. (New York State Department of Health, New York State Breast Cancer Research and Education Fund), and by Q.W. (Mr. William H. Goodwin and Mrs. Alice Goodwin and the Commonwealth Foundation for Cancer Research, and the Experimental Therapeutics Center, SKI). We thank Dr. George Sukenick, Ms. Sylvi Rusli, and Ms. Hui Fang of the Sloan-Kettering Institute’s NMR core facility for mass spectral and NMR spectroscopic analysis (SKI core grant no. CA02848). We thank Ms. Rebecca Wilson for editorial counsel.
Footnotes
Supporting Information Available. Experimental procedures and spectroscopic and analytical data for all new compounds. This material is available free of charge via the internet at http://pubs.acs.org.
References
- (1)(a).See preceding two papers: Tan Z, Shang S, Halkina T, Yuan Y, Danishefsky SJ. J. Am. Chem. Soc. 2009;131 doi: 10.1021/ja808704m.Yuan Y, Chen J, Wan Q, Tan Z, Chen G, Kan C, Danishefsky SJ. J. Am. Chem. Soc. 2009;131 doi: 10.1021/ja808705v.
- (2)(a).Wang Z-G, Zhang X, Visser M, Live D, Zatorski A, Iserloh U, Lloyd KO, Danishefsky SJ. Angew. Chem. Int. Ed. 2001;40:1728–1732. [PubMed] [Google Scholar]; (b) Miller JS, Dudkin VY, Lyon GJ, Muir TW, Danishefsky SJ. Angew. Chem. Int. Ed. 2003;42:431–434. doi: 10.1002/anie.200390131. [DOI] [PubMed] [Google Scholar]; (c) Wu B, Chen J, Warren JD, Chen G, Hua Z, Danishefsky SJ. Angew. Chem. Int. Ed. 2006;45:4116–4125. doi: 10.1002/anie.200600538. [DOI] [PubMed] [Google Scholar]; (d) Wu B, Tan Z, Chen G, Chen J, Hua Z, Wan Q, Ranganathan K, Danishefsky SJ. Tetrahedron Lett. 2006;47:8009–8011. doi: 10.1016/j.tetlet.2006.09.045. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Chen J, Chen G, Wu B, Wan Q, Tan Z, Hua Z, Danishefsky SJ. Tetrahedron Lett. 2006;47:8013–8016. doi: 10.1016/j.tetlet.2006.09.045. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Chen G, Wan Q, Tan Z, Kan C, Hua Z, Ranganathan K, Danishefsky SJ. Angew. Chem. Int. Ed. 2007;46:7383–7387. doi: 10.1002/anie.200702865. [DOI] [PubMed] [Google Scholar]; (g) Wan Q, Danishefsky SJ. Angew. Chem. Int. Ed. 2007;46:9248–9252. doi: 10.1002/anie.200704195. [DOI] [PubMed] [Google Scholar]; (h) Wan Q, Chen J, Yuan Y, Danishefsky SJ. J. Am. Chem. Soc. 2008;130:15814–15816. doi: 10.1021/ja804993y. [DOI] [PMC free article] [PubMed] [Google Scholar]; (i) Warren JD, Miller JS, Keding SJ, Danishefsky SJ. J. Am. Chem. Soc. 2004;126:6576–6578. doi: 10.1021/ja0491836. [DOI] [PubMed] [Google Scholar]; (j) Wilson RM, Danishefsky SJ. Pure Appl. Chem. 2007;79:2189–2216. [Google Scholar]
- (3)(a).For general reviews on EPO, see: Sytkowski AJ. Erythropoietin. Wiley-VCH Verlag GmbH & Co. KGaA; Weinheim: 2004. Jelkmann W. Intern. Med. 2004;43:649–659. doi: 10.2169/internalmedicine.43.649.
- (4).Higuchi M, Masayoshi O, Kuboniwa H, Tomonoh K, Shimonaka Y, Ochi N. J. Biol. Chem. 1992;267:7703–7709. [PubMed] [Google Scholar]
- (5)(a).Egrie JC, Grant JR, Gillies DK, Aoki KH, Strickland TW. Glycoconjugate J. 1993;10:263. [Google Scholar]; (b) Egrie JC, Browne JK. Br. J. Cancer. 2001;84(S1):3–10. doi: 10.1054/bjoc.2001.1746. and references therein. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Kochendoerfer GG, Chen S-Y, Mao F, Cressman S, Traviglia S, Shao H, Hunter CL, Low DW, Cagle EN, Carnevali M, Gueriguian V, Keogh PJ, Porter H, Stratton SM, Wiedeke MC, Wilken J, Tang J, Levy JJ, Miranda LP, Crnogorac MM, Kalbag S, Botti P, Schindler-Horvat J, Savatski L, Adamson JW, Kung A, Kent SBH, Bradburne JA. Science. 2003;299:884–887. doi: 10.1126/science.1079085. [DOI] [PubMed] [Google Scholar]
- (7)(a).Kornfeld R, Kornfeld S. Annu. Rev. Biochem. 1985;54:631–664. doi: 10.1146/annurev.bi.54.070185.003215. [DOI] [PubMed] [Google Scholar]; (b) Roth J. Chem. Rev. 2002;102:285–303. doi: 10.1021/cr000423j. [DOI] [PubMed] [Google Scholar]; (c) Rudd PM, Dwek RA. Crit. Rev. Biochem. Mol. Biol. 1997;32:1–100. doi: 10.3109/10409239709085144. [DOI] [PubMed] [Google Scholar]
- (8).Dawson PE, Muir TW, Clark-Lewis I, Kent SBH. Science. 1994;266:776–779. doi: 10.1126/science.7973629. [DOI] [PubMed] [Google Scholar]
- (9)(a).For the first example of glycoprotein chemical synthesis using NCL, see: Shin Y, Winans KA, Backes BJ, Kent SBH, Ellman JA, Bertozzi CR. J. Am. Chem. Soc. 1999;121:11684–11689.For recent examples of protein synthesis using NCL, see: Torbeev VY, Kent SBH. Angew. Chem. Int. Ed. 2007;46:1667–1670. doi: 10.1002/anie.200604087.Durek T, Torbeev VY, Kent SBH. Proc. Natl. Acad. Sci. U.S.A. 2007;104:4846–64851. doi: 10.1073/pnas.0610630104.For a review on NCL, see Dawson PE, Kent SBH. Annu. Rev. Biochem. 2000;69:923–960. doi: 10.1146/annurev.biochem.69.1.923.
- (10).Note that the sialyloligosaccharide was isolated from hen’s egg yolk and does not contain any fucose unit. Yamamoto N, Tanabe Y, Okamoto R, Dawson PE, Kajihara Y. J. Am. Chem. Soc. 2008;130:501–510. doi: 10.1021/ja072543f.
- (11)(a).For other direct condensation methods, see: Blake J. Int. J. Pept. Protein Res. 1981;17:273–274. doi: 10.1111/j.1399-3011.1981.tb01992.x.Aimoto S, Mizoguchi N, Hojo H, Yoshimura S. Bull. Chem. Soc. Jpn. 1989;62:524–531.Aimoto S. Biopolymers. 1999;51:247–265. doi: 10.1002/(SICI)1097-0282(1999)51:4<247::AID-BIP2>3.0.CO;2-W.Payne RJ, Ficht S, Greenberg WA, Wong C-H. Angew. Chem. Int. Ed. 2008;47:4411–4415. doi: 10.1002/anie.200705298.
- (12).Okamoto R, Kajihara Y. Angew. Chem. Int. Ed. 2008;47:5402–5406. doi: 10.1002/anie.200801097. [DOI] [PubMed] [Google Scholar]
- (13)(a).For other examples of auxiliary-based cysteine-free ligations, see: Canne LE, Bark SJ, Kent SBH. J. Am. Chem. Soc. 1996;118:5891–5896.Botti P, Carrasco MR, Kent SBH. Tetrahedron Lett. 2001:1831–1833.Marinzi C, Bark SJ, Offer J, Dawson PE. Bioorg. Med. Chem. 2001;9:2323–2328. doi: 10.1016/s0968-0896(01)00136-5.Low DW, Hill MG, Carrasco MR, Kent SBH, Botti P. Proc. Natl. Acad. Sci. U.S.A. 2001;98:6554–6559. doi: 10.1073/pnas.121178598.Offer J, Boddy CNC, Dawson PE. J. Am. Chem. Soc. 2002;124:4642–4646. doi: 10.1021/ja016731w.Macmillan D, Anderson DW. Org. Lett. 2004;6:4659–4662. doi: 10.1021/ol048145o.Payne RJ, Ficht S, Tang S, Brik A, Yang Y-Y, Case DA, Wong C-H. J. Am. Chem. Soc. 2007;129:13527–13536. doi: 10.1021/ja073653p. and references therein.
- (14).Li X, Danishefsky SJ. J. Am. Chem. Soc. 2008;130:5446–5448. doi: 10.1021/ja800612r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15)(a).For other studies on alanine ligations, see: Yan LZ, Dawson PE. J. Am. Chem. Soc. 2001;123:526–533. doi: 10.1021/ja003265m. and references therein.Pentelute B, Kent SBH. Org. Lett. 2007;9:687–690. doi: 10.1021/ol0630144.
- (16).Chen J, Wan Q, Yuan Y, Zhu J, Danishefsky SJ. Angew. Chem. Int. Ed. 2008;47:8521–8524. doi: 10.1002/anie.200803523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).For other studies on valine ligations, see: Haase C, Rohde H, Seitz O. Angew. Chem. Int. Ed. 2008;47:6807–6810. doi: 10.1002/anie.200801590.
- (18).Unpublished results.
- (19).For other studies on homocysteine ligations, see: Tam JP, Yu Q. Biopolymers. 1998;46:5402–5406. doi: 10.1002/(SICI)1097-0282(19981015)46:5<319::AID-BIP3>3.0.CO;2-S.
- (20)(a).In addition to alanine, valine, and homocysteine ligations, a phenylalanine ligation has also been reported. See: Botti P, Tchertchian S. 2006. WO 133962.Crich D, Banerjee A. J. Am. Chem. Soc. 2007;129:10064–10065. doi: 10.1021/ja072804l.
- (21).Following completion of our studies, C.-H. Wong and co-workers published a direct aminolysis method that could, in theory, be applied to the synthesis of our glycopeptide fragment. See reference 11d.
- (22).Wu B, Hua Z, Warren JD, Ranganathan K, Wan Q, Chen G, Tan Z, Chen J, Endo A, Danishefsky SJ. Tetrahedron Lett. 2006;47:5577–5579. doi: 10.1016/j.tetlet.2006.09.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).The synthesis of the EPO(1–28) peptide via solid phase peptide synthesis (SPPS) required the use of a commercially available pseudoproline-protected dipeptide for residues Asp8 and Ser9 (Fmoc-Asp(OtBu)-Ser (ψMe,Mepro)-OH) to avoid unwanted aspartimide formation.
- (24)(a).Anisfeld ST, Lansbury PT., Jr. J. Org. Chem. 1990;55:5560–5562. [Google Scholar]; (b) Cohen-Anisfeld ST, Lansbury PT., Jr. J. Am. Chem. Soc. 1993;115:10531–10537. [Google Scholar]
- (25).Alternatively, trypsin could be used to provide the short peptide fragment YLLEAK, which should be readily resolved if Ala19 has epimerized. While we cannot show that Ala19 has not epimerized, model studies with the corresponding disaccharide substrates indicate formation of a separable mixture of diastereomers in a four to one ratio.
- (26).Villain M, Gaertner H, Botti P. Eur. J. Org. Chem. 2003:3267–3272. [Google Scholar]
- (27).Peptide 19 was prepared in two steps from 33, see Supporting Information for synthesis.
- (28)(a).Other examples of competitive intramolecular NCL have been observed. See: Bang D, Pentelute BL, Kent SBH. Angew. Chem. Int. Ed. 2006;45:3985–3988. doi: 10.1002/anie.200600702.Torbeev VY, Kent SBH. Angew. Chem. Int. Ed. 2007;46:1667–1670. doi: 10.1002/anie.200604087.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.