

Abstract
Background and purpose:
Combining statin and fibrate in clinical practice provides a greater reduction of triglycerides than either drug given alone, but the mechanism for this effect is poorly understood. Apolipoprotein AV (apoAV) has been implicated in triglyceride metabolism. This study was designed to investigate the effect of the combination of statin and fibrate on apoAV and the underlying mechanism(s).
Experimental approach:
Hypertriglyceridaemia was induced in rats by giving them 10% fructose in drinking water for 2 weeks. They were then treated with atorvastatin, fenofibrate or the two agents combined for 4 weeks, and plasma triglyceride and apoAV measured. We also tested the effects of these two agents on triglycerides and apoAV in HepG2 cells in culture. Western blot and reverse transcription polymerase chain reaction was used to measure apoAV and peroxisome proliferator-activated receptor-α (PPARα) expression.
Key results:
The combination of atorvastatin and fenofibrate resulted in a greater decrease in plasma triglycerides and a greater increase in plasma and hepatic apoAV than either agent given alone. Hepatic expression of the PPARα was also more extensively up-regulated in rats treated with the combination. A similar, greater increase in apoAV and a greater decrease in triglycerides were observed following treatment of HepG2 cells pre-exposed to fructose), with the combination. Adding an inhibitor of PPARα (MK886) abolished the effects of atorvastatin on HepG2 cells.
Conclusions and implications:
A combination of atorvastatin and fenofibrate increased apoAV and decreased triglycerides through up-regulation of PPARα.
Keywords: apolipoprotein AV, statin and fibrate, triglyceride, PPARα
Introduction
Elevated plasma triglycerides are acknowledged as an independent risk factor for cardiovascular disease (Criqui et al., 1993). In the presence of severe hypertriglyceridemia or mixed hyperlipidemia, the combination of statins and fibrates is a possible treatment (Miller et al., 2008), as this combination induces a greater reduction of triglycerides (Ooi et al., 1997). However, the mechanism(s) for the greater hypotriglyceridemic effect remains to be clarified.
The recently identified apolipoprotein AV (apoAV) is implicated in triglyceride metabolism (Pennacchio et al., 2001; Van der Vliet et al., 2001). Deficiency of the apoAV gene results in an increase of plasma triglycerides in mice, whereas its overexpression leads to a reduction of triglycerides (Pennacchio et al., 2001; Van der Vliet et al., 2001). Similarly, inherited human apoAV deficiency contributes to severe hypertriglyceridemia in the affected subjects (Priore Oliva et al., 2005).
Fibrates are a class of lipid-lowering drug, especially effective in decreasing triglycerides. Their hypotriglyceridemic action involves reduced hepatic triglyceride synthesis and increased circulating triglyceride clearance (Pennacchio et al., 2002). Of note, recent data showed that fibrates induce apoAV expression by activating the peroxisome proliferator-activated receptor-α (PPARα) (Prieur et al., 2003; Vu-Dac et al., 2003). Interestingly, the hypotriglyceridemic effect of statins is also associated with up-regulation of PPARα (Roglans et al., 2002; Landrier et al., 2004; Paumelle and Staels, 2007).
As the hypotriglyceridemic effects of both the fibrates and the statins involve the PPARα signal pathway, this study was designed to investigate the effects of a statin–fibrate combination on apoAV in hypertriglyceridemic rats and the underlying mechanism(s).
Methods
Animals and experimental design
All animal care and procedures were conducted in accordance with the principles of the local Committee of Animal Experimentation. The animal experiments were performed as described previously by Roglans et al. (2002), with modifications. Forty 8-week-old male Sprague-Dawley rats (Shanghai Slac, Shanghai, China) were randomized into five groups (n= 8 each group): control, fructose, atorvastatin, fenofibrate and combination (atorvastatin plus fenofibrate) groups. For the fructose group and the three drug-treated groups, we first established the hypertriglyceridemic animal model with fructose supplied as a 10% solution in drinking water for 2 weeks. Then, the different treatments were given to these four groups as follows: (i) the fructose group continued on 10% fructose in drinking water for 4 weeks; and (ii) the three drug groups were given the 10% fructose in drinking water plus either atorvastatin (Pfizer, New York, NY, USA; 10 mg·kg−1·d−1), fenofibrate (Sigma, St. Louis, MO, USA; 100 mg·kg−1·d−1) or the two drugs together at the same doses (combined treatment), by daily oral gavage for 4 weeks. At the end of 4 weeks, the animals were killed by decapitation under anesthesia with sodium pentobarbital (50 mg·kg−1, i.p.). Blood samples were collected at death in 5% ethylenediamine tetraacetic acid and stored at −80 °C until analysis. Plasma apoAV was measured by an ELISA method (Santa Cruz, CA, USA). Liver samples were immediately frozen and stored at −80°C until needed.
Cell experiment
The cell experiments were performed as described previously (Wilcox et al., 1999), with the following modifications. All chemicals were dissolved in dimethyl sulphoxide (DMSO), and treatments were added directly to cell culture media, and the final concentration of DMSO in media was maintained at 0.1% (v/v). HepG2 cells were divided into six groups: (i) control group, treated with 0.1% DMSO alone; (ii) fructose group, with 100 µM fructose; (iii) atorvastatin group, with 100 µM atorvastatin and 100 µM fructose; (iv) fenofibrate group, with 100 µM atorvastatin and 100 µM fructose; (v) atorvastatin plus fenofibrate group, with both drugs and 100 µM fructose; and (vi) atorvastatin plus MK886, 100 µM atorvastatin and 10 µM MK886 (a selective inhibitor of PPARα, Wako Pure Chemicals, Osaka, Japan) and 100 µM fructose. After 24 h incubation, the cells were washed, and cellular triglycerides were measured using enzymatic reagents (Boehringer Mannheim, Indianapolis, IN, USA) as described previously (Wilcox et al., 1999). Cellular triglyceride results were reported as µg cellular triglycerides (mg cell protein)−1. Expression of apoAV and PPARα were determined by RT-PCR and western blot analysis.
Reverse transcription polymerase chain reaction (RT-PCR) analysis
The expressions of apoAV and PPARα mRNAs were assessed by RT-PCR. All RT-PCR buffers and reagents used from a reverse transcription system kit (A3500, Promega, Madison, WI, USA), and all procedures were performed in a 2720 Thermal Cycler (Applied Biosystems, Framingham, MA, USA) according to the product instructions. The sequences of the sense and antisense primers used for amplification were shown in Table 1. The GAPDH was used as internal control in the PCR reaction. At the end of RT-PCR, 5 µL of each PCR sample was analysed by electrophoresis on an 1% agarose gel containing nucleic acid stain (GoldView, Mannheim, Germany), and was scanned by Bio-Rad Gel Doc 2000 Imaging System (Hercules, CA, USA) and analyzed for optical density through Quantity One 4.03 analysis software (Bio-Rad, Hercules, CA, USA). The results for APOAV or PPARα mRNA levels were presented relative to the expression of GAPDH.
Table 1.
Primers used for the PCR reaction
| Gene | Source | GenBank™ no. | Primer sequences | PCR product | Amplification cycles |
|---|---|---|---|---|---|
| PPARα | Rat | M88592 | Forward: 5′-TGAAAGATTCGGAAACTGC-3′ | 110 bp | 38 |
| Reverse: 5′-TCCTGCGAGTATGACCC-3′ | |||||
| PPARα | Human | NM001001928 | Forward: 5′-ACTTATCCTGTGGTCCCCGG-3′ | 252 bp | 38 |
| Reverse: 5′-CCGACAGAAAGGCACTTGTGA-3′ | |||||
| ApoAV | Rat | NM080576 | Forward: 5′-AGTCAAAGAACTCTTCCACC-3′ | 455 bp | 40 |
| Reverse: 5′-CCTTATTACTGTCTGAGTGT-3′ | |||||
| ApoAV | Human | BC101789 | Forward: 5′-ACGCACGCATCCAGCAGAAC-3′ | 109 bp | 40 |
| Reverse: 5′-CCTTATTACTGTCTGAGTGT-3′ |
ApoAV, apolipoprotein AV; PPARα, peroxisome proliferator-activated receptor-α; PCR, polymerase chain reaction.
Western blot analysis
The protein expression of apoAV and PPARα was quantified by Western blot analysis. Briefly, proteins from a 50 µg sample were separated on 10% sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and transferred onto PVDF membranes. The membrane was incubated overnight with mouse monoclonal anti-apoAV antibody (Santa Cruz) or rabbit monoclonal anti-PPARα antibody (Abcam, Cambridge, MA, USA) at 4°C for overnight. After incubation with secondary antibody, immunoreactive bands were visualized using the enhanced chemiluminescence detection system. Data were quantified by densitometry after scanning using the TINA software (Raytest, Straubenhardt, Germany).
Statistical analyses
Data were analysed with the use of SPSS 15.0 (SPSS Inc., Chicago, IL, USA) and presented as mean ± SD unless otherwise indicated. Log transformations were carried out for distribution-dependent analyses. Differences between intragroup and intergroup means were analysed by Student t-test or one-way anova. Coefficients of correlation (r and r2) were calculated by the Pearson correlation analysis. P-values < 0.05 were considered statistically significant.
Results
Characteristics of rats at end of the study
The characteristics of the rats at the end of this study (at week 14), including body weight, total cholesterol, high density lipoprotein cholesterol, low density lipoprotein cholesterol, and fasting plasma insulin and glucose, are shown in Table 2. Our results were consistent with previous data, showing that such fructose water-fed rats were hypertriglyceridemic, but normoglycemic, normoinsulinemic and non-obese (Park et al., 1997; Roglans et al., 2002).
Table 2.
Characteristics of rats at the end of study (at week 14)
| Control | Fructose | Atorvastatin | Fenofibrate | Combined | |
|---|---|---|---|---|---|
| Body weight (g) | 303 ± 29 | 309 ± 30 | 305 ± 24 | 305 ± 27 | 306 ± 29 |
| Total cholesterol (mM) | 1.35 ± 0.09 | 2.30 ± 0.18* | 1.78 ± 0.13*,# | 1.86 ± 0.16*,# | 1.68 ± 0.15*,# |
| HDL cholesterol (mM) | 0.59 ± 0.11 | 0.58 ± 0.09 | 0.62 ± 0.11 | 0.60 ± 0.17 | 0.63 ± 0.18 |
| LDL cholesterol (mM) | 1.33 ± 0.15 | 1.38 ± 0.21 | 1.09 ± 0.17*,# | 1.17 ± 0.15# | 1.05 ± 0.25*,# |
| Insulin (mU·L−1) | 17.62 ± 3.50 | 18.02 ± 4.46 | 17.90 ± 4.15 | 17.81 ± 3.93 | 17.69 ± 4.23 |
| Blood glucose (mM) | 5.39 ± 0.55 | 5.44 ± 0.64 | 5.42 ± 0.43 | 5.40 ± 0.42 | 5.39 ± 0.43 |
Data are the mean ± SD (n= 8).
Values significantly different from control.
Values significantly different from fructose (P < 0.05).
LDL, low density lipoprotein; HDL, high density lipoprotein.
Combination treatment more effectively decreased plasma triglycerides and increased apoAV in rats than monotherapy
The plasma triglycerides and apoAV levels in rats are shown in Figure 1. There were no significant differences between any groups for plasma triglycerides and apoAV levels at baseline. However, fructose-fed animals had higher plasma triglycerides than controls (P < 0.001), showing that we were able to induce hypertriglyceridemia in our animal model. However, these fructose-fed hypertriglyceridemic animals had lower plasma apoAV than in controls, that is, rats without fructose. After treatment with the combination, rats had lower plasma triglycerides than after either monotherapy (both P < 0.05). Conversely, plasma apoAV in the group treated with the combination was significantly increased over the levels in either monotherapy group (both P < 0.05).
Figure 1.
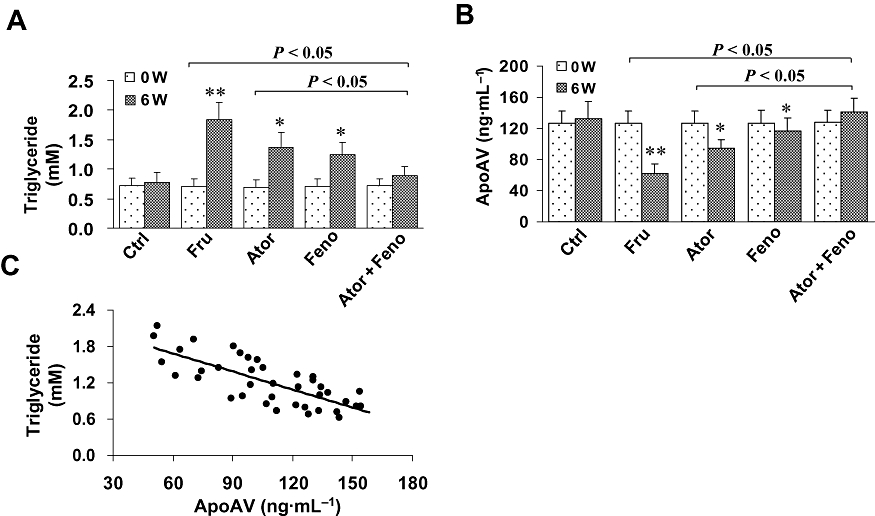
Plasma apolipoprotein AV (apoAV) and triglycerides in rats and their relationship. Rats were randomized into five groups: (i) control group (Ctrl); (ii) fructose only group (Fru); (iii) atorvastatin group (Ator); (iv) fenofibrate group (Feno); and (v) combination group (Ator + Feno). (A and B) There were no significant differences in plasma triglyceride and apoAV at baseline between all groups. However, after 6 weeks, fructose-treated animals had higher triglycerides and lower apoAV than in the control rats (both P < 0.05). Atorvastatin and/or fenofibrate treated groups exhibited lower plasma triglycerides and higher apoAV level (all P < 0.05), but these changes were greater in the group given the combined therapy (both P < 0.05). (C) After pooling all data together at week 6, an inverse correlation between triglyceride and apoAV was found (r=−0.78, P < 0.001). *P < 0.05, **P < 0.001, compared with controls. Data shown as mean ± SD.
To test the relationship between apoAV and triglycerides, we performed a correlation analysis after pooling all data together. A strong inverse correlation between these two variables were found at baseline (r=−0.74, r2= 0.54, P < 0.001) that still remain at week 14 (r=−0.78, r2= 0.61, P < 0.001) (Figure 1C).
Combination treatment more effectively increased hepatic apoAV and PPARα expression than monotherapy
RT-PCR analysis showed that fructose-fed animals exhibited a 50% reduction of APOAV gene expression, relative to that in controls (Figure 2A). Conversely, all drug-treated rats had higher APOAV expression than the fructose-fed rats (all P < 0.05), and this increase was greater after combination therapy than after the statin or fibrate as monotherapy (both P < 0.05). Similar observations were made by the Western blot analysis.
Figure 2.
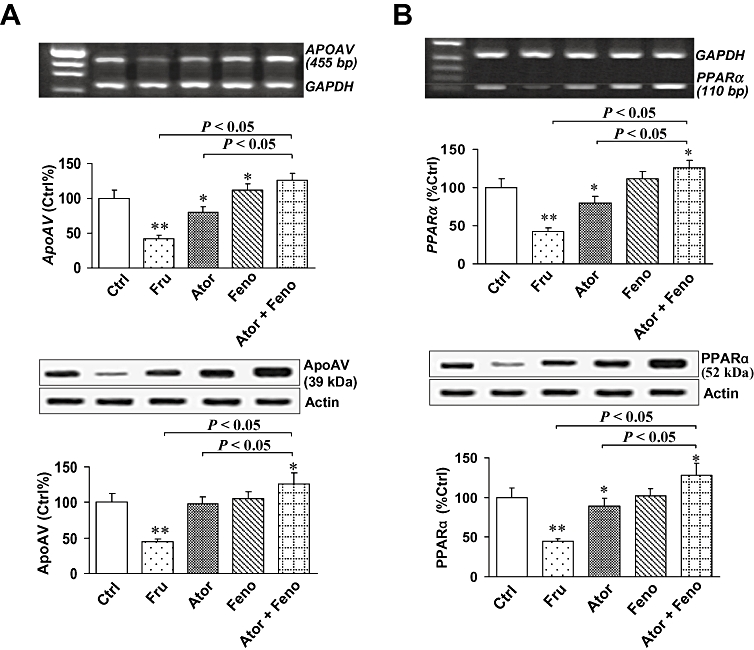
Hepatic apolipoprotein AV (apoAV) and peroxisome proliferator-activated receptor-α (PPARα) expression in rats. (A) By reverse transcription polymerase chain reaction (RT-PCR) analysis, hepatic apoAV gene expression in fructose-fed animals decreased by 50% relative to expression in control rats (P < 0.001). Conversely, all drug-treated rats showed higher apoAV expression than fructose-fed rats (all P < 0.05), and this increase was greater in the combination group (both P < 0.05). By western blot analysis, similar effects were found. (B) By RT-PCR and western blot analyses, we also found comparable changes in hepatic PPARα expression. *P < 0.05, **P < 0.001, compared with controls. Summary data shown as mean ± SD.
We also analysed hepatic PPARα expression by RT-PCR and western blot (Figure 2B). As compared with controls, hepatic PPARα expression in fructose-fed animals was markedly decreased (P < 0.001). However, this decrease of hepatic PPARα expression was reversed by monotherapy of fructose-fed rats (P < 0.05), and combined therapy raised hepatic PPARα expression even further (both P < 0.05).
Atorvastatin and fenofibrate decreased triglycerides and increased apoAV through up-regulation of PPARα in HepG2 cells
To further investigate whether the hypotriglyceridemic effect involved activation of PPARα, we used MK886, a selective inhibitor of PPARα in our experiments with HepG2 cells.
We found that triglyceride levels were raised 1.25 fold in HepG2 cells incubated with fructose (100 µM), relative to levels in control cells without fructose (P < 0.01) (Figure 3A). When atorvastatin or fenofibrate were added in the presence of fructose, the effect of the fructose was almost completely reversed, and triglyceride levels fell to values close to those in control cells. Combined treatment with both atorvastatin and fenofibrate induced further falls in triglycerides to levels below control (P < 0.05). However, when fructose-exposed cells were treated with atorvastatin and the PPARα antagonist, MK886, the effect of atorvastatin on triglycerides was totally reversed (P < 0.01).
Figure 3.
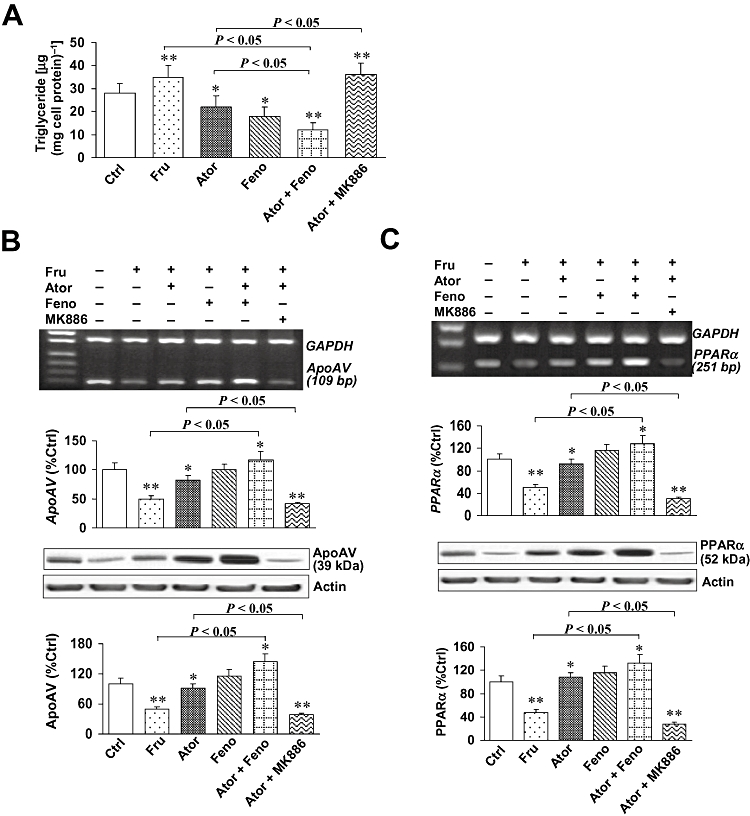
Triglycerides and apolipoprotein AV (apoAV) and peroxisome proliferator-activated receptor-α (PPARα) expression in HepG2 cells. HepG2 cells were divided into six groups: (i) control group (Ctrl); (ii) fructose group (Fru); (iii) atorvastatin group (Ator); (iv) fenofibrate group (Feno); (v) atorvastatin plus fenofibrate group (Ator + Feno); and (vi) atorvastatin plus MK886 group (Ator + MK886). (A) Triglycerides in HepG2 cells. We found that triglycerides increased in fructose-treated cells relative to levels in control cells (P < 0.01). When atorvastatin or fenofibrate was added, cells had lower triglycerides than fructose-treated cells (all P < 0.05). This decrease was greater in the group treated with the combination (P < 0.05). However, when atorvastatin was combined with MK886, the effect of the statin on cellular triglycerides was lost (P < 0.01). (B and C) The mRNA and protein expression of apoAV and PPARα in cells. By reverse transcription polymerase chain reaction and western blotting analysis, we found lower apoAV and PPARα expression in fructose-treated cells than in controls (both P < 0.01). Atorvastatin or fenofibrate induced an up-regulation of apoAV, and PPARα expression that was more pronounced in cells treated with the combination (P < 0.05). However, when MK886 was added to atorvastatin, PPARα expression was repressed (P < 0.05), and apoAV expression decreased (P < 0.01). *P < 0.05, **P < 0.001, compared with controls. Data shown as mean ± SD.
Then, we determined the expression of apoAV and PPARα in HepG2 cells by RT-PCR and Western blotting analysis (Figure 3B and C). We found lower expression of apoAV and PPARα in fructose-treated cells than in control cells (both P < 0.01). Adding atorvastatin or fenofibrate to the fructose treatment reversed the fructose-induced falls in apoAV and PPARα expression and this reversal was more pronounced in the cells receiving the combination of statin and fibrate (all P < 0.05). However, when atorvastatin was combined with the PPARα antagonist, MK886, the effects of the statin on PPARα expression (P < 0.05), and apoAV expression were completely blocked (P < 0.01).
Discussion
Clinically, the combination of a statin and a fibrate induces a greater hypotriglyceridemic effects than does monotherapy (Ooi et al., 1997; Miller et al., 2008). However, the mechanism underlying the hypotriglyceridemic effects of the combination remains poorly understood. The present study showed that these two hypolipidemic agents increase levels of apoAV by up-regulation of PPARα expression, and thus provided a molecular explanation for the greater effects of a combination of statin and fibrate, in treatment of hypertriglyceridemia.
ApoAV is a novel member of the apolipoprotein family involved in triglyceride homeostasis (Pennacchio et al., 2001; Van der Vliet et al., 2001). The triglyceride-lowering role of apoAV has been demonstrated earlier (Pennacchio et al., 2001; 2002; Van der Vliet et al., 2001), and our findings in the present study were consistent with these earlier results. To date, three triglyceride-lowering pathways of apoAV have been proposed: (i) inhibitory effects on production and secretion of very low density lipoprotein (VLDL) (Schaap et al., 2004); (ii) stimulation of lipoprotein lipase (LPL)-mediated triglyceride hydrolysis (Grosskopf et al., 2005); and (iii) acceleration of hepatic uptake of VLDL (Nilsson et al., 2007).
PPAR-α, a ligand-activated transcription factor, has been implicated in the up-regulation of apoAV gene expression, and a functional PPAR response element in the proximal apoAV promoter has been detected using deletion and mutagenesis analyses (Prieur et al., 2003; Vu-Dac et al., 2003). These authors demonstrated that fibrate, an agonist of PPARα, enhanced APOAV gene expression. Up-regulation of PPARα, and hence of apoAV, by fibrate were also observed in the study, resulting in a proportional reduction of triglycerides. Thus, it appears that one mechanism of the hypotriglyceridemic action of fibrates could be by up-regulating apoAV via the PPARα pathway.
Interestingly, we also found that atorvastatin increased hepatic and plasma apoAV, and decreased plasma triglycerides as a consequence. This finding provides a new mechanistic rationale for the hypotriglyceridemic effects of statins. Such effects have previously attributed to a variety of mechanisms, including inhibition of hepatic VLDL synthesis (Kasim et al., 1992; Scharnagl and März, 2005), promotion of circulating VLDL clearance (Kasim et al., 1992), and acceleration of LPL-mediated triglyceride hydrolysis (Yokoyama et al., 2007). Of note, statins share common hypotriglyceridemic pathways with apoAV (Schaap et al., 2004; Grosskopf et al., 2005; Nilsson et al., 2007). Therefore, it is possible that statins could initially affect apoAV levels and then reduce triglycerides via apoAV-mediated hypotriglyceridemic pathways.
As the pleiotropic, anti-atherogenic properties of statins, including lipid modulatory, anti-inflammatory, anti-oxidative activities and endothelial protection, all involve the PPARα signal pathway (Paumelle and Staels, 2007), we investigated whether statin-mediated regulation of apoAV was also associated with PPARα, and found that atorvastatin increased apoAV in a PPARα-dependent manner.
Previously, investigators have demonstrated that statins enhanced PPARα expression, resulting in a reduction of non-esterified fatty acids and an increase of liver fatty acid-binding protein, which contributes to the lowering of triglycerides (Roglans et al., 2002; Landrier et al., 2004). Therefore, we conclude that PPARα-mediated up-regulation of apoAV would also contribute to the hypotriglyceridemic effect of statins. Subsequently, we investigated whether the hypotriglyceridemic effects of combining atorvastatin and fenofibrate involved apoAV. We observed that the combination treatment induced a greater response of apoAV and of triglycerides than either drug given as monotherapy. Further, the effects of the statin were eliminated when PPARα was blocked with a selective antagonist. Thus, these findings provided evidence for our previous speculation (Bai et al., 2008) that statin and fibrate could act together to increase apoAV and thus decrease triglycerides through up-regulation of PPARα.
In summary, our study demonstrated that atorvastatin and fenofibrate increased apoAV and hence decreased triglycerides, and this resulted from their common up-regulation of PPARα. Our findings provide a new mechanistic rationale for the clinical combination of statins and fibrates to obtain greater hypotriglyceridemic effects. On the other hand, human studies are needed to determine whether similar therapeutic effects will be achieved in humans.
Acknowledgments
This study was supported by Key Clinical Projects for Affiliated Hospitals of Ministry of Health of China (Grant 2007-2009NHCN) (Dr Shui-ping Zhao), National Natural Science Foundation of China (Grant 30770857) (Dr Shui-ping Zhao) and Innovation Projects in Central South University (Grant 2008CSU) (Dr Xian-sheng Huang).
Glossary
Abbreviations:
- ApoAV
apolipoprotein AV
- DMSO
dimethyl sulphoxide
- EDTA
ethylenediamine tetraacetic acid
- GAPDH
glyceraldehyde phosphate dehydrogenase
- HDL
high density lipoprotein
- LDL
low density lipoprotein
- PPARα
peroxisome proliferator-activated receptor-α
- PVDF
polyvinylidene difluoride
- RT-PCR
reverse transcription polymerase chain reaction
- VLDL
very low density lipoprotein
Conflicts of interest
None.
References
- Bai L, Zhao SP, Muang XS, Zhang Q. Upregulating APOAV expression by statins via PPAR-α activated pathway possibly contribute to their triglyceride-lowering effect. Bioscience Hypotheses. 2008;1:292–294. [Google Scholar]
- Criqui MH, Heiss G, Cohn R, Cowan LD, Suchindran CM, Bangdiwala S, et al. Plasma triglyceride level and mortality from coronary heart disease. N Engl J Med. 1993;328:1220–1225. doi: 10.1056/NEJM199304293281702. [DOI] [PubMed] [Google Scholar]
- Grosskopf I, Baroukh N, Lee SJ, Kamari Y, Harats D, Rubin EM, et al. Apolipoprotein A-V deficiency results in marked hypertriglyceridemia attributable to decreased lipolysis of triglyceride-rich lipoproteins and removal of their remnants. Arterioscler Thromb Vasc Biol. 2005;25:2573–2579. doi: 10.1161/01.ATV.0000186189.26141.12. [DOI] [PubMed] [Google Scholar]
- Kasim SE, LeBoeuf RC, Khilnani S, Tallapaka L, Dayananda D, Jen KL. Mechanisms of triglyceride-lowering effect of an HMG-CoA reductase inhibitor in a hypertriglyceridemic animal model, the Zucker obese rat. J Lipid Res. 1992;33:1–7. [PubMed] [Google Scholar]
- Landrier JF, Thomas C, Grober J, Duez H, Percevault F, Souidi M, et al. Statin induction of liver fatty acid-binding protein (L-FABP) gene expression is peroxisome proliferator-activated receptor-alpha-dependent. J Biol Chem. 2004;279:45512–45518. doi: 10.1074/jbc.M407461200. [DOI] [PubMed] [Google Scholar]
- Miller M, Cannon CP, Murphy SA, Qin J, Ray KK, Braunwald E, PROVE IT-TIMI 22 Investigators Impact of triglyceride levels beyond low-density lipoprotein cholesterol after acute coronary syndrome in the PROVE IT-TIMI 22 trial. J Am Coll Cardiol. 2008;51:724–730. doi: 10.1016/j.jacc.2007.10.038. [DOI] [PubMed] [Google Scholar]
- Nilsson SK, Lookene A, Beckstead JA, Gliemann J, Ryan RO, Olivecrona G. Apolipoprotein A-V interaction with members of the low density lipoprotein receptor gene family. Biochemistry. 2007;46:3896–3904. doi: 10.1021/bi7000533. [DOI] [PubMed] [Google Scholar]
- Ooi TC, Heinonen T, Alaupovic P, Davignon J, Leiter L, Lupien PJ, et al. Efficacy and safety of a new hydroxymethylglutaryl- coenzyme A reductase inhibitor, atorvastatin, in patients with combined hyperlipidemia: comparison with fenofibrate. Arterioscler Thromb Vasc Biol. 1997;17:1793–1799. doi: 10.1161/01.atv.17.9.1793. [DOI] [PubMed] [Google Scholar]
- Park J, Lemieux S, Lewis GF, Kuksis A, Steiner G. Chronic exogenous insulin and chronic carbohydrate supplementation increase de novo VLDL triglyceride fatty acid production in rats. J Lipid Res. 1997;38:2529–2536. [PubMed] [Google Scholar]
- Paumelle R, Staels B. Peroxisome proliferator-activated receptors mediate pleiotropic actions of statins. Circ Res. 2007;100:1394–1395. doi: 10.1161/01.RES.0000269334.42814.d2. [DOI] [PubMed] [Google Scholar]
- Pennacchio LA, Olivier M, Hubacek JA, Cohen JC, Cox DR, Fruchart JC, et al. An apolipoprotein influencing triglycerides in humans and mice revealed by comparative sequencing. Science. 2001;294:169–173. doi: 10.1126/science.1064852. [DOI] [PubMed] [Google Scholar]
- Pennacchio LA, Olivier M, Hubacek JA, Krauss RM, Rubin EM, Cohen JC. Two independent apolipoprotein A5 haplotypes influence human plasma triglyceride levels. Hum Mol Genet. 2002;11:3031–3038. doi: 10.1093/hmg/11.24.3031. [DOI] [PubMed] [Google Scholar]
- Prieur X, Coste H, Rodriguez JC. The human apolipoprotein AV gene is regulated by peroxisome proliferator-activated receptor-alpha and contains a novel farnesoid X-activated receptor response element. J Biol Chem. 2003;278:25468–25480. doi: 10.1074/jbc.M301302200. [DOI] [PubMed] [Google Scholar]
- Priore Oliva C, Pisciotta L, Li Volti G, Sambataro MP, Cantafora A, Bellocchio A, et al. Inherited apolipoprotein A-V deficiency in severe hypertriglyceridemia. Arterioscler Thromb Vasc Biol. 2005;25:411–417. doi: 10.1161/01.ATV.0000153087.36428.dd. [DOI] [PubMed] [Google Scholar]
- Roglans N, Sanguino E, Peris C, Alegret M, Vázquez M, Adzet T, et al. Atorvastatin treatment induced peroxisome proliferator-activated receptor alpha expression and decreased plasma nonesterified fatty acids and liver triglyceride in fructose-fed rats. J Pharmacol Exp Ther. 2002;302:232–239. doi: 10.1124/jpet.302.1.232. [DOI] [PubMed] [Google Scholar]
- Schaap FG, Rensen PC, Voshol PJ, Vrins C, van der Vliet HN, Chamuleau RA, et al. ApoAV reduces plasma triglycerides by inhibiting very low density lipoprotein-triglyceride (VLDL-TG) production and stimulating lipoprotein lipase-mediated VLDL-TG hydrolysis. J Biol Chem. 2004;279:27941–27947. doi: 10.1074/jbc.M403240200. [DOI] [PubMed] [Google Scholar]
- Scharnagl H, März W. New lipid-lowering agents acting on LDL receptors. Curr Top Med Chem. 2005;5:233–242. doi: 10.2174/1568026053544524. [DOI] [PubMed] [Google Scholar]
- Van der Vliet HN, Sammels MG, Leegwater AC, Levels JH, Reitsma PH, Boers W, et al. Apolipoprotein A-V: a novel apolipoprotein associated with an early phase of liver regeneration. J Biol Chem. 2001;276:4512–4520. doi: 10.1074/jbc.M106888200. [DOI] [PubMed] [Google Scholar]
- Vu-Dac N, Gervois P, Jakel H, Nowak M, Bauge E, Dehondt H, et al. Apolipoprotein A5, a crucial determinant of plasma triglyceride levels, is highly responsive to peroxisome proliferator-activated receptor alpha activators. J Biol Chem. 2003;278:17982–17985. doi: 10.1074/jbc.M212191200. [DOI] [PubMed] [Google Scholar]
- Wilcox LJ, Barrett PH, Huff MW. Differential regulation of apolipoprotein B secretion from HepG2 cells by two HMG-CoA reductase inhibitors, atorvastatin and simvastatin. J Lipid Res. 1999;40:1078–1089. [PubMed] [Google Scholar]
- Yokoyama M, Seo T, Park T, Yagyu H, Hu Y, Son NH, et al. Effects of lipoprotein lipase and statins on cholesterol uptake into heart and skeletal muscle. J Lipid Res. 2007;48:646–655. doi: 10.1194/jlr.M600301-JLR200. [DOI] [PubMed] [Google Scholar]