

Abstract
Background and purpose:
Natural compounds obtained from marine organisms have received considerable attention as potential sources of novel drugs for treatment of human inflammatory diseases. Capnellene, isolated from the marine soft coral Capnella imbricate, 4,4,6a-trimethyl-3-methylene-decahydro-cyclopenta[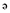 ]pentalene-2,3a-diol (GB9) exhibited anti-inflammatory actions on activated macrophages in vitro. Here we have assessed the anti-neuroinflammatory properties of GB9 and its acetylated derivative, acetic acid 3a-hydroxy-4,4,6a-trimethyl-3-methylene-decahydro-cyclopenta[
]pentalene-2,3a-diol (GB9) exhibited anti-inflammatory actions on activated macrophages in vitro. Here we have assessed the anti-neuroinflammatory properties of GB9 and its acetylated derivative, acetic acid 3a-hydroxy-4,4,6a-trimethyl-3-methylene-decahydro-cyclopenta[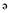 ]pentalen-2-yl ester (GB10).
]pentalen-2-yl ester (GB10).
Experimental approach:
Effects of GB9 or GB10 on the expression of inducible nitric oxide synthase (iNOS), and cyclooxygenase-2 (COX-2) in interferon-γ (IFN-γ)-stimulated mouse microglial BV2 cells were measured by Western blot. The in vivo effects of these compounds were examined in the chronic constriction injury (CCI) rat model of neuropathic pain, measuring thermal hyperalgesia, and microglial activation and COX-2 protein in lumbar spinal cord, by immunohistochemistry.
Key results:
In BV2 cells, GB9 and GB10 inhibited the expression of iNOS and COX-2, stimulated by IFN-γ. Intrathecal administration of GB9 and GB10 inhibited CCI-induced nociceptive sensitization and thermal hyperalgesia in a dose-dependent manner. Intraperitoneal injection of GB9 inhibited CCI-induced thermal hyperalgesia and also inhibited CCI-induced activation of microglial cells and up-regulation of COX-2 in the dorsal horn of the lumbar spinal cord ipsilateral to the injury.
Conclusion and implications:
Taken together, these data indicate that the marine-derived capnellenes, GB9 and GB10, had anti-neuroinflammatory and anti-nociceptive properties in IFN-γ-stimulated microglial cells and in neuropathic rats respectively. Therefore, capnellene may serve as a useful lead compound in the search for new therapeutic agents for treatment of neuroinflammatory diseases.
Keywords: neuroinflammation, microglial cells, neuropathy, cyclooxygenase-2, marine, inducible nitric oxide synthase
Introduction
Capnellene, a tricyclic sesquiterpene, was first isolated from the soft coral Capnella imbricate in Indonesia and its chemical structure described by Kaisin et al. (1974). One of the compounds used in the present study, Δ9(12)-capnellene-8β, 10α-diol (4,4,6a-trimethyl-3-methylene-decahydro-cyclopenta[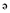 ]pentalene-2,3a-diol; GB9; Figure 1), was first isolated from the soft coral Capnella imbricate by Sheikh et al. (1976) and later from the same species of soft coral at Green Island near Taiwan (Chang et al., 2008). GB9 and its acetylated form, 8a-acetoxy-Δ9(12)-capnellene-10a-01 (acetic acid 3a-hydroxy-4,4,6a-trimethyl-3-methylene-decahydro-cyclopenta[
]pentalene-2,3a-diol; GB9; Figure 1), was first isolated from the soft coral Capnella imbricate by Sheikh et al. (1976) and later from the same species of soft coral at Green Island near Taiwan (Chang et al., 2008). GB9 and its acetylated form, 8a-acetoxy-Δ9(12)-capnellene-10a-01 (acetic acid 3a-hydroxy-4,4,6a-trimethyl-3-methylene-decahydro-cyclopenta[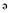 ]pentalen-2-yl ester; GB10; Figure 1) were found to exert anti-inflammatory activity in vitro (Chang et al., 2008). Both compounds significantly down-regulated the lipopolysaccharide-induced expression of the pro-inflammatory proteins, inducible nitric oxide synthase (iNOS) and cyclooxygenase-2 (COX-2), in macrophage cells, at a concentration of 10 µM. Inflammation is a pathophysiological state usually associated with pain, which can be alleviated by many substances with anti-inflammatory properties.
]pentalen-2-yl ester; GB10; Figure 1) were found to exert anti-inflammatory activity in vitro (Chang et al., 2008). Both compounds significantly down-regulated the lipopolysaccharide-induced expression of the pro-inflammatory proteins, inducible nitric oxide synthase (iNOS) and cyclooxygenase-2 (COX-2), in macrophage cells, at a concentration of 10 µM. Inflammation is a pathophysiological state usually associated with pain, which can be alleviated by many substances with anti-inflammatory properties.
Figure 1.
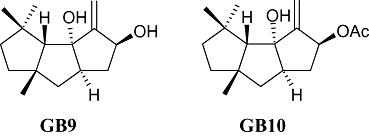
The chemical structures of capnellene, GB9 (4,4,6a-trimethyl-3-methylene-decahydro-cyclopenta[ ]pentalene-2,3a-diol) and GB10 (acetic acid 3a-hydroxy-4,4,6a-trimethyl-3-methylene-decahydro-cyclopenta[
]pentalene-2,3a-diol) and GB10 (acetic acid 3a-hydroxy-4,4,6a-trimethyl-3-methylene-decahydro-cyclopenta[ ]pentalen-2-yl ester).
]pentalen-2-yl ester).
Inflammatory states within the central nervous system (CNS) are termed neuroinflammation, which can give rise to nerve damage affecting peripheral or central nerves and leading to pathological nociceptive transmission, which is referred to as neuropathic pain (Myers et al., 2006). Patients with neuropathic pain often develop hyperalgesia (an increased response to painful stimuli), allodynia (pain evoked by non-painful stimuli) and a resistance to opioids and other analgesics, including non-steroidal anti-inflammatory drugs (Arner and Meyerson, 1988; Kalso and Vainio, 1990; Clark and Lee, 1995; Ossipov and Porreca, 2005). Neuropathic pain afflicts people worldwide and severely affects the patient's quality of life. Thus, new, efficacious and safe analgesics for neuropathic pain are urgently needed. In clinical settings, morphine has been the therapeutic mainstay in pain management. However, an increasing number of animal and human studies have shown reduction of the opiate analgesic effect in neuropathic pain (Arner and Meyerson, 1988; Courteix et al., 1993; Zurek et al., 2001). It has been suggested that higher doses of opioids may attenuate neuropathic pain syndromes (Gordon and Love, 2004). However, the high doses required to obtain adequate analgesia may be accompanied by unacceptable or very serious side effects (Portenoy et al., 1990; McQuay et al., 1992).
In the CNS, microglial cells play the most important role in neuroinflammatory processes. Microglial cells are the primary immunoresponsive cells in the CNS and are involved in signalling cascades that are associated with proinflammatory cytokines and chemokines and their receptor systems (Ladeby et al., 2005). In the normal adult CNS, microglia constitute relatively stable cell populations with little turnover and proliferation of the resident cells, also referred to as ‘resting’ microglia (Vilhardt, 2005; Hains and Waxman, 2006). Thus far, no functions have been attributed to resting microglia. However, microglia are very sensitive to any disturbance of the CNS environment and rapidly transform to an activated state characterized by amoeboid morphology (Vilhardt, 2005; Hains and Waxman, 2006). Activated microglial cells are capable of synthesizing and releasing various proinflammatory mediators, such as NO, prostaglandin and tumour necrosis factor-α (Watkins and Maier, 2002), which can promote nociceptive transmission by causing activation of dorsal horn neurones (Myers et al., 2006). Many studies have indicated that inhibition of microglial activation attenuates the development of neuropathy (Raghavendra et al., 2003; Hains and Waxman, 2006). Very few studies have directly examined the ability of marine-derived natural products to inhibit inflammatory responses in microglia or elucidated their possible analgesic mechanisms in an in vivo model of neuropathy.
Using an in vitro neuroinflammatory system, the present study found that the marine-derived compounds GB9 and GB10 significantly inhibited interferon-γ (IFN-γ)-induced up-regulation of the proinflammatory proteins iNOS and COX-2 in a mouse microglial cell line (BV2 cells). We have also tested these marine-derived compounds in vivo for their ability to attenuate nociceptive sensitization in a chronic constriction injury (CCI) model of neuropathic pain. The results demonstrated that GB9 and GB10 not only significantly reversed hyperalgesic behaviours but also attenuated microglial cell activation in the spinal cord in neuropathic rats. These marine-derived tricyclic sesquiterpene compounds could be potential therapeutic drugs for neuropathic pain syndromes, despite their inhibition of neuroinflammation in microglial cells.
Methods
Microglia culture and in vitro anti-inflammatory assay
The mouse microglial cell line BV2, generated from primary mouse microglia transfected with a v-raf/v-myc oncogene (Blasi et al., 1990) and was maintained at 37°C in Dulbecco's modified Eagle's medium/F12 medium (Life Technologies, Grand Island, NY, USA) with 5% heat-inactivated fetal bovine serum (HyClone, Logan, UT, USA), 50 U·mL−1 penicillin and 50 mg·mL−1 streptomycin (Sigma Chemical, St Louis, MO, USA) under a humidified atmosphere of 5% CO2/95% air. Inflammatory microglial cells were induced by incubating them for 16 h in a medium containing IFN-γ (10 U·mL−1). The dosage of IFN-γ for induction of BV2 cell inflammation was determined according to Kim et al. (2003). Murine IFN-γ was purchased from R & D (Minneapolis, MN, USA). For the anti-neuroinflammatory activity assay, GB9 or GB10 (1, 5, 10, 20 or 30 µM) was added to the cells 10 min before IFN-γ challenge. After 16-h incubation, the cells were washed with ice-cold phosphate-buffered saline (PBS), lysed in ice-cold lysis buffer (50 mM Tris, pH 7.5, 150 mM NaCl, 1% Triton X-100, 100 µg·mL−1 phenylmethylsulphonyl fluoride, 1 µg·mL−1 aprotinin), and then centrifuged at 20 000× g for 30 min at 4°C. The supernatant was decanted from the pellet and retained for Western blot analysis of iNOS and COX-2. Protein concentrations were determined using the DC protein assay kit (Bio-Rad, Hercules, CA, USA) modified from the method of Lowry et al. (1951). Western blotting was performed as described previously (Jean et al., 2008). An equal volume of sample buffer (2% sodium dodecyl sulphate (SDS), 10% glycerol, 0.1% bromophenol blue, 2% 2-mercaptoethanol and 50 mM Tris-HCl, pH 7.2) was added to the sample, which was then loaded onto a tricine SDS-polyacrylamide gel and electrophoresed at 150 V for 90 min. The proteins were transferred to a polyvinylidene difluoride membrane (Immobilon-P, 0.45-µm pore size; Millipore, Bedford, MA, USA) at 125 mA overnight at 4°C in transfer buffer (50 mM Tris-HCl, 380 mM glycine, 1% SDS and 20% methanol). The membrane was blocked for 50 min at room temperature with 5% non-fat dry milk in Tris-buffered saline containing 0.1% Tween (TTBS; 0.1% Tween 20, 20 mM Tris-HCl, 137 mM NaCl, pH 7.4), and then incubated for 180 min at room temperature with antibodies against iNOS (polyclonal antibody, 1:1000 dilution; BD Pharmingen, San Diego, CA, USA; Catalogue No. 6103322), or COX-2 (polyclonal antibody, 1:1000 dilution; Cayman Chemical, Ann Arbor, MI, USA; Catalogue No. 160106) proteins. The iNOS and COX-2 antibodies recognized bands at ∼135 and ∼70 kDa respectively. The immunoreactive bands were visualized using enhanced chemiluminescence (ECL kit; Millipore). The images were visualized using the UVP BioChemi Imaging System, and relative densitometric quantification was performed using LabWorks 4.0 software (UVP, Upland, CA, USA). Monoclonal antibody against β-actin (Sigma, St Louis, MO, USA) was used as the internal control for protein loading, and data are expressed as a ratio of the protein of interest to β-actin. Relative variations between the bands of the various treatment samples and of the control group were calculated using the same image. Relative variations between the bands of the drug-treatment samples and the IFN-γ samples were calculated using the same image.
Animals
All animal care and experimental use of animals conformed to the Guiding Principles in the Care and Use of Animals of the American Physiology Society and was approved by the National Sun Yat-sen University Animal Care and Use Committee. Every effort was made to minimize both the number of animals used and their suffering. Male Wistar rats weighing 260–285 g were used throughout the experiments. The rats were maintained in Plexiglas cages in a temperature-controlled (22 ± 1°C) room, under a 12-h light/dark cycle, and given free access to food and water. Each rat was used only once during the study.
Implantation of intrathecal catheters
As described previously (Wu et al., 2007), under isoflurane (2%) anaesthesia, intrathecal (i.t.) catheters (PE5 tubes: 9 cm, 0.008-inch inner diameter, 0.014-inch outer diameter; Spectranetics, Colorado Springs, CO, USA) were inserted via the atlanto-occipital membrane into the i.t. space at the level of the lumbar enlargement of the spinal cord and externalized and fixed to the cranial aspect of the head. The dead volume of the i.t. catheter was 3.5 µL. The rats were then returned to their home cages for a 5-day recovery period. Each rat was housed individually under a 12-h light/dark daily cycle with food and water freely available. Rats that showed evidence of gross neurological injury or the presence of fresh blood in the cerebrospinal fluid (CSF) were excluded from the study.
Induction of peripheral mononeuropathy and thermal hyperalgesia testing
Five days after catheterization, the rats were anaesthetized with isoflurane (2%). A CCI of the right common sciatic nerve was performed, according to the method described by Bennett and Xie (1988). Briefly, the rat's right common sciatic nerve was exposed and a 5-mm-long nerve segment was then dissected. Four loose ligatures (4–0 chromic gut) were placed around the sciatic nerve at 1-mm intervals. The skin incision was closed with 4–0 silk sutures. All rats received one postoperative injection of Veterin (cefazolin) intramuscularly to prevent infection. Thermal hyperalgesia was assessed by placing the hind paw on a radiant heat source and measuring the paw withdrawal latency (PWL) at low-intensity heat (active intensity = 25) set to a cut-off time of 30 s using an IITC analgesiometer (IITC, Woodland Hills, CA, USA). The PWL was measured as described previously by Hargreaves et al. (1988) as the average of two measurements per paw.
Effects of i.t. or systemic delivery of GB9 or GB10 on CCI-induced neuropathy
On day 14 post CCI surgery, the CCI rats received one i.t. bolus injection (10 µL) of GB9 (5, 10, 25, 50, 100 µg) or GB10 (5, 10, 25, 50, 100 µg) via the surgically implanted i.t. catheter. GB9 and GB10 were dissolved in 20% DMSO and delivered in a volume of 10 µL. The control group received a bolus i.t. injection (10 µL) of 20% DMSO in artificial CSF (aCSF; composition: 151.1 mM Na+, 2.6 mM K+, 122.7 mM Cl-, 21.0 mM HCO3-, 0.9 mM Mg2+, 1.3 mM Ca2+, 2.5 mM HPO42− and 3.5 mM dextrose and bubbled with 5% CO2 in 95% O2 to adjust the final pH to 7.3). All i.t. injections in the rats were followed by an i.t. aCSF flush injection (10 µL) to ensure complete drug delivery before nociceptive behavioural testing was performed. The anti-thermal hyperalgesia of GB9 or GB10 was performed at the following times: 30, 60, 90 and 180 min after drug injection. PWL (s) was also transformed into the percentage of maximum possible effect (%MPE) using the following formula: % MPE = (post-drug latency − baseline)/(cut off − baseline) × 100%, where the post-drug latency is the response measured 30, 60, 90 or 180 min after injection of the compound or saline, the baseline is the response measured immediately prior to test injection, and the cut-off time is 30 s. For statistical analysis, the area under the curve (AUC) for the plot of PWL versus time was calculated using the trapezoidal method (Rowland and Tozer, 1995) from 0 to 180 min after compounds or vehicle injection. In the study of the systemic effects of the compounds on neuropathy, GB9 (10 mg·kg−1) or 20% DMSO (100 µL) was injected intraperitoneally (i.p.) into neuropathic rats on the 14th day after CCI surgery. The effects of i.t. or systemic administration of compounds on neurological activity were evaluated using the Basso, Beattie and Bresnahan (BBB) locomotor scale (Basso et al., 1996). In the hot water immersion test (52 ± 0.5°C), tail-flick latency was used to measure the acute anti-nociceptive effect in naïve rats. The rats were placed in a plastic restrainer for drug injection and testing. At this temperature, the mean tail-flick latency was approximately 2.4 ± 0.4 s in the control group. An automatic cut-off was set at 10 s to prevent tissue injury (Wen et al., 2004).
Spinal immunohistofluorescence
Spinal tissue was collected, 6h after treatments, from the lumbar enlargement (L2–L4) of animals from the following groups: naïve rats, CCI plus i.p. vehicle, CCI plus i.p. GB9 (10 mg·kg−1), or GB9 (10 mg·kg−1) alone. Rats were deeply anaesthetized with isoflurane 5% and were intracardially perfused with 500 mL of cold PBS containing heparin (0.2 U·mL−1), then with 4% paraformaldehyde in 500 mL of 0.1 M PBS (pH 7.4). The tissues were then removed and post-fixed in the same fixative for 2 h and then transferred to a 30% sucrose solution overnight at 4°C. Spinal cord tissues from the various groups were then mounted on the same block in OCT compound, using a modified method from a previous study (Sung et al., 2003; Chen et al., 2008) to decrease the variation in immunohistochemical procedures. Immunoreactivity of the control group was presented as 100%. Sections (10 µm) were cut together on a cryostat (Microm, HM550, Waldorf, Germany) at −30°C and processed for immunofluorescence. After drying at room temperature for 1 h, the sections were immersed in ethanol/acetone (1:1) for 3 min at 4°C, and pre-incubated (1 h) with 4% normal goat serum diluted in 0.01% Triton X-100 in PBS. After washing in ice-cold PBS, the sections were incubated overnight at 4°C with monoclonal antibody OX42 (CD11b, microglia marker, 1:200; Serotec, Oxford, England), polyclonal anti-iNOS antibody (1:50; BD Pharmingen, San Diego, CA, USA) or anti-COX-2 (1:100 dilution; Cayman Chemical, Ann Arbor, MI, USA), and in 0.01% Triton X-100, 2% normal goat serum in PBS. The sections were then reacted for 1 h at room temperature with Alexa Fluor 488 (green fluorescence) or rhodamine (red fluorescence)-conjugated secondary antibody (1:300; Jackson ImmunoResearch Laboratories, West Grove, PA, USA). For the double immunofluorescent staining, spinal sections were incubated with a mixture of COX-2 and OX42, or COX-2 and neuronal-specific nuclear protein (NeuN) (neuronal marker, 1:100, Alexa Fluor 488 conjugated antibody, Chemicon, Temecula, CA, USA) antibodies overnight at 4°C, followed by a mixture of Alexa Fluor 488-conjugated and rhodamine-conjugated secondary antibodies for 1 h at room temperature. For the immunostaining analysis, four spinal sections were randomly selected and scanned using a Leica DM-2500 fluorescence microscope (Leica, Wetzlar, Germany). The images were captured with a SPOT CCD RT-slider integrating camera (Diagnostic Instruments, Sterling Heights, MI, USA). The laser wavelength was set at 488 nm for Alexa Fluor 488 green fluorescence and 568 nm for rhodamine red fluorescence. Control experiments, performed without the primary antibody, were run for each immunohistofluorescent experiment. Our control experiments revealed only background levels of signal. For the quantification of immunofluorescence staining, in each rat, every fourth section was selected from a series of consecutive lumbar spinal cord sections, and four successive sections were measured. Five rats were included in each group. Measurements and images were made on the medial half portion of the ipsilateral dorsal horn for quantitative analysis under a 20× objective. The size of the image was kept the same in all conditions. The images were then quantified using Image J software (National Institutes of Health, Bethesda, MD, USA) by an observer unaware of experimental conditions. The pixel measurement and analysis function was used for counting the density-slicing area in the image of the positive area of the dorsal horn of the spinal cord. Then, the percentage changes in staining density between CCI and CCI plus GB9 and the corresponding control group were calculated using the following equation: (control pixels − treatment pixels)/(control pixels). The criteria for resting and activated microglia were as described previously (Hains and Waxman, 2006). Briefly, the resting morphology is characterized by small compact somata bearing long, thin, ramified processes, and activated morphology is characterized by a process length less than the diameter of the soma compartment. Immunohistochemical data were expressed as the percentage change compared with untreated control animals or the contralateral side (non-injury side), which were considered to be 100%. All samples from all groups were numbered randomly, and these numbers were used to identify the samples through the evaluation to prevent bias. The double immunohistochemical observations were evaluated at 630× magnification by a investigator unaware of the treatment groups, using three sections per rat.
Data and statistical analysis
All data are presented as the mean ± standard error on the mean (SEM). For the immunoreactivity data, the intensity of each test band is expressed as the integrated optical density, calculated with respect to the average optical density of the corresponding control (IFN-γ-only treatment) band. To simplify data analysis, values derived from the temporal determination of PWL were transformed to AUC. The AUCs for the time–response curves for PWL were calculated for individual animals using SigmaPlot software, with time on the x-axis and response on the y-axis. For statistical analysis, all of the data were analysed with a one-way analysis of variance, followed by the Student-Newman-Keuls post hoc test for multiple comparisons. A significant difference was defined as P < 0.05.
Results
Effects of GB9 and GB10 on IFN-γ-induced expression of iNOS and COX-2 protein in microglial cells
Up-regulation of the proinflammatory, 130-kDa iNOS and 71-kDa COX-2 proteins in the INFγ-stimulated microglial cells was evaluated using Western blot analysis. The analysis was carried out on whole cell lysates using antibodies against mouse iNOS and COX-2. The cells were pretreated with GB9 or GB10 for 10 min and stimulated with IFN-γ for 16 h. Dose–response curves for inhibition of IFN-γ-induced iNOS and COX-2 protein expression by GB9 and GB10 are shown in Figures 2 and 3 respectively. Concentrations of 1–30 µM of GB9 and GB10 significantly inhibited IFN-γ-induced iNOS protein expression; the IC50 values for GB9 and GB10 were 17.1 ± 2.8 and 6.2 ± 2.8 µM respectively. Concentrations of 5–30 µM of GB9 and GB10 significantly inhibited COX-2 protein expression after IFN-γ challenge; the IC50 values for GB9 and GB10 were 6.21 ± 2.5 and 17.9 ± 2.9 µM respectively. Both GB9 and GB10 almost completely eliminated IFN-γ-induced COX-2 protein expression in microglial cells at a concentration of 30 µM. Both GB9 and GB10 significantly inhibited IFN-γ-induced levels of iNOS and COX-2 in a concentration-dependent manner. Only the vehicle (DMSO), GB9 and GB10 (1–30 µM) did not induce significant up-regulation of iNOS and COX-2 protein expression in the microglial cells. Furthermore, neither GB9 nor GB10 (1–30 µM) induced cytotoxicity in the microglial cells, as determined through Trypan blue staining.
Figure 2.
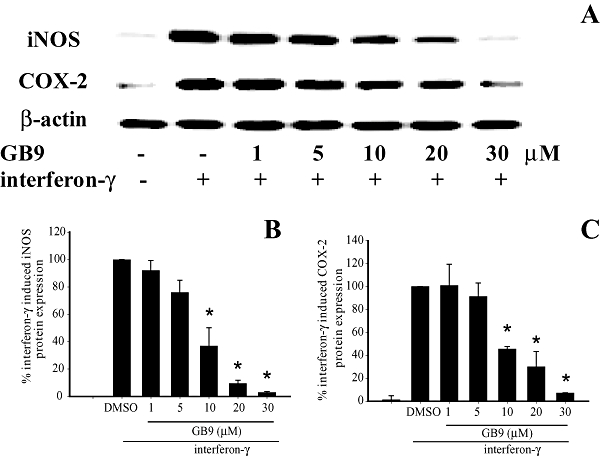
Effects of GB9 on expression of inducible nitric oxide synthase (iNOS) and cyclooxygenase-2 (COX-2) proteins in microglial cells (BV2 cells), induced by interferon-γ (IFN-γ). (A) Western blots for iNOS, COX-2 and β-actin proteins from microglial cells. (B) Relative density of iNOS immunoblot. (C) Relative density of COX-2 immunoblot. The relative intensity of the IFN-γ-stimulated group was taken to be 100%. Band intensities were quantified by densitometry and are indicated as a percentage change relative to that of the IFN-γ-stimulated group. Western blotting with β-actin was performed to verify that equivalent amounts of protein were loaded in each lane. The experiment was repeated five times. *, significantly different from the IFN-γ-stimulated group (P < 0.05).
Figure 3.
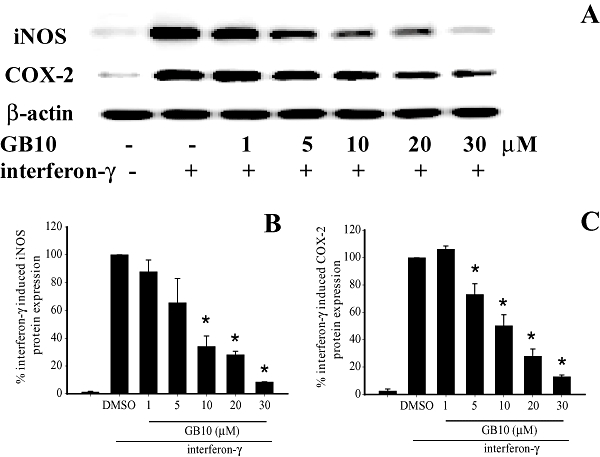
Effects of GB10 on expression of inducible nitric oxide synthase (iNOS) and cyclooxygenase-2 (COX-2) proteins in microglial cells induced by interferon-γ (IFN-γ). (A) Western blots for iNOS, COX-2 and β-actin proteins from microglial cells. (B) Relative density of iNOS immunoblot. (C) Relative density of COX-2 immunoblot. The relative intensity of the IFN-γ-stimulated group was taken to be 100%. Band intensities were quantified by densitometry and are indicated as a percentage change relative to that of the IFN-γ-stimulated group. Western blotting with β-actin was performed to verify that equivalent amounts of protein were loaded in each lane. The experiment was repeated five times. *, significantly different from the IFN-γ-stimulated group (P < 0.05).
Anti-nociceptive effects of i.t. GB9 and GB10 on neuropathic rats
There were no significant differences in the PWL baseline among the experimental groups before sciatic nerve ligature surgery. The average baseline PWL in the thermal nociceptive test was 29.7 ± 0.3 s (n= 36). As expected, thermal hyperalgesia (PWL = 11.4 ± 1.7) was present in the CCI hind paw, 14 days after surgery. Figure 4A and B present the time course of %MPE for GB9 and GB10, given i.t., at doses of 5–100 µg. The compounds showed rapid anti-nociceptive effect with maximum action at 30 min after i.t. injection. The duration of the anti-nociceptive effect, represented by the area under the %MPE versus time curve (AUC), extended from 30 to 180 min after injection (Figure 4C). Both GB9 and GB10 produced significant and dose-dependent anti-nociceptive effects in neuropathic rats by i.t. injection. I.t. injection of the vehicle (20% DMSO) did not affect CCI-induced thermal hyperalgesia. Locomotor function was analysed using the BBB rating scale (Basso et al., 1996) to assess the motor effects of the compounds injected. In the present experiments, control rats treated with i.t. GB9 or GB10 at doses of 5–100 µg exhibited a normal neurological profile (BBB score = 25).
Figure 4.
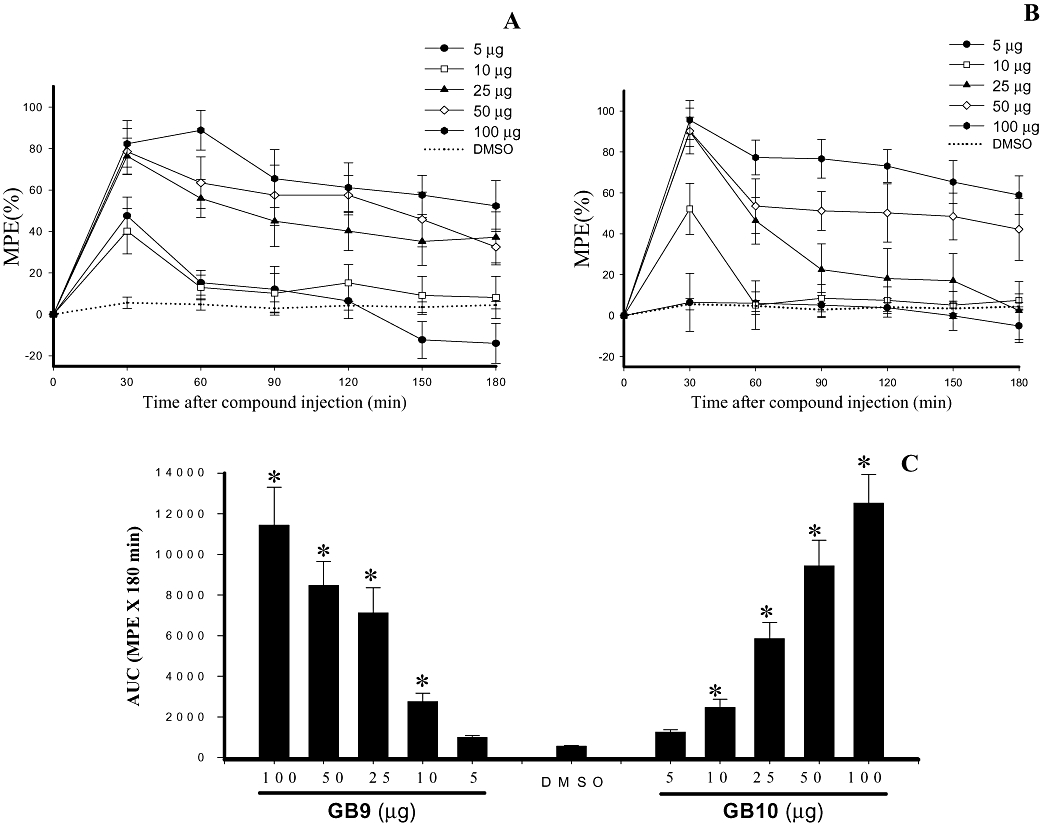
Effects of capnellenes (GB9 and GB10), given i.t., on thermal hyperalgesia evoked by chronic constriction injury (CCI) in rats. (A) Time course for i.t. GB9 over a range of doses. (B) Time course for i.t. GB10 over a range of doses. The x-axis shows the time in minutes from drug injection, the y-axis shows %MPE calculated as a mean of five to six animals per dose. (C) Area under the analgesic effect–time curve (%MPE-time curve ± SEM) for i.t. DMSO (20%) and 5, 10, 25, 50 and 100 µg of GB9 and GB10. The results for the different doses of capnellenes were significantly different from the vehicle (DMSO) group (*P < 0.05) and were dose-dependent.
Anti-nociceptive effect of systemic GB9 on neuropathy
In general, i.t. doses of drugs are approximately 1–2% of their systemic dose, and the doses used in this study were insufficient for a systemic dose–response anti-nociceptive study. Therefore, only GB9 at 10 mg·kg−1 was selected for examination of systemic effects. Results are shown in Figure 5 for day 14 after CCI surgery. The PWL for CCI rats treated with one bolus i.p. injection of GB9 (10 mg·kg−1) was significantly increased to approximately 6–8 s within 1–3 h. Systemic GB9 (10 mg·kg−1) significantly inhibited CCI-induced thermal hyperalgesia behaviour for at least 6 h. I.p. GB9 alone did not produce anti-nociceptive effects or locomotor dysfunction, as determined by the hot water immersion test at 52°C and the BBB rating scale respectively (n= 4).
Figure 5.
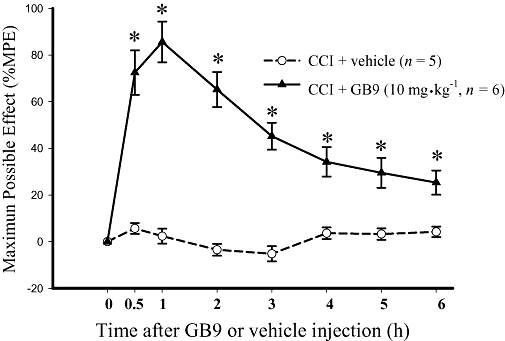
Time course showing the analgesic effect of systemic capnellene, GB9 on chronic constriction injury (CCI)-induced thermal hyperalgesia. GB9 (10 mg·kg−1, i.p.) was administered 14 days after sciatic nerve injury. GB9 significantly inhibited CCI-induced thermal hyperalgesia. * indicates a significant difference compared with the same time point in the CCI plus vehicle group (P < 0.05). Each point or bar represents the mean ± SEM.
Effects of GB9 on CCI-induced changes in spinal microglial cells
As shown in Figure 6, immunohistochemistry was performed using the OX-42 antibody, which labels cells with the microglial surface marker CD11b. The distribution of OX-42 immunoreactivity in the dorsal part of the lumbar spine of the control (Figure 6A), CCI (Figure 6B), and CCI + GB9 (Figure 6C) rats is shown. Fourteen days after CCI, OX-42 immunoreactivity was apparently increased in the dorsal horn on the side ipsilateral to the injury (Figure 6B). At 6 h, i.p. GB9 inhibited CCI-induced up-regulation of OX-42 immunoreactivity (Figure 6C). Higher-magnification images of OX-42 immunoreactivity in the dorsal horn on the ipsilateral side in the control (Figure 6D), CCI (Figure 6E) and CCI + GB9 (Figure 6F) rats are taken from Figure 6A, Figure 6B and Figure 6C respectively. In control rats, OX-42 immunoreactivity was homogeneously distributed throughout the spinal grey matter (Figure 6D). The morphology of microglial cells exhibited the resting-type shape, which has small compact somata bearing many long, thin, ramified processes (Figure 6D, inset). For both immunoreactivity and morphology of OX-42, there were no significant differences between the left and right sides of the spinal cord in the control group. As observed in previous studies of CCI rats, OX-42 immunoreactivity was significantly up-regulated and microglia exhibited an activated phenotype, showing hypertrophy and retraction of cytoplasmic processes, in the superficial dorsal horn (laminae I–III) on the side ipsilateral to the injury (Figure 6E and E, inset). Compared with the contralateral (non-injury) side, a significant shift from resting to activated morphology was found on side ipsilateral to the sciatic nerve injury. In the CCI group, the contralateral dorsal horn showed an immunostaining pattern similar to sham-operated rats. I.p. treatment with GB9 (10 mg·kg−1) after 6 h inhibited CCI-induced up-regulation of OX-42 immunoreactivity and reduced the proportion of the activated phenotype in microglial cells in the ipsilateral dorsal horn of the spinal cord (Figure 6F and F, inset). Quantification of OX-42 immunoreactivity showed that i.p. GB9 significantly inhibited CCI-induced up-regulation of OX-42 immunoreactivity in the lumbar dorsal horn on the side ipsilateral to the injury (Figure 6G). Moreover, compared with the control group, i.p. GB9 alone did not affect OX-42 immunoreactivity and microglial morphology.
Figure 6.
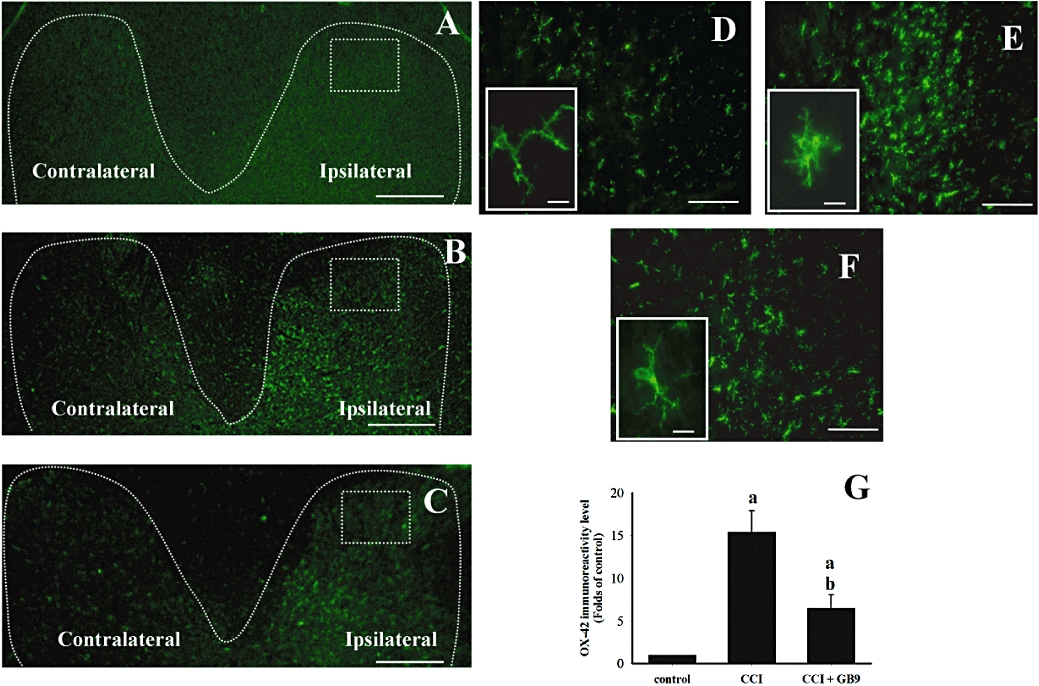
I.p. capnellene, GB9 (10 mg·kg−1, i.p.) inhibits chronic constriction injury (CCI)-induced microglial cell activation on the ipsilateral side of L4-5 spinal cord. The immunostaining images show cells labelled with OX-42 (a microglial cell-specific marker; green) for the spinal cord sections (10 µm) from the control (A and D), CCI (B and E) and CCI plus GB9 (C and F) groups. Panels D, E and F show OX-42 immunoreactivity on the ipsilateral side of the spinal cord from square marked in A, B and C respectively. During the same time period, immunohistochemistry indicates a substantial increase in OX-42 immunoreactivity in the ipsilateral dorsal horn at day 14 after CCI surgery. The CCI-induced up-regulation of OX-42 immunoreactivity was inhibited by 6-h i.p. capnellene. Basal levels of OX-42 signal were observed within the lumbar dorsal horn of the control group (D). The high-magnification inset in D show the typical resting microglial morphology with ramified processes and small soma diameter. In CCI animals (E, inset), microglia exhibited the activated phenotype, marked cellular hypertrophy and retraction of processes compared with the control (D, inset) and CCI + GB9 (F, inset) groups. Quantification of OX-42 immunoreactivity indicated that i.p. significantly but not completely inhibited CCI-induced microglial activation in the ipsilateral dorsal horn of the spinal cord (G). Scale bars: A–C, 200 µm; D–F, 100 µm; D inset, E inset and F inset, 10 µm. a, P < 0.05 compared with the control; b, P < 0.05 compare with the CCI-only group.
Effects of GB9 on CCI-induced up-regulation of COX-2 expression in neuronal and microglial cells
The distribution of COX-2 immunoreactivity in the ipsilateral side of the spinal cord in control (Figure 7A), CCI (Figure 7B), CCI + GB9 (Figure 7C) and GB9 (Figure 7D) rats is shown in Figure 7. Within the laminae (I–IV) of the dorsal horn ipsilateral to the injury, COX-2 was up-regulated after postoperative day 14. The CCI-induced up-regulation of COX-2 was significantly inhibited by i.p. GB9 (10 mg·kg−1) after 6 h (Figure 7E). Compared with the control group, GB9 alone did not change COX-2 immunoreactivity in the spinal dorsal horn (Figure 7E). To identify the cell types that up-regulated COX-2 protein expression after CCI, double immunofluorescence staining of COX-2 was conducted with a neuronal (NeuN) or microglial (OX-42) cell-specific marker. The cellular specificity of COX-2 expression was confirmed, as demonstrated by immunofluorescent staining to detect NeuN (green, Figure 8A, D and G) and COX-2 (red, Figure 8B, E and H) in the dorsal horn in the side of the spinal cord ipsilateral to the injury. The merged images (yellow, Figure 8C, F and I) indicated that COX-2 was colocalized with neuronal cells. Moreover, in the CCI group, clear up-regulation of COX-2 was observed in the neuronal cells compared with the control and the CCI + GB9 group. As shown in Figure 9, CCI-induced COX-2 expression was confirmed in microglial cells in the side of the spinal cord ipsilateral to the injury. Immunofluorescent staining of the microglial cell-specific marker OX-42 (green, Figure 9A, D and G) and COX-2 (red, Figure 9B, E and H) was observed in the dorsal horn in the side of the spinal cord ipsilateral to the injury in the control, CCI and CCI + GB9 groups. The merged images show whether COX-2 was colocalized with microglial cell in the spinal cord (Figure 9C, F and I). In the control and the CCI + GB9 groups, COX-2-positive microglial cells revealed extremely weak immunostaining (Figure 9C and I). In the control group, COX-2 was almost completely colocalized with NeuN, but not with OX-42. However, in the CCI group, not only NeuN, but also OX-42, was colocalized with COX-2 (Figure 9F). I.p. injection of GB9 clearly inhibited CCI-induced COX-2 expression in microglial cells.
Figure 7.
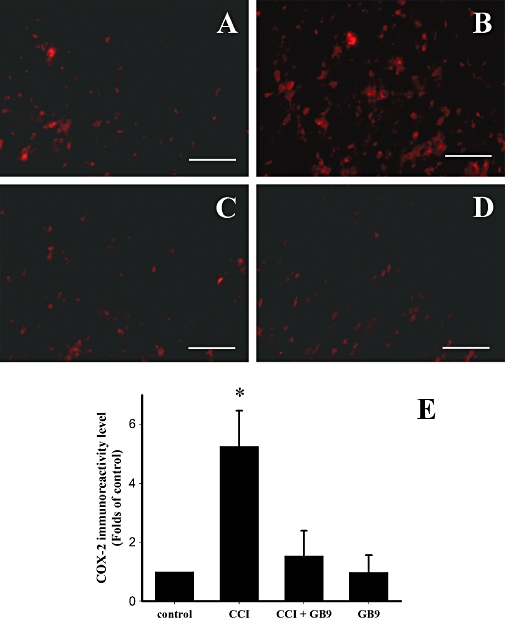
I.p. capnellene, GB9 (10 mg·kg−1, i.p.) inhibited chronic constriction injury (CCI)-induced up-regulation of cyclooxygenase-2 (COX-2) immunoreactivity on the ipsilateral side of the lumbar spinal cord. COX-2 immunoreactivity in the control, CCI, CCI plus GB9, and GB9 groups are shown on panels A, B, C and D respectively. (E) Quantification of the COX-2 immunoreactive-positive area on the ipsilateral side of the lumbar spinal cord. The results showed that 6 h of i.p. GB9 significantly inhibited COX-2 up-regulation on day 14 after CCI. *, significantly different from the CCI group (P < 0.05). Scale bars: 50 µm for all images.
Figure 8.
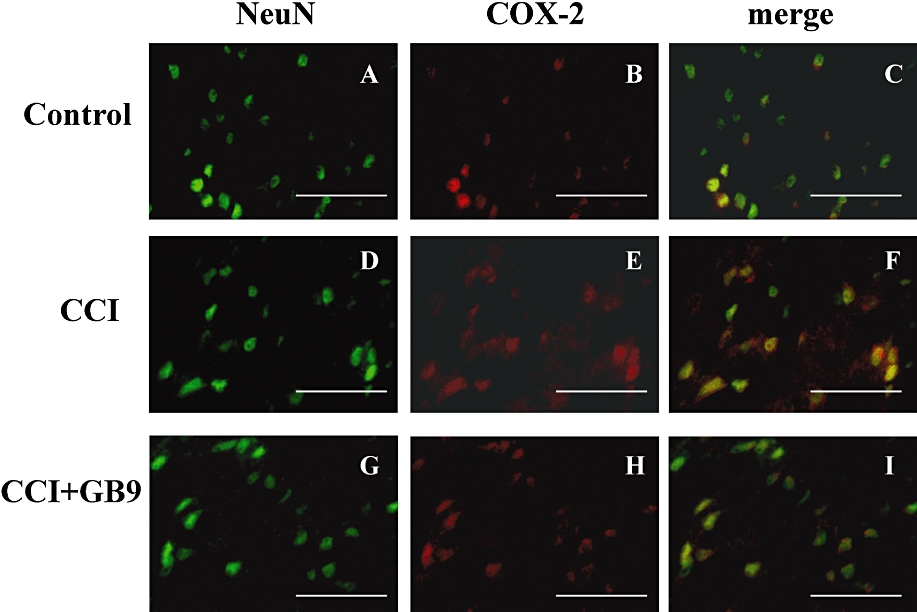
Double-labelled immunofluorescent staining of NeuN (green) and cyclooxygenase-2 (COX-2) (red) in the dorsal region of the lumbar spinal cord ipsilateral to the injury after i.p. GB9 (10 mg·kg−1, i.p.) administration, showing spinal cord sections from the control (A–C), chronic constriction injury (CCI) (D–F), and CCI + GB9 (G–I) groups. The images represent multiple fields examined for each group from three independent immunofluorescence observations. The immunostaining images show cells labelled with NeuN (green) and COX-2 (red) in the spinal cord. The merged images of C, F and I (yellow) indicate colocalization of COX-2 and NeuN (neuronal specific marker) immunoreactive cells in the spinal cord. The results of double immunofluorescent staining from the control, CCI and CCI + GB9 groups all showed that COX-2 was colocalized with NeuN (C, F and I). COX-2 was stronger in the CCI group than in the control and CCI + GB9 groups. Scale bars: 50 µm for all images.
Figure 9.
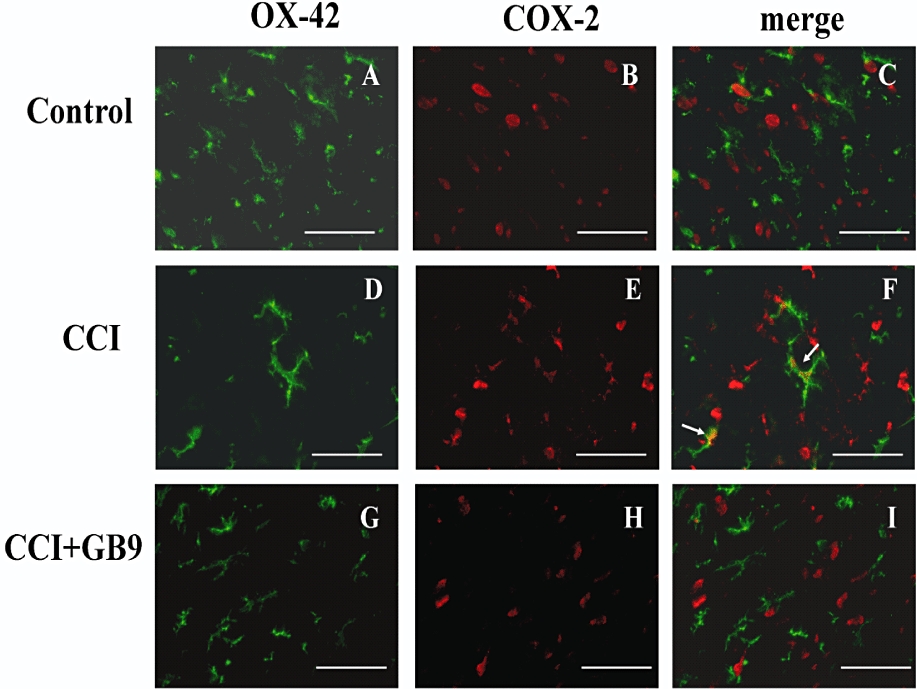
Double-labelled immunofluorescent staining of OX-42 (green) and cyclooxygenase-2 (COX-2) (red) in the dorsal region of the lumbar spinal cord ipsilateral to the injury after i.p. GB9 (10 mg·kg−1, i.p.) administration, showing spinal cord sections from the control (A–C), chronic constriction injury (CCI) (D–F) and CCI + GB9 (G–I) groups. The images represent multiple fields examined for each group from three independent immunofluorescence observations. The immunostaining images show cells labelled with OX-42 (green) and COX-2 (red) in the spinal cord. The merged images of C, F and I indicate colocalization of COX-2 and OX-42 (microglia-specific marker) immunoreactive cells in the spinal cord. The results of double immunofluorescent staining showed that COX-2 is colocalized with OX-42 in the CCI group (F, white arrow). For both the control and CCI + GB9 groups, the OX-42-positive cells were not highly colocalized with COX-2 (C and I). Scale bars: 50 µm for all images.
Discussion
The present study demonstrates that IFN-γ-stimulated microglial cells are an in vitro model for evaluating the effects of anti-neuroinflammatory compounds. The marine-derived capnellene (GB9) and its acetylated derivative (GB10) were able to down-regulate the expression of proinflammatory iNOS and COX-2 protein in IFN-γ-stimulated microglial cells. Also, central administration of capnellene significantly inhibited CCI-induced thermal hyperalgesia in a dose-dependent manner in rats. The immunohistochemical results showed CCI-induced microglia activation and up-regulation of COX-2 protein expression in neuronal and microglial cells in the spinal dorsal horn on the side ipsilateral to the injury. Systemically injected GB9 not only prevented CCI-induced pathological changes in the spinal cord, but also inhibited a neuropathic pain symptom, thermal hyperalgesia.
Two COX isozymes, COX-1 and COX-2, catalyse the rate-limiting steps of prostaglandin and thromboxane synthesis. Prostaglandins are critically involved in peripheral and spinal nociceptive sensitization (Schaible and Schmidt, 1988; Baba et al., 2001; Vanegas and Schaible, 2001). It is well known that in the CNS, both COX-1 and COX-2 are constitutively expressed (Willingale et al., 1997; Ebersberger et al., 1999). The present immunohistochemical results demonstrate that COX-2 immunoreactivity is present in the spinal cord of naïve rats (Figure 7). However, unlike COX-1, COX-2 is an inducible enzyme that increases in the peripheral and central nervous systems following injury or inflammation (Seibert et al., 1994; Zhao et al., 2000), and as such, plays a more important role in neuropathology than COX-1. Inhibition of COX-2, but not COX-1, by selective inhibitors attenuates hyperalgesia in neuropathic rats (Matsunaga et al., 2007). Broom et al. (2004) found that COX-2 protein in the dorsal horn increased in a spared nerve injury model of neuropathic pain. The present immunohistochemical observations also showed a significant increase in COX-2 immunoreactivity in the dorsal horn of the spinal cord ipsilateral to the injured site in CCI rats. This CCI-induced increase in COX-2 and nociceptive sensitization was inhibited by capnellene. These results suggest that capnellene produces an analgesic effect on neuropathy via inhibition of the expression of COX-2 protein in the spinal cord.
There is growing evidence indicating that activation of spinal glia plays a critical role in development and maintenance of pathological pain (Colburn et al., 1999; Fu et al., 1999; Hashizume et al., 2000). Up-regulation of spinal COX, especially COX-2, is involved in the development and/or maintenance of pathological pain states (Zhao et al., 2000; Takeda et al., 2005). However, it remains controversial which cell types in the spinal cord express COX-2 and mediate nociceptive transmission after peripheral nerve injury. The current study, using colocalization immunostaining, was useful in confirming the identity of COX-2 immunoreactive cell types. The present results showed that most of the COX-2 immunoreactivity was colocalized with neuronal, but not microglial, cells in naïve rats. Moreover, significant up-regulation of COX-2 was found in neuronal and microglial cells of the dorsal part of the lumbar spine in CCI rats. In the current study, the immunohistochemical findings for microglia were similar to previous results (Durrenberger et al., 2004); a significantly increased number of COX-2 immunoreactive microglial cells were observed in the ipsilateral dorsal horn of the spinal cord after peripheral injury. However, there is some conflicting evidence about the role of COX-2 in spinal microglial cells, in relation to nociceptive sensitization. In the incision injury and nerve injury models, microglial activation and COX-1 up-regulation in the spinal cord have also been found (Zhu and Eisenach, 2003; Zhu et al., 2003). Zhang et al. (2007) also indicated that spinal microglial activation may play a role in pain processing through the increased expression of COX-1 after formalin injection into the hind paw. Two COX isozymes have been are known to catalyse the rate-limiting steps of prostaglandins synthesis, and subsequently to modulate nociceptive sensitization. However, COX-1 requires a higher arachidonic acid concentration than COX-2 for catalysis (Versteeg et al., 1999). In our unpublished in vitro observations, neither IFN-γ nor capnellene significantly changed the expression of COX-1 protein in microglial cells. We suggest therefore that the anti-nociceptive effects of capnellene are related to decreased COX-2 expression in microglia or neurones.
In the present study, the activation and localization of COX-2 in astrocytes were not examined. The relative contributions of microglia and astrocytes to neuropathic pain are still under investigation. Peripheral nerve injury activates not only microglia but also astrocytes. The activation of spinal astrocytes also plays an important role in the maintenance of nociceptive sensitization in various neuropathic pain models (Colburn et al., 1997; Marchand et al., 2005; Ji et al., 2006; Zhuang et al., 2006; Vega-Avelaira et al., 2007). Similar to microglial activation, activated astrocytes exhibit hypertrophied cell bodies and increased production/release of proinflammatory mediators (Raghavendra et al., 2004; Watkins and Maier, 2002). Recent studies suggest that microglia are more important in the development of nociceptive hypersensitivity (Cui et al., 2006; 2008; Mika et al., 2007; 2009;). Minghetti and Levi (1995) found that microglial cells are able to release higher levels of the proinflammatory mediators PGE2, PGD2 and TXB2 than astrocytes, after stimulation by a 100-fold lower concentration of endotoxin. Moreover, they also suggested that COX-2 is the major isoform expressed in activated microglial cells, as a source of prostanoids in the CNS (Levi et al., 1998; Minghetti and Levi, 1998). It will be of considerable interest to examine the role of capnellene in CCI-induced astrocyte activation in future studies.
The pharmacological inhibition of microglial activation attenuated nociceptive hypersensitivity in neuropathy (Raghavendra et al., 2003; Ledeboer et al., 2005; Cui et al., 2006; 2008; Mika et al., 2007; 2009;). However, an inhibitor of microglial activation, minocycline, was effective in preventing, but not reducing, established allodynia and hyperalgesia (Raghavendra et al., 2003; Ledeboer et al., 2005). Minocycline inhibited microglial activation of microglia, without affecting astrocytes or neurones (Tikka and Koistinaho, 2001; Wu et al., 2002). Microglia are cells in the CNS that exhibit an early response after peripheral injuries, and the consequently released pro-inflammatory products lead to astrocyte and neurone activation, which, in turn, maintains a long-term pathological state (Svensson et al., 1993; Kreutzberg, 1996). In the present study, capnellene significantly inhibited proinflammatory protein expression in IFN-γ-stimulated microglia, and also inhibited the CCI-induced elevation of microglial and neuronal COX-2 in the spinal cord. Moreover, the CCI-induced activation of spinal microglia was significantly inhibited by systemic treatment with capnellene GB9. The inhibitory effect of this capnellene on CCI-induced changes in astrocytes was not investigated. The present results provide strong evidence that the pharmacological disruption of microglial cell activation can produce potential anti-neuroinflammatory and anti-nociceptive effects in a model of neuropathic pain.
The inducible isoforms of NOS, iNOS, which produces large amounts of NO, participates in the pathophysiology of neuropathic pain (Meller et al., 1994; Levy et al., 2001). However, the central expression of iNOS in the neuropathic pain model remains controversial. In the present study, we did not find any positive staining of iNOS in the spinal cord sections of the experimental groups. This result was similar to that of De Alba et al. (2006), who found no central immunostaining for iNOS in CCI-induced neuropathic pain. Moreover, the lack of central iNOS expression in a model of peripheral inflammation is consistent with published reports (Goff et al., 1998; Dolan et al., 2000; De Alba et al., 2006). Many studies have demonstrated that iNOS is expressed locally in neuropathic pain or the carrageenan-induced inflammatory pain models (Salvemini et al., 1996; Levy and Zochodne, 1998; Handy and Moore, 1998; Levy et al., 1999; Levy et al., 2001). In an earlier study, using the present anti-iNOS antibody, we found local iNOS immunostaining in carrageenan-injected paw tissue in rats (Jean et al., 2008). The above results suggested that the NO derived from iNOS would have a predominantly peripheral effect and that it plays a role in nociceptive sensitization. The present and previous in vitro data showed that capnellene inhibited iNOS protein expression in IFN-γ-stimulated microglia and lipopolysaccharide-stimulated macrophage cells (Chang et al., 2008). The systemic administration of capnellene attenuated thermal hyperalgesia in neuropathic rats. Thus, the possibility that capnellene could attenuate the expression of peripheral iNOS protein cannot be excluded.
Today, few pharmacological agents are effective in alleviating neuropathic pain. Opioids provide some relief, but are limited by tolerance and undesirable side effects. Furthermore, non-selective COX inhibitors are clinically limited by adverse events, especially gastrointestinal and renal dysfunction. Recently, ziconotide, a marine-derived drug, has shown potential for inhibition of neuropathy-induced nociceptive behaviours and no opioid-like withdrawal syndrome has been reported. However, ziconotide must be administered intrathecally (Penn and Paice, 2000). The present study showed that not only i.t. but also systemic capnellene significantly attenuated nociceptive sensitization in neuropathic rats. Moreover, i.p. capnellene did not produce anti-nociceptive effects and neurological dysfunctions in normal rats. Capnellene, with a chemical structure that lacks a steroid or opioid backbone, was observed to exhibit anti-neuroinflammatory activity in in vitro and in vivo studies. Therefore, capnellene may be suitable for the development of orally or intrathecally administered drugs. We propose that capnellene can be considered a member of the group of drugs referred to as an anti-inflammatory compounds unrelated to steroids, which have significant therapeutic advantages.
In summary, the present study demonstrates that capnellene significantly inhibits expression of two proinflammatory proteins, iNOS and COX-2, in IFN-γ-stimulated microglial cells, in a dose-dependent manner. Our in vivo neuroinflammatory study found that both centrally and systemically administered capnellene can reverse thermal hyperalgesia induced by sciatic nerve injury (CCI). An increasing number of studies have shown that pathological pain states can be blocked by drugs that disrupt microglial activation (Sweitzer et al., 2001; Milligan et al., 2003; Ledeboer et al., 2005). We consider the attenuation of neuropathic pain by capnellene is likely to be associated with its inhibitory effects on activated microglial cells and inflammatory neurones in the spinal cord, which are implicated in the development and maintenance of nociceptive hypersensitization. Additionally, capnellene alone did not produce any neurological dysfunction or nociceptive behaviour in normal rats. The present results suggest that the marine-derived natural compound, capnellene, could be a potent therapeutic agent for neuroinflammatory disease and neuropathic pain.
Acknowledgments
The study was partly supported by the Asia-Pacific Ocean Research Center, National Sun Yat-sen University (97C031701) and the National Science Council of Taiwan (NSC 96-2313-B-110-001-MY3).
Glossary
Abbreviations:
- CCI
chronic constriction injury
- COX
cyclooxygenase
- iNOS
inducible nitric oxide synthase
- IFN-γ
interferon-γ
- MPE
maximum possible effect
- PWL
paw withdrawal latency
Conflict of interest
None.
References
- Arner S, Meyerson BA. Lack of analgesic effect of opioids on neuropathic and idiopathic forms of pain. Pain. 1988;33:11–23. doi: 10.1016/0304-3959(88)90198-4. [DOI] [PubMed] [Google Scholar]
- Baba H, Kohno T, Moore KA, Woolf CJ. Direct activation of rat spinal dorsal horn neurons by prostaglandin E2. J Neurosci. 2001;21:1750–1756. doi: 10.1523/JNEUROSCI.21-05-01750.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basso DM, Beattie MS, Bresnahan JC. Graded histological and locomotor outcomes after spinal cord contusion using the NYU weight-drop device versus transection. Exp Neurol. 1996;139:244–256. doi: 10.1006/exnr.1996.0098. [DOI] [PubMed] [Google Scholar]
- Bennett GJ, Xie YK. A peripheral mononeuropathy in rat that produces disorders of pain sensation like those seen in man. Pain. 1988;33:87–107. doi: 10.1016/0304-3959(88)90209-6. [DOI] [PubMed] [Google Scholar]
- Blasi E, Barluzzi R, Bocchini V, Mazzolla R, Bistoni F. Immortalization of murine microglial cells by a v-raf/v-myc carrying retrovirus. J Neuroimmunol. 1990;27:229–237. doi: 10.1016/0165-5728(90)90073-v. [DOI] [PubMed] [Google Scholar]
- Broom DC, Samad TA, Kohno T, Tegeder I, Geisslinger G, Woolf CJ. Cyclooxygenase 2 expression in the spared nerve injury model of neuropathic pain. Neuroscience. 2004;124:891–900. doi: 10.1016/j.neuroscience.2004.01.003. [DOI] [PubMed] [Google Scholar]
- Chang CH, Wen ZH, Wang SK, Duh CY. Capnellenes from the Formosan soft coral Capnella imbricata. J Nat Prod. 2008;71:619–621. doi: 10.1021/np0706116. [DOI] [PubMed] [Google Scholar]
- Chen WF, Jean YH, Sung CS, Huang SY, Wu GJ Ho JT, et al. Intrathecally injected granulocyte colony-stimulating factor produced neuroprotective effects in spinal cord ischemia via the mitogen-activated protein kinase and Akt pathways. Neuroscience. 2008;153:31–43. doi: 10.1016/j.neuroscience.2008.01.062. [DOI] [PubMed] [Google Scholar]
- Clark CM, Jr, Lee DA. Prevention and treatment of the complications of diabetes mellitus. N Engl J Med. 1995;332:1210–1217. doi: 10.1056/NEJM199505043321807. [DOI] [PubMed] [Google Scholar]
- Colburn RW, DeLeo JA, Rickman AJ, Yeager MP, Kwon P, Hickey WF. Dissociation of microglial activation and neuropathic pain behaviours following peripheral nerve injury in the rat. J Neuroimmunol. 1997;79:163–175. doi: 10.1016/s0165-5728(97)00119-7. [DOI] [PubMed] [Google Scholar]
- Colburn RW, Rickman AJ, DeLeo JA. The effect of site and type of nerve injury on spinal glial activation and neuropathic pain behavior. Exp Neurol. 1999;157:289–304. doi: 10.1006/exnr.1999.7065. [DOI] [PubMed] [Google Scholar]
- Courteix C, Eschalier A, Lavarenne J. Streptozocin-induced diabetic rats: behavioural evidence for a model of chronic pain. Pain. 1993;53:81–88. doi: 10.1016/0304-3959(93)90059-X. [DOI] [PubMed] [Google Scholar]
- Cui Y, Chen Y, Zhi JL, Guo RX, Feng JQ, Chen PX. Activation of p38 mitogen-activated protein kinase in spinal microglia mediates morphine antinociceptive tolerance. Brain Res. 2006;1069:235–243. doi: 10.1016/j.brainres.2005.11.066. [DOI] [PubMed] [Google Scholar]
- Cui Y, Liao XX, Liu W, Guo RX, Wu ZZ, Zhao CM, et al. A novel role of minocycline: attenuating morphine antinociceptive tolerance by inhibition of p38 MAPK in the activated spinal microglia. Brain Behav Immun. 2008;22:114–123. doi: 10.1016/j.bbi.2007.07.014. [DOI] [PubMed] [Google Scholar]
- De Alba J, Clayton NM, Collins SD, Colthup P, Chessell I, Knowles RG. GW274150, a novel and highly selective inhibitor of the inducible isoform of nitric oxide synthase (iNOS), shows analgesic effects in rat models of inflammatory and neuropathic pain. Pain. 2006;120:170–181. doi: 10.1016/j.pain.2005.10.028. [DOI] [PubMed] [Google Scholar]
- Dolan S, Field LC, Nolan AM. The role of nitric oxide and prostaglandin signaling pathways in spinal nociceptive processing in chronic inflammation. Pain. 2000;86:311–320. doi: 10.1016/S0304-3959(00)00262-1. [DOI] [PubMed] [Google Scholar]
- Durrenberger PF, Facer P, Gray RA, Chessell IP, Naylor A, Bountra C, et al. Cyclooxygenase-2 (Cox-2) in injured human nerve and a rat model of nerve injury. J Peripher Nerv Syst. 2004;9:15–25. doi: 10.1111/j.1085-9489.2004.09104.x. [DOI] [PubMed] [Google Scholar]
- Ebersberger A, Grubb BD, Willingale HL, Gardiner NJ, Nebe J, Schaible HG. The intraspinal release of prostaglandin E2 in a model of acute arthritis is accompanied by an up-regulation of cyclo-oxygenase-2 in the spinal cord. Neuroscience. 1999;93:775–781. doi: 10.1016/s0306-4522(99)00164-5. [DOI] [PubMed] [Google Scholar]
- Fu KY, Light AR, Matsushima GK, Maixner W. Microglial reactions after subcutaneous formalin injection into the rat hind paw. Brain Res. 1999;825:59–67. doi: 10.1016/s0006-8993(99)01186-5. [DOI] [PubMed] [Google Scholar]
- Goff JR, Burkey AR, Goff DJ, Jasmin L. Reorganization of the spinal dorsal horn in models of chronic pain: correlation with behaviour. Neuroscience. 1998;82:559–574. doi: 10.1016/s0306-4522(97)00298-4. [DOI] [PubMed] [Google Scholar]
- Gordon DB, Love G. Pharmacologic management of neuropathic pain. Pain Manag Nurs. 2004;5:19–33. doi: 10.1016/j.pmn.2004.10.005. [DOI] [PubMed] [Google Scholar]
- Hains BC, Waxman SG. Activated microglia contribute to the maintenance of chronic pain after spinal cord injury. J Neurosci. 2006;26:4308–4317. doi: 10.1523/JNEUROSCI.0003-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Handy RL, Moore PK. A comparison of the effects of L-NAME, 7-NI and L-NIL on carrageenan-induced hindpaw oedema and NOS activity. Br J Pharmacol. 1998;123:1119–1126. doi: 10.1038/sj.bjp.0701735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hargreaves K, Dubner R, Brown F, Flores C, Joris J. A new and sensitive method for measuring thermal nociception in cutaneous hyperalgesia. Pain. 1988;32:77–88. doi: 10.1016/0304-3959(88)90026-7. [DOI] [PubMed] [Google Scholar]
- Hashizume H, DeLeo JA, Colburn RW, Weinstein JN. Spinal glial activation and cytokine expression after lumbar root injury in the rat. Spine. 2000;25:1206–1217. doi: 10.1097/00007632-200005150-00003. [DOI] [PubMed] [Google Scholar]
- Ji RR, Kawasaki Y, Zhuang ZY, Wen YR, Decosterd I. Possible role of spinal astrocytes in maintaining chronic pain sensitization: review of current evidence with focus on bFGF/JNK pathway. Neuron Glia Biol. 2006;2:259–269. doi: 10.1017/S1740925X07000403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jean YH, Chen WF, Duh CY, Huang SY, Hsu CH, Lin CS, et al. Inducible nitric oxide synthase and cyclooxygenase-2 participate in anti-inflammatory and analgesic effects of the natural marine compound lemnalol from Formosan soft coral Lemnalia cervicorni. Eur J Pharmacol. 2008;578:323–331. doi: 10.1016/j.ejphar.2007.08.048. [DOI] [PubMed] [Google Scholar]
- Kaisin M, Sheikh YM, Durham LJ, Djerassi C, Tursch B, Daloze D, et al. Capnellane – a new tricyclic sesquiterpene skeleton from the soft coral Capnella imbricata. Tetrahedron Lett. 1974;15:2239–2242. [Google Scholar]
- Kalso E, Vainio A. Morphine and oxycodone hydrochloride in the management of cancer pain. Clin Pharmacol Ther. 1990;47:639–646. doi: 10.1038/clpt.1990.85. [DOI] [PubMed] [Google Scholar]
- Kim HY, Park EJ, Joe EH, Jou I. Curcumin suppresses Janus kinase-STAT inflammatory signaling through activation of Src homology 2 domain-containing tyrosine phosphatase 2 in brain microglia. J Immunol. 2003;171:6072–6079. doi: 10.4049/jimmunol.171.11.6072. [DOI] [PubMed] [Google Scholar]
- Kreutzberg GW. Microglia: a sensor for pathological events in the CNS. Trends Neurosci. 1996;19:312–318. doi: 10.1016/0166-2236(96)10049-7. [DOI] [PubMed] [Google Scholar]
- Ladeby R, Wirenfeldt M, Garcia-Ovejero D, Fenger C, Dissing-Olesen L, Dalmau I, et al. Microglial cell population dynamics in the injured adult central nervous system. Brain Res Brain Res Rev. 2005;48:196–206. doi: 10.1016/j.brainresrev.2004.12.009. [DOI] [PubMed] [Google Scholar]
- Ledeboer A, Sloane EM, Milligan ED, Frank MG, Mahony JH, Maier SF, et al. Minocycline attenuates mechanical allodynia and proinflammatory cytokine expression in rat models of pain facilitation. Pain. 2005;115:71–83. doi: 10.1016/j.pain.2005.02.009. [DOI] [PubMed] [Google Scholar]
- Levi G, Minghetti L, Aloisi F. Regulation of prostanoid synthesis in microglial cells and effects of prostaglandin E2 on microglial functions. Biochimie. 1998;80:899–904. doi: 10.1016/s0300-9084(00)88886-0. [DOI] [PubMed] [Google Scholar]
- Levy D, Zochodne DW. Local nitric oxide synthase activity in a model of neuropathic pain. Eur J Neurosci. 1998;10:1846–1855. doi: 10.1046/j.1460-9568.1998.00186.x. [DOI] [PubMed] [Google Scholar]
- Levy D, Hoke A, Zochodne DW. Local expression of inducible nitric oxide synthase in an animal model of neuropathic pain. Neurosci Lett. 1999;260:207–209. doi: 10.1016/s0304-3940(98)00982-3. [DOI] [PubMed] [Google Scholar]
- Levy D, Kubes P, Zochodne DW. Delayed peripheral nerve degeneration, regeneration, and pain in mice lacking inducible nitric oxide synthase. J Neuropathol Exp Neurol. 2001;60:411–421. doi: 10.1093/jnen/60.5.411. [DOI] [PubMed] [Google Scholar]
- Lowry OH, Rosebrough NJ, Farr AL, Randall RJ. Protein measurement with the Folin phenol reagent. J Biol Chem. 1951;193:265–275. [PubMed] [Google Scholar]
- Marchand F, Perretti M, McMahon SB. Role of the immune system in chronic pain. Nat Rev Neurosci. 2005;6:521–532. doi: 10.1038/nrn1700. [DOI] [PubMed] [Google Scholar]
- Matsunaga A, Kawamoto M, Shiraishi S, Yasuda T, Kajiyama S, Kurita S, et al. Intrathecally administered COX-2 but not COX-1 or COX-3 inhibitors attenuate streptozotocin-induced mechanical hyperalgesia in rats. Eur J Pharmacol. 2007;554:12–17. doi: 10.1016/j.ejphar.2006.09.072. [DOI] [PubMed] [Google Scholar]
- McQuay HJ, Jadad AR, Carroll D, Faura C, Glynn CJ, Moore RA, et al. Opioid sensitivity of chronic pain: a patient-controlled analgesia method. Anaesthesia. 1992;47:757–767. doi: 10.1111/j.1365-2044.1992.tb03253.x. [DOI] [PubMed] [Google Scholar]
- Meller ST, Dykstra C, Grzybycki D, Murphy S, Gebhart GF. The possible role of glia in nociceptive processing and hyperalgesia in the spinal cord of the rat. Neuropharmacology. 1994;33:1471–1478. doi: 10.1016/0028-3908(94)90051-5. [DOI] [PubMed] [Google Scholar]
- Mika J, Wawrzczak-Bargiela A, Osikowicz M, Makuch W, Przewlocka B. Attenuation of morphine tolerance by minocycline and pentoxifylline in naive and neuropathic mice. Brain Behav Immun. 2009;23:75–84. doi: 10.1016/j.bbi.2008.07.005. [DOI] [PubMed] [Google Scholar]
- Mika J, Osikowicz M, Makuch W, Przewlocka B. Minocycline and pentoxifylline attenuate allodynia and hyperalgesia and potentiate the effects of morphine in rat and mouse models of neuropathic pain. Eur J Pharmacol. 2007;560:142–149. doi: 10.1016/j.ejphar.2007.01.013. [DOI] [PubMed] [Google Scholar]
- Milligan ED, Twining C, Chacur M, Biedenkapp J, O'Connor K, Poole S, et al. Spinal glia and proinflammatory cytokines mediate mirror-image neuropathic pain in rats. J Neurosci. 2003;23:1026–1040. doi: 10.1523/JNEUROSCI.23-03-01026.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minghetti L, Levi G. Induction of prostanoid biosynthesis by bacterial lipopolysaccharide and isoproterenol in rat microglial cultures. J Neurochem. 1995;65:2690–2698. doi: 10.1046/j.1471-4159.1995.65062690.x. [DOI] [PubMed] [Google Scholar]
- Minghetti L, Levi G. Microglia as effector cells in brain damage and repair: focus on prostanoids and nitric oxide. Prog Neurobiol. 1998;54:99–125. doi: 10.1016/s0301-0082(97)00052-x. [DOI] [PubMed] [Google Scholar]
- Myers RR, Campana WM, Shubayev VI. The role of neuroinflammation in neuropathic pain: mechanisms and therapeutic targets. Drug Discov Today. 2006;11:8–20. doi: 10.1016/S1359-6446(05)03637-8. [DOI] [PubMed] [Google Scholar]
- Ossipov MH, Porreca F. Challenges in the development of novel treatment strategies for neuropathic pain. NeuroRx. 2005;2:650–661. doi: 10.1602/neurorx.2.4.650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penn RD, Paice JA. Adverse effects associated with the intrathecal administration of ziconotide. Pain. 2000;85:291–296. doi: 10.1016/s0304-3959(99)00254-7. [DOI] [PubMed] [Google Scholar]
- Portenoy RK, Foley KM, Inturrisi CE. The nature of opioid responsiveness and its implications for neuropathic pain: new hypotheses derived from studies of opioid infusions. Pain. 1990;43:273–286. doi: 10.1016/0304-3959(90)90025-9. [DOI] [PubMed] [Google Scholar]
- Raghavendra V, Tanga F, DeLeo JA. Inhibition of microglial activation attenuates the development but not existing hypersensitivity in a rat model of neuropathy. J Pharmacol Exp Ther. 2003;306:624–630. doi: 10.1124/jpet.103.052407. [DOI] [PubMed] [Google Scholar]
- Raghavendra V, Tanga FY, DeLeo JA. Complete Freunds adjuvant-induced peripheral inflammation evokes glial activation and proinflammatory cytokine expression in the CNS. Eur J Neurosci. 2004;20:467–473. doi: 10.1111/j.1460-9568.2004.03514.x. [DOI] [PubMed] [Google Scholar]
- Rowland M, Tozer TN. Assessment of AUC. In: Balado D, editor. Clinical Pharmacokinetics: Concepts and Applications. Media, PA: Williams and Wilkins; 1995. pp. 469–470. [Google Scholar]
- Salvemini D, Wang ZQ, Bourdon DM, Stern MK, Currie MG, Manning PT. Evidence of peroxynitrite involvement in the carrageenan-induced rat paw edema. Eur J Pharmacol. 1996;303:217–220. doi: 10.1016/0014-2999(96)00140-9. [DOI] [PubMed] [Google Scholar]
- Schaible HG, Schmidt RF. Excitation and sensitization of fine articular afferents from cat's knee joint by prostaglandin E2. J Physiol. 1988;403:91–104. doi: 10.1113/jphysiol.1988.sp017240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seibert K, Zhang Y, Leahy K, Hauser S, Masferrer J, Perkins W, et al. Pharmacological and biochemical demonstration of the role of cyclooxygenase 2 in inflammation and pain. Proc Natl Acad Sci USA. 1994;91:12013–12017. doi: 10.1073/pnas.91.25.12013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheikh YM, Singy G, Kaisin M, Eggert H, Djerassi C, Tursch B, et al. Chemical studies of marine invertebrates-XIV. Four representatives of a novel sesquiterpene class – the capnellane skeleton. Tetrahedron. 1976;32:1171–1178. [Google Scholar]
- Sung B, Lim G, Mao J. Altered expression and uptake activity of spinal glutamate transporters after nerve injury contribute to the pathogenesis of neuropathic pain in rats. J Neurosci. 2003;23:2899–2910. doi: 10.1523/JNEUROSCI.23-07-02899.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svensson M, Eriksson P, Persson JK, Molander C, Arvidsson J, Aldskogius H. The response of central glia to peripheral nerve injury. Brain Res Bull. 1993;30:499–506. doi: 10.1016/0361-9230(93)90284-i. [DOI] [PubMed] [Google Scholar]
- Sweitzer SM, Schubert P, DeLeo JA. Propentofylline, a glial modulating agent, exhibits antiallodynic properties in a rat model of neuropathic pain. J Pharmacol Exp Ther. 2001;297:1210–1217. [PubMed] [Google Scholar]
- Takeda K, Sawamura S, Tamai H, Sekiyama H, Hanaoka K. Role for cyclooxygenase 2 in the development and maintenance of neuropathic pain and spinal glial activation. Anesthesiology. 2005;103:837–844. doi: 10.1097/00000542-200510000-00023. [DOI] [PubMed] [Google Scholar]
- Tikka TM, Koistinaho JE. Minocycline provides neuroprotection against N-methyl-D-aspartate neurotoxicity by inhibiting microglia. J Immunol. 2001;166:7527–7533. doi: 10.4049/jimmunol.166.12.7527. [DOI] [PubMed] [Google Scholar]
- Vanegas H, Schaible HG. Prostaglandins and cyclooxygenases in the spinal cord. Prog Neurobiol. 2001;64:327–363. doi: 10.1016/s0301-0082(00)00063-0. [DOI] [PubMed] [Google Scholar]
- Vega-Avelaira D, Moss A, Fitzgerald M. Age-related changes in the spinal cord microglial and astrocytic response profile to nerve injury. Brain Behav Immun. 2007;21:617–623. doi: 10.1016/j.bbi.2006.10.007. [DOI] [PubMed] [Google Scholar]
- Versteeg HH, van Bergen en Henegouwen PM, van Deventer SJ, Peppelenbosch MP. Cyclooxygenase-dependent signalling: molecular events and consequences. FEBS Lett. 1999;445:1–5. doi: 10.1016/s0014-5793(99)00105-2. [DOI] [PubMed] [Google Scholar]
- Vilhardt F. Microglia: phagocyte and glia cell. Int J Biochem Cell Biol. 2005;37:17–21. doi: 10.1016/j.biocel.2004.06.010. [DOI] [PubMed] [Google Scholar]
- Watkins LR, Maier SF. Beyond neurons: evidence that immune and glial cells contribute to pathological pain states. Physiol Rev. 2002;82:981–1011. doi: 10.1152/physrev.00011.2002. [DOI] [PubMed] [Google Scholar]
- Wen ZH, Chang YC, Cherng CH, Wang JJ, Tao PL, Wong CS. Increasing of intrathecal CSF excitatory amino acids concentration following morphine challenge in morphine-tolerant rats. Brain Res. 2004;995:253–259. doi: 10.1016/j.brainres.2003.10.008. [DOI] [PubMed] [Google Scholar]
- Willingale HL, Gardiner NJ, McLymont N, Giblett S, Grubb BD. Prostanoids synthesized by cyclo-oxygenase isoforms in rat spinal cord and their contribution to the development of neuronal hyperexcitability. Br J Pharmacol. 1997;122:1593–1604. doi: 10.1038/sj.bjp.0701548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu DC, Jackson-Lewis V, Vila M, Tieu K, Teismann P, Vadseth C, et al. Blockade of microglial activation is neuroprotective in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine mouse model of Parkinson disease. J Neurosci. 2002;22:1763–1771. doi: 10.1523/JNEUROSCI.22-05-01763.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu GJ, Chen WF, Sung CS, Jean YH, Shih CM, Shyu CY, et al. Preventive effects of intrathecal methylprednisolone administration on spinal cord ischemia in rats: the role of excitatory amino acid metabolizing systems. Neuroscience. 2007;147:294–303. doi: 10.1016/j.neuroscience.2007.04.040. [DOI] [PubMed] [Google Scholar]
- Zhang FY, Wan Y, Zhang ZK, Light AR, Fu KY. Peripheral formalin injection induces long-lasting increases in cyclooxygenase 1 expression by microglia in the spinal cord. J Pain. 2007;8:110–117. doi: 10.1016/j.jpain.2006.06.006. [DOI] [PubMed] [Google Scholar]
- Zhao Z, Chen SR, Eisenach JC, Busija DW, Pan HL. Spinal cyclooxygenase-2 is involved in development of allodynia after nerve injury in rats. Neuroscience. 2000;97:743–748. doi: 10.1016/s0306-4522(00)00052-x. [DOI] [PubMed] [Google Scholar]
- Zhu X, Conklin D, Eisenach JC. Cyclooxygenase-1 in the spinal cord plays an important role in postoperative pain. Pain. 2003;104:15–23. doi: 10.1016/s0304-3959(02)00465-7. [DOI] [PubMed] [Google Scholar]
- Zhu X, Eisenach JC. Cyclooxygenase-1 in the spinal cord is altered after peripheral nerve injury. Anesthesiology. 2003;99:1175–1179. doi: 10.1097/00000542-200311000-00026. [DOI] [PubMed] [Google Scholar]
- Zhuang ZY, Wen YR, Zhang DR, Borsello T, Bonny C, Strichartz GR, et al. A peptide c-Jun N-terminal kinase (JNK) inhibitor blocks mechanical allodynia after spinal nerve ligation: respective roles of JNK activation in primary sensory neurons and spinal astrocytes for neuropathic pain development and maintenance. J Neurosci. 2006;26:3551–3560. doi: 10.1523/JNEUROSCI.5290-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zurek JR, Nadeson R, Goodchild CS. Spinal and supraspinal components of opioid antinociception in streptozotocin induced diabetic neuropathy in rats. Pain. 2001;90:57–63. doi: 10.1016/s0304-3959(00)00386-9. [DOI] [PubMed] [Google Scholar]