

Abstract
Background and purpose:
Diphenyleneiodonium (DPI) is often used as an NADPH oxidase inhibitor, but is increasingly being found to have unrelated side effects. We investigated its effects on smooth muscle contractions and the related mechanisms.
Experimental approach:
We studied isometric contractions in smooth muscle strips from bovine trachea. Cholinesterase activity was measured using a spectrophotometric assay; internal Ca2+ pump activity was assessed by Ca2+ uptake into smooth muscle microsomes.
Key results:
Contractions to acetylcholine were markedly enhanced by DPI (10−4 M), whereas those to carbachol (CCh) were not, suggesting a possible inhibition of cholinesterase. DPI markedly suppressed contractions evoked by CCh, KCl and 5-HT, and also unmasked phasic activity in otherwise sustained responses. Direct biochemical assays confirmed that DPI was a potent inhibitor of acetylcholinesterase and butyrylcholinesterase (IC50∼8 × 10−6 M and 6 × 10−7 M, respectively), following a readily reversible, mixed non-competitive type of inhibition. The inhibitory effects of DPI on CCh contractions were not mimicked by another NADPH oxidase inhibitor (apocynin), nor the Src inhibitors PP1 or PP2, ruling out an action through the NADPH oxidase signalling pathway. Several features of the DPI-mediated suppression of agonist-evoked responses (i.e. suppression of peak magnitudes and unmasking of phasic activity) are similar to those of cyclopiazonic acid, an inhibitor of the internal Ca2+ pump. Direct measurement of microsomal Ca2+ uptake revealed that DPI modestly inhibits the internal Ca2+ pump.
Conclusions and implications:
DPI inhibits cholinesterase activity and the internal Ca2+ pump in tracheal smooth muscle.
Keywords: airway smooth muscle, contraction, NADPH oxidase, cholinesterase, SERCA
Introduction
The primary function of smooth muscle is the conversion of biochemical energy into physical tension, leading to forceful cell shortening. The central mechanism in this energy conversion is the interaction between actin and myosin filaments (the ‘sliding filament theory’) powered by the ATPase activity of myosin and triggered by phosphorylation of myosin by myosin light chain kinase. However, an increasing number and variety of other signalling events are being found to modulate this excitation–contraction coupling. One recently identified player is c-src (Wijetunge and Hughes 1996; 2007; Ibitayo et al., 1998; Ishida et al., 1999; Brandt et al., 2002; Tolloczko et al., 2002; Hirshman et al., 2005; Krymskaya et al., 2005).
Src or c-src is a family of proto-oncogenic tyrosine kinases originally discovered in chickens as close relatives of the cancer-forming retroviruses v-src, but which have since been shown to play roles in many other cell types and in many other cellular processes: one of these includes excitation–contraction coupling in smooth muscle. In rat tracheal smooth muscle (TSM), Src modulates serotonin-evoked Ca2+ signalling (by regulating phosphatidylinositol-3,4-bisphosphate levels and Ca2+ influx) and contraction (Tolloczko et al., 2002). In human airway smooth muscle cells, Src mediates thromboxane- (Suzuki et al., 2004) and leukotriene D4- (Ravasi et al., 2006) induced DNA synthesis and cell proliferation. In guinea pig airway smooth muscle, Src may contribute to airway hyper-responsiveness via a mechanism involving increased Ca2+ sensitization (Oguma et al., 2007). Src appears to be important in excitation–contraction coupling of other non-airway tissues, including gastrointestinal (Ross et al., 2007) and vascular (Wijetunge and Hughes, 2007) smooth muscles.
Upstream from Src is the oxygen radical-producing enzyme, NADPH oxidase. Peroxide-mediated activation of Src has been investigated in pulmonary arterial myocytes, where it contributes to the regulation of [Ca2+]i and excitation–contraction coupling (Rathore et al., 2008). Superoxide derived from NADPH oxidase up-regulates the expression of phosphodiesterase 5, and thus increases the excitability of human vascular smooth muscle (Muzaffar et al., 2008).
Thus, there is increasing interest in the roles of NADPH oxidase and Src in smooth muscle function, and increasing need for pharmacological tools to study this signalling pathway. One tool which is frequently used for this purpose is diphenyleneiodonium (DPI) (Cross and Jones, 1986). However, DPI inhibits other flavin-containing enzymes, including nitric oxide synthase (Stuehr et al., 1991; Dodd-o et al., 1997), xanthine oxidase (Sanders et al., 1997), P-450 NADPH reductase (Tew, 1993) and mitochondrial respiratory chain complex I (Majander et al., 1994), raising concerns about its selectivity and about interpretation of data collected using this compound. Recently, in our study of the role of NADPH oxidase signalling in bronchodilator responses in airway smooth muscle, we uncovered a mixture of effects of DPI (unpublished observations). Here, we describe these mixed effects and find them to be mediated by cellular targets other than NADPH oxidase, namely, cholinesterases and the internal Ca2+ pump in smooth muscle.
Methods
Preparation of isolated tissues
All experimental procedures were approved by the McMaster University Animal Care Committee, the McMaster University Biosafety Committee and the St. Joseph's Healthcare Research Ethics Board, and conform to the guidelines set out by the Canadian Council on Animal Care. Tracheas were obtained from cows (200–500 kg) killed at a local abattoir and transported to the laboratory in ice-cold Krebs buffer [in mM: NaCl, 116; KCl, 4.2; CaCl2, 2.5; NaH2PO4, 1.6; MgSO4, 1.2; NaHCO3, 22; d-glucose, 11 (pH 7.4)]. In the laboratory, the epithelium was removed and strips of smooth muscle were excised (∼2–3 mm wide, ∼10 mm long); these were used immediately or stored in Krebs buffer at 4°C for use up to 48 h later.
Muscle bath technique
The strips of TSM were mounted vertically in organ baths using silk suture (Ethicon 4-O, Ethicon, Somerville, NJ, USA) tied to a Grass FT.03 force transducer (Harvard Apparatus, Holliston, MA, USA) on one end, and to a glass rod which served as an anchor on the other end. These were bathed in Krebs buffer bubbled with 95% O2/5% CO2, maintained at 37°C and supplemented with indomethacin (10 µM) and N-ω-nitro-l- arginine (l-NNA; 10−4 M) in order to block the production of inhibitory prostaglandins and nitric oxide, respectively, which would suppress mechanical responses. Tissues were passively stretched to impose a preload tension of ≈1 g, and isometric changes in tension were digitized (two samples per second) and recorded on-line (DigiMed System Integrator, MicroMed, Louisville, KY, USA) for plotting on the computer. Tissues were equilibrated for 1 h before starting the experiments, during which time they were challenged with 60 mM KCl three times to assess the functional state of each tissue.
In the first set of experiments, tissues were pretreated for 60 min with DPI (10−5, 3 × 10−5 or 10−4 M) or DMSO alone, after which the magnitude and time-course of the responses to carbachol (CCh; 3 × 10−5 M) were examined.
In the second set of experiments, tissues were challenged repeatedly at 30 min intervals (10 min exposure, then 20 min wash period) with either 5-HT (10−6 M), potassium chloride (60 mM), CCh (10−6 M) or acetylcholine (ACh; 10−6 M). These challenges were given three times before supplementing the Krebs media with DPI (10−4 M). The peak magnitudes of these agonist-evoked contractions were then quantified and compared.
Quantification of cholinesterase activities
Acetylcholinesterase (AChE) and butyrylcholinesterase (BChE) activities were measured spectrophotometrically (Cary 3Bio, Varian, Darmstadt, Germany) at 412 nm with a modified Ellman assay (Worek et al., 1999; Eyer et al., 2003). The standard assay mixture (3.16 mL) contained 0.45 mM acetyl thiocholine (AChE) or 1.0 mM butyryl thiocholine (BChE) as substrate, and 0.3 mM 5,5'-dithio-bis-2-nitrobenzoic acid as chromogen in 0.1 M phosphate buffer (pH 7.4). Assays were run at 37°C. Haemoglobin-free human erythrocyte ghosts were used as source of AChE and were prepared according to Dodge et al. (1963) with minor modifications (Worek et al., 2002). Isolated human BChE was suspended in phosphate buffer (0.1 M; pH 7.4).
The inhibitory potency of DPI was assessed by incubation of AChE and BChE with 10−6 to 10−4 M and 10−7 to 10−4 M DPI, respectively, for 5 min before adding substrate to determine enzyme activity: IC50 values were then determined by non-linear regression analysis of the [DPI]-AChE/BChE relationships. For the determination of the inhibition mode, AChE and BChE activities were quantified after adding different substrate concentrations (2–8 × 10−4 M) in the presence of 0–20 × 10−6 M and 0–2.5 × 10−6 M DPI, respectively. The reversibility of enzyme inhibition by DPI was tested by incubation of AChE and BChE with 8 × 10−5 M DPI followed by extensive dilution (316-fold) and determination of enzyme activity.
Ca2+ uptake
A radiometric assay described previously (Grover and Samson, 1997) was used to quantify Ca2+ uptake into crude arterial microsomes prepared from porcine coronary arteries obtained from a local abattoir. In brief, pig coronary artery smooth muscle cells were isolated and plated in Dulbecco's modified Eagle's medium (DMEM) supplemented with 0.5 mM N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid (pH 7.4), 2 mM glutamine, 50 mg·L−1 gentamicin, 0.125 mg·L−1 amphotericin B and 10% fetal bovine serum, and then allowed to grow to confluence. Cells were passaged by trypsinization (0.25% trypsin, 1 mM EDTA in Ca2+- and Mg2+-free Hanks's balanced salt solution) for 4 min at 37°C and then replating. After the fifth passage, the cells were homogenized, and 25–75 µg protein was added to 150 µL of a 45Ca2+ uptake medium so that the final composition of the solutions was 30 mM imidazole–HCl (pH 6.8 at 37°C), 100 mM KCl, 1 mM MgCl2, 5 mM sodium azide, 1 mM EGTA, 0.85 mM CaCl2 (plus trace amounts of 45CaCl2), 5 mM ATP, 10 mM creatine phosphate, 50–80 U/mL creatine kinase and 5 mM oxalate. The samples were incubated for 30 min at 37°C in the presence or absence of DPI (3 × 10−5 M or 10−4 M) or thapsigargin (10−6 M) and then filtered through 0.45 µm nitrocellulose filters, and washed three times with a chilled solution containing 8% sucrose, 0.5 mM EGTA and 40 mM imidazole–HCl (pH 7.0). The filters were then placed in vials containing Beckman Ready Safe cocktail, and the amount of radioactivity in the filters was determined with a scintillation counter.
Data analysis
Contractions were expressed as a percentage of the response to 60 mM KCl added during the equilibration period (immediately before onset of the experiment). AChE and BChE activities were expressed as percentage of control activity. Data are reported as mean ± SEM; n refers to the number of animals. Statistical comparisons were made using analysis of variance (with Bonferroni post hoc test); P < 0.05 was considered statistically significant.
Materials
Names of drugs and molecular targets conform to guidelines in Alexander et al. (2008). All chemicals were obtained from Sigma Chemical Company and prepared as 10 mM stock solutions, either as aqueous solutions (KCl; ACh; CCh; 5-HT; acetyl thiocholine, butyryl thiocholine), DMSO (DPI) or ethanol (1H-(1,2,4) oxadiazole(4,3-α)quinoxaline-1-one (ODQ); apocynin). Aliquots were then added to the muscle baths; the final bath concentration of solvents did not exceed 0.1%, which we have found elsewhere to have little or no effect on mechanical activity.
Results
DPI directly antagonizes excitatory responses
We first investigated the effects of DPI on cholinergic contractions. Following the equilibration period, tissues were pretreated for 60 min with DPI (10−5, 3 × 10−5, 10−4 M or DMSO alone), then challenged with CCh (3 × 10−5 M). Vehicle-treated controls exhibited a brisk and sustained contraction to CCh. At 10−4 M, however, DPI had a marked inhibitory effect on CCh-evoked contractions (Figure 1A,B): the latter were markedly reduced in peak magnitude and became highly transient in nature with phasic activity and spike-like oscillations in tone (Figure 1A). At times, DPI alone raised baseline tone on its own, before any challenge with CCh (not shown).
Figure 1.
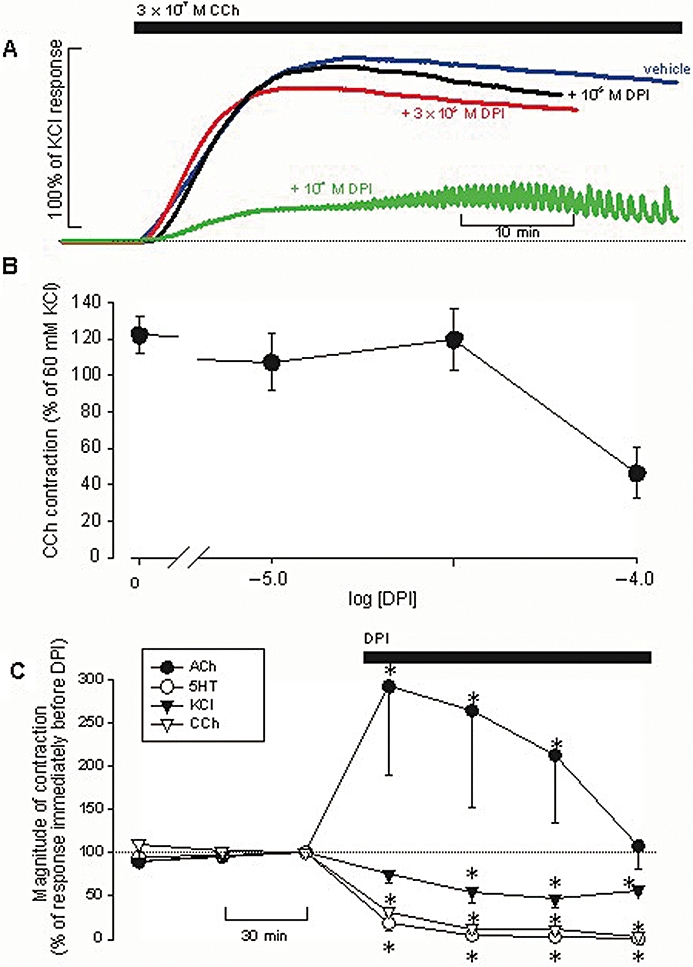
Effects of diphenyleneiodonium (DPI) on mechanical activity in bovine tracheal smooth muscle. (A) Representative tracings showing the increase in tone evoked by 3 × 10−7 M CCh in the absence or presence of DPI (concentrations as indicated); responses are standardized as a % of the response to KCl evoked earlier in the experiment. (B) Concentration–response relationship of the inhibitory effect of DPI on carbachol (CCh)-evoked contractions; symbols indicate mean (±SEM) magnitudes of CCh-evoked responses. (C) Mean (±SEM) responses to four successive additions of acetylcholine (ACh), CCh, 5-HT (all 10−6 M) or KCl (60 mM), as indicated, before and after addition of 10−4 M DPI to the Krebs solution; n= 6 for all. *Significantly different from values before DPI addition.
Next, we examined whether this inhibitory effect of DPI was specific to CCh, or also affected excitatory responses to other spasmogens. Tissues were challenged repeatedly at 30 min intervals with 5-HT (10−6 M), KCl (60 mM), CCh (10−6 M) or ACh (10−6 M; washes out more readily than CCh) three times before and four more times after introduction of DPI (10−4 M; n= 6 for all). Contractions evoked by 5-HT and CCh were relatively quickly reduced in magnitude by DPI, being nearly abolished by the second challenge in the presence of DPI (Figure 1C). KCl-evoked contractions were somewhat more resistant, being reduced only ∼50% by the fourth challenge in the presence of DPI (Figure 1C). On the other hand, ACh-evoked responses were immediately and markedly augmented in the presence of DPI (more than doubled), although the magnitude of this augmentation diminished over the course of the assay (Figure 1C).
Does DPI suppress excitatory responses through action on NADPH oxidase or c-src?
Given that DPI is believed to be a powerful inhibitor of NADPH oxidase, which in turn transduces its effects through generation of superoxide and activation of Src, we examined whether the effect of DPI on CCh contractions could be mimicked by the NADPH oxidase inhibitor apocynin (Stolk et al., 1994) or the Src inhibitors PP1 or PP2 (Hanke et al., 1996).
Tissues were pretreated for 60 min with vehicle (n= 9), DPI (10−4 M; n= 8), apocynin (10−4; n= 5), PP1 (10−5 M; n= 6) or PP2 (10−5 M; n= 6) before challenging with 3 × 10−7 M CCh. Only DPI had any statistically significant effect on the magnitude of contractions so evoked (Figure 2). We would therefore conclude that the inhibitory effect of DPI on excitatory responses in bovine TSM is not exerted through NADPH oxidase.
Figure 2.

Comparison of effects of various inhibitors on responses to carbachol (CCh). Tissues were pretreated with diphenyleneiodonium (DPI) (10−4 M), as well as with ODQ (10−4 M; n= 5), apocynin (10−4 M; n= 5), PP1 (10−5 M; n= 6) or PP2 (10−5 M; n= 6) for 30 min before recording the peak contractile response to CCh (3 × 10−7 M). *indicates significantly different from control.
Does DPI suppress excitatory responses through activation of guanylate cyclase?
DPI has also been described as an inhibitor of nitric oxide synthase (Dodd-o et al., 1997). However, we would note that our data were collected in the presence of 10−4 M l-NNA, and therefore, presume that nitric oxide synthase is already fully inhibited. Nonetheless, we compared the effect of DPI with and without the soluble guanylyl cyclase inhibitor, ODQ. Tissues were pretreated with 10−4 M ODQ, with or without 10−4 M DPI, for 30 min before testing the response to CCh (n= 5). ODQ alone did not reduce cholinergic responses (again, in the presence of l-NNA); more importantly, there was no statistically significant difference between the magnitude of cholinergic responses obtained in the presence of DPI alone and those in the presence of DPI plus ODQ (Figure 2).
Does DPI suppress excitatory responses through inhibition of sarcoplasmic/endoplasmic reticulum Ca2+ ATPase (SERCA)?
Our observations that DPI exerts the same effects – modest increase in baseline tone, suppression of peak magnitude of agonist-evoked responses and unmasking of phasic activity and oscillations in what are otherwise sustained contractions – as does cyclopiazonic acid, an inhibitor of the SERCA (the internal Ca2+ pump) (Janssen et al., 1997; 2001; Helli et al., 2005) – led us to conjecture whether DPI inhibits the internal Ca2+ pump. Microsomes were prepared from pig coronary artery (n= 6), supplied with ATP (to provide energy to the Ca2+ pump) and oxalate (stimulates Ca2+ retention in the sarcoplasmic reticulum), and used to evaluate Ca2+ uptake in the presence or absence of DPI using previously published methods (Grover and Samson, 1997). A comparison was made with thapsigargin, a well-described SERCA inhibitor (Low et al., 1991). The data from these experiments (summarized in Table 1) confirmed that DPI at the concentrations used in this study partially inhibited SERCA activity.
Table 1.
Ca2+ uptake into microsomes
| Ca2+uptake (µmol·g−1) | % Inhibition | |
|---|---|---|
| Microsomes alone | 45 ± 1.0 | NA |
| Microsomes + thapsigargin (10−6M) | 13.2 ± 0.6 | 71 |
| Microsomes + DPI (3 × 10−5 M) | 36.8 ± 0.7 | 18 |
| Microsomes + DPI (10−4 M) | 30.5 ± 0.9 | 32 |
Microsomes from pig coronary artery were incubated for 30 min in the presence of ATP (5 mM), oxalate (5 mM) and blockers (as indicated), and uptake of 45Ca2+ was measured.
Values shown in the table are means ± SEM, from six microsomal preparations.
DPI, diphenyleneiodonium.
Does DPI augment ACh-evoked responses through inhibition of AChE?
We also considered the mechanism underlying the transient enhancement of ACh-evoked contractions shown in Figure 1C. Given that ACh responses were augmented, but not those to CCh, we hypothesized that DPI acts by inhibiting activity of AChE. To test this, the activity of human AChE was quantified (see Methods) in the presence of varying concentrations of DPI (10−6 to 10−4 M); a comparison was made with the effect of DPI (10−7 to 10−4 M) on the activity of human BChE. DPI was indeed a potent inhibitor of both enzymes, with a concentration for half maximal inhibition (IC50) of 8 µM (95% confidence interval: 6.9–8.6 µM) and 0.6 µM (95% confidence interval: 0.4–0.8 µM), respectively (n= 4 for both; Figure 3). In another experiment, we assessed the nature of this inhibition by measuring enzyme activities over a range of concentrations of substrate (200–800 µM acetyl thiocholine or butyryl thiocholine) and inhibitor (DPI; 0–20 µM) (Figure 4; n= 4): the data indicated a mixed, non-competitive type of inhibition. Finally, we assessed reversibility of this inhibition by incubating AChE or BChE with 80 µM DPI (or solvent), then transferring an aliquot to a cuvette for determination of enzyme activity after dilution (by a factor of 316; n= 4; data not shown) and found that the dilution resulted in a complete recovery of enzyme activity.
Figure 3.

Effect of diphenyleneiodonium (DPI) on cholinesterase activities. The inhibitory potency of DPI was determined by incubation of human AChE and BChE with 10−6 to 10−4 M and 10−7 to 10−4 M DPI, respectively, for 5 min before adding substrate to determine enzyme activity. Each point represents the means ± SD of four experiments.
Figure 4.
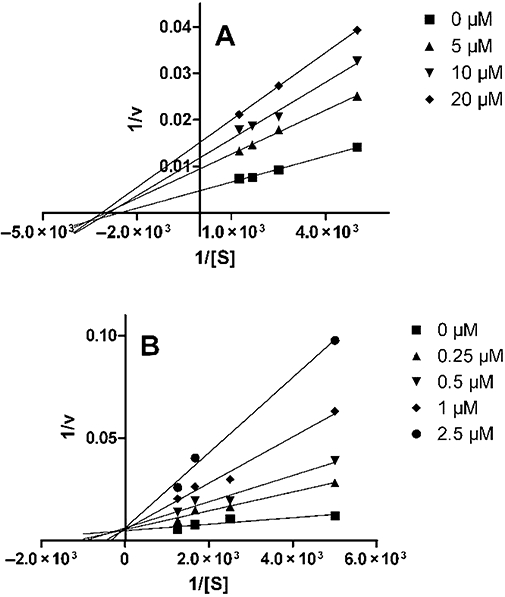
Determination of type of inhibition of cholinesterases by diphenyleneiodonium (DPI). Lineweaver–Burk plots for the inhibition of human AChE (A) and BChE (B) by DPI (concentrations given at right in figure) in the presence of different substrate concentrations (200–800 µM). Data are given as means (n= 4).
Consistent with an inhibitory action of DPI against AChE, we found DPI did not augment ACh-evoked contractions in TSM strips which had already been pretreated for 30 min with the AChE inhibitor neostigmine (10−4 M; data not shown); instead, only a gradual reduction in ACh-evoked contractions was seen, much like the changes summarized in Figure 1C for CCh and 5-HT.
Discussion
As outlined above, NADPH oxidase and Src are increasingly found to play important roles in various aspects of cell function, and for this reason pharmacological tools which modulate this signalling pathway are becoming more valuable. However, as with all pharmacological tools, it is important to recognize the limitations of these tools, including their actions on other cellular targets. DPI is no exception. Although often used in the past as a selective NADPH oxidase inhibitor, we here present data which highlight its potent actions against cholinesterases, and less potent actions against SERCA, adding to a long list of actions on other enzymes including nitric oxide synthase (Stuehr et al., 1991; Dodd-o et al., 1997), xanthine oxidase (Sanders et al., 1997), P-450 NADPH reductase (Tew, 1993) and mitochondrial respiratory chain complex I (Majander et al., 1994).
The inhibition of AChE (and BChE) was rapid, leading to an immediate augmentation of ACh-evoked contractions, which correlated with the results from the inhibition assays. DPI was shown to be a potent, readily reversible, mixed non-competitive type inhibitor of both human AChE and BChE, with IC50 values in the low micromolar (AChE) and high nanomolar range (BChE). At the concentrations used in our muscle bath studies, DPI would have completely inhibited ACh degradation, which explains the marked augmentation of ACh-evoked contractions (but not of CCh-, 5-HT or KCl-evoked contractions).
Although the DPI-induced enhancement of responses to ACh could also be explained by an inhibitory effect on nitric oxide synthase (Stuehr et al., 1991; Dodd-o et al., 1997; Kesler et al., 2002), which otherwise opposes contraction through the synthesis of nitric oxide, our experiments were all performed in the presence of the nitric oxide synthesis inhibitor l-NNA (10−4 M), and so this enzyme should already be inhibited. Besides, inhibition of nitric oxide synthase would have effects on all contractor responses, not just those evoked by ACh.
The inhibitory effect on excitation–contraction coupling, on the other hand, was only seen at relatively high concentrations (10−4 M, but not 3 × 10−5 M or lower), and shared many characteristics with that of the SERCA inhibitor, cyclopiazonic acid (Amoako et al., 1996). First, the effect was not agonist specific, reducing responsiveness to Ach, CCh, 5-HT and KCl. Moreover, the receptor-mediated responses were essentially abolished, while responses to KCl were only partially reduced (contractions to KCl involve voltage-dependent Ca2+ influx and activation of Rho-kinase (Janssen et al., 2004; Liu et al., 2005) rather than release of internal Ca2+). Also, this effect took a considerable amount of time to develop, presumably due to the nature of the underlying mechanism. That is, there is an ongoing ‘leak’ of Ca2+ from the internal store through a number of pathways (ryanodine- and inositol trisphosphate-gated receptor/channels) which is compensated by the action of SERCA: inhibition of the latter leads to unmasking of the leak with transient elevation of [Ca2+]i (accounting for the transient increase in baseline tone) and gradual depletion of the store (accounting for the concomitant loss of responsiveness). Finally, both cyclopiazonic acid and DPI unmasked phasic activity (Janssen and Sims, 1993; Janssen and Nana, 1997; Janssen et al., 1997; 2001;). Consistent with our conjecture that DPI was acting to suppress SERCA activity, we did indeed find it to inhibit Ca2+ uptake into smooth muscle microsomes. Even though ACh and CCh act on the same cholinergic receptor, the ACh-responses did not appear to decrease to the same level as the CCh responses even by the fourth challenge. It is possible that they would have if we had extended the duration of these experiments, i.e. it may take much longer for this inhibitory effect on SERCA to overcome the enhancement by inhibition of AChE activity). Alternatively, it may be that ACh and CCh couple somewhat differently to the muscarinic receptor in that the former more powerfully activates the RhoA/ROCK pathway, which may explain the additional tone at the end of these experiments.
Overall, we found that DPI – which is often used as an inhibitor of NADPH oxidase – is also a highly potent inhibitor of cholinesterase and SERCA activities. Less germane to the focus of this study, we did not find that either NADPH oxidase or Src contributed to contractor responses in bovine TSM, as the latter were not inhibited by another NADPH oxidase inhibitor (apocynin) nor the Src inhibitors PP1 and PP2. In contrast, Src seems to be important in 5-HT-evoked contraction of rat TSM (Tolloczko et al., 2002), as well as ATP-induced airway hyper-responsiveness in guinea pig TSM (Oguma et al., 2007). The explanation for the discrepancy between these findings and ours is unclear, but may include species-related differences (i.e. unique to rodents).
Acknowledgments
The authors are grateful to SE Samson and AK Grover for data pertaining to the effects of DPI on Ca2+ uptake into microsomes, and to M Pusch for technical assistance with cholinesterase assays. These studies were supported by funds provided by the Canadian Institutes of Health Research.
Glossary
Abbreviations:
- AChE
acetylcholinesterase
- ATCh
acetylthiocholine iodide
- ASM
airway smooth muscle
- BChE
butyrylcholinesterase
- BTCh
S-butyrylthiocholine iodide
- CCh
carbachol
- CPA
cyclopiazonic acid
- DPI
diphenyleneiodonium
- l-NNA
N-ω-nitro-l-arginine
- ODQ
1H-(1,2,4)oxadiazole(4,3-α)quinoxaline-1-one
- SERCA
sarcoplasmic/endoplasmic reticulum Ca2+ ATPase (internal Ca2+ pump)
- TSM
tracheal smooth muscle
References
- Alexander SP, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC) Br J Pharmacol. (3rd edition) 2008;153(Suppl. 2):S1–209. doi: 10.1038/sj.bjp.0707746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amoako D, Qian Y, Kwan CY, Bourreau JP. Probing excitation–contraction coupling in trachealis smooth muscle with the mycotoxin cyclopiazonic acid. Clin Exp Pharmacol Physiol. 1996;23:733–737. doi: 10.1111/j.1440-1681.1996.tb01768.x. [DOI] [PubMed] [Google Scholar]
- Brandt D, Gimona M, Hillmann M, Haller H, Mischak H. Protein kinase C induces actin reorganization via a src- and rho- dependent. Pathway J Biol Chem. 2002;277:20903–20910. doi: 10.1074/jbc.M200946200. [DOI] [PubMed] [Google Scholar]
- Cross AR, Jones OT. The effect of the inhibitor diphenylene iodonium on the superoxide-generating system of neutrophils. Specific labelling of a component polypeptide of the oxidase. Biochem J. 1986;237:111–116. doi: 10.1042/bj2370111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dodd-o JM, Zheng G, Silverman HS, Lakatta EG, Ziegelstein RC. Endothelium-independent relaxation of aortic rings by the nitric oxide synthase inhibitor diphenyleneiodonium. Br J Pharmacol. 1997;120:857–864. doi: 10.1038/sj.bjp.0701014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dodge JT, Mitchell C, Hanahan DJ. The preparation and chemical characteristics of hemoglobin-free ghosts of human erythrocytes. Arch Biochem Biophys. 1963;100:119–130. doi: 10.1016/0003-9861(63)90042-0. [DOI] [PubMed] [Google Scholar]
- Eyer P, Worek F, Kiderlen D, Sinko G, Stuglin A, Simeon-Rudolf V, et al. Molar absorption coefficients for the reduced Ellman reagent: reassessment. Anal Biochem. 2003;312:224–227. doi: 10.1016/s0003-2697(02)00506-7. [DOI] [PubMed] [Google Scholar]
- Grover AK, Samson SE. Peroxide resistance of ER Ca2+ pump in endothelium: implications to coronary artery function. Am J Physiol. 1997;273:C1250–C1258. doi: 10.1152/ajpcell.1997.273.4.C1250. [DOI] [PubMed] [Google Scholar]
- Hanke JH, Gardner JP, Dow RL, Changelian PS, Brissette WH, Weringer EJ, et al. Discovery of a novel, potent, and Src family-selective tyrosine kinase inhibitor. Study of Lck- and FynT-dependent T cell activation. J Biol Chem. 1996;271:695–701. doi: 10.1074/jbc.271.2.695. [DOI] [PubMed] [Google Scholar]
- Helli PB, Pertens E, Janssen LJ. Cyclopiazonic acid activates a Ca2+-permeable, non-selective cation conductance in porcine and bovine tracheal smooth muscle. J Appl Physiol. 2005;99:1759–1768. doi: 10.1152/japplphysiol.00242.2005. [DOI] [PubMed] [Google Scholar]
- Hirshman CA, Zhu D, Pertel T, Panettieri RA, Emala CW. Isoproterenol induces actin depolymerization in human airway smooth muscle cells via activation of an Src kinase and GS. Am J Physiol Lung Cell Mol Physiol. 2005;288:L924–L931. doi: 10.1152/ajplung.00463.2004. [DOI] [PubMed] [Google Scholar]
- Ibitayo AI, Tsunoda Y, Nozu F, Owyang C, Bitar KN. Src kinase and PI 3-kinase as a transduction pathway in ceramide-induced contraction of colonic smooth muscle. Am J Physiol. 1998;275:G705–G711. doi: 10.1152/ajpgi.1998.275.4.G705. [DOI] [PubMed] [Google Scholar]
- Ishida T, Ishida M, Suero J, Takahashi M, Berk BC. Agonist-stimulated cytoskeletal reorganization and signal transduction at focal adhesions in vascular smooth muscle cells require c-Src. J Clin Invest. 1999;103:789–797. doi: 10.1172/JCI4189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janssen LJ, Nana R. Na+/K+ ATPase mediates rhythmic spontaneous relaxations in canine airway smooth muscle. Respir Physiol. 1997;108:187–194. doi: 10.1016/s0034-5687(97)00023-6. [DOI] [PubMed] [Google Scholar]
- Janssen LJ, Sims SM. Emptying and refilling of Ca2+ store in tracheal myocytes as indicated by ACh-evoked currents and contraction. Am J Physiol. 1993;265:C877–C886. doi: 10.1152/ajpcell.1993.265.4.C877. [DOI] [PubMed] [Google Scholar]
- Janssen LJ, Walters DK, Wattie J. Regulation of [Ca2+]i in canine airway smooth muscle by Ca2+-ATPase and Na+/Ca2+ exchange mechanisms. Am J Physiol. 1997;273:L322–L330. doi: 10.1152/ajplung.1997.273.2.L322. [DOI] [PubMed] [Google Scholar]
- Janssen LJ, Wattie J, Lu-Chao H, Tazzeo T. Muscarinic excitation–contraction coupling mechanisms in tracheal and bronchial smooth muscles. J Appl Physiol. 2001;91:1142–1151. doi: 10.1152/jappl.2001.91.3.1142. [DOI] [PubMed] [Google Scholar]
- Janssen LJ, Tazzeo T, Zuo J, Pertens E, Keshavjee S. KCl evokes contraction of airway smooth muscle via activation of RhoA and Rho-kinase. Am J Physiol Lung Cell Mol Physiol. 2004;287:L852–L858. doi: 10.1152/ajplung.00130.2004. [DOI] [PubMed] [Google Scholar]
- Kesler BS, Mazzone SB, Canning BJ. Nitric oxide-dependent modulation of smooth-muscle tone by airway parasympathetic nerves. Am J Respir Crit Care Med. 2002;165:481–488. doi: 10.1164/ajrccm.165.4.2004005. [DOI] [PubMed] [Google Scholar]
- Krymskaya VP, Goncharova EA, Ammit AJ, Lim PN, Goncharov DA, Eszterhas A, et al. Src is necessary and sufficient for human airway smooth muscle cell proliferation and migration. FASEB J. 2005;19:428–430. doi: 10.1096/fj.04-2869fje. [DOI] [PubMed] [Google Scholar]
- Liu C, Zuo J, Pertens E, Helli PB, Janssen LJ. Regulation of Rho/ROCK signaling in airway smooth muscle by membrane potential and [Ca2+]i. Am J Physiol Lung Cell Mol Physiol. 2005;289:L574–L582. doi: 10.1152/ajplung.00134.2005. [DOI] [PubMed] [Google Scholar]
- Low AM, Gaspar V, Kwan CY, Darby PJ, Bourreau JP, Daniel EE. Thapsigargin inhibits repletion of phenylephrine-sensitive intracellular Ca2+ pool in vascular smooth muscles. J Pharmacol Exp Ther. 1991;258:1105–1113. [PubMed] [Google Scholar]
- Majander A, Finel M, Wikstrom M. Diphenyleneiodonium inhibits reduction of iron–sulfur clusters in the mitochondrial NADH–ubiquinone oxidoreductase (complex I) J Biol Chem. 1994;269:21037–21042. [PubMed] [Google Scholar]
- Muzaffar S, Shukla N, Bond M, Sala-Newby GB, Newby AC, Angelini GD, et al. Superoxide from NADPH oxidase upregulates type 5 phosphodiesterase in human vascular smooth muscle cells: inhibition with iloprost and NONOate. Br J Pharmacol. 2008;155:847–856. doi: 10.1038/bjp.2008.300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oguma T, Ito S, Kondo M, Makino Y, Shimokata K, Honjo H, et al. Roles of P2X receptors and Ca2+ sensitization in extracellular adenosine triphosphate-induced hyperresponsiveness in airway smooth muscle. Clin Exp Allergy. 2007;37:893–900. doi: 10.1111/j.1365-2222.2007.02719.x. [DOI] [PubMed] [Google Scholar]
- Rathore R, Zheng YM, Niu CF, Liu QH, Korde A, Ho YS, et al. Hypoxia activates NADPH oxidase to increase [ROS]i and [Ca2+]i through the mitochondrial ROS-PKCepsilon signaling axis in pulmonary artery smooth muscle cells. Free Radic Biol Med. 2008;45:1223–1231. doi: 10.1016/j.freeradbiomed.2008.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravasi S, Citro S, Viviani B, Capra V, Rovati GE. CysLT1 receptor-induced human airway smooth muscle cells proliferation requires ROS generation, EGF receptor transactivation and ERK1/2 phosphorylation. Respir Res. 2006;7:42. doi: 10.1186/1465-9921-7-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross GR, Kang M, Shirwany N, Malykhina AP, Drozd M, Akbarali HI. Nitrotyrosylation of Ca2+ channels prevents c-Src kinase regulation of colonic smooth muscle contractility in experimental colitis. J Pharmacol Exp Ther. 2007;322:948–956. doi: 10.1124/jpet.107.123075. [DOI] [PubMed] [Google Scholar]
- Sanders SA, Eisenthal R, Harrison R. NADH oxidase activity of human xanthine oxidoreductase – generation of superoxide anion. Eur J Biochem. 1997;245:541–548. doi: 10.1111/j.1432-1033.1997.00541.x. [DOI] [PubMed] [Google Scholar]
- Stolk J, Hiltermann TJ, Dijkman JH, Verhoeven AJ. Characteristics of the inhibition of NADPH oxidase activation in neutrophils by apocynin, a methoxy-substituted catechol. Am J Respir Cell Mol Biol. 1994;11:95–102. doi: 10.1165/ajrcmb.11.1.8018341. [DOI] [PubMed] [Google Scholar]
- Stuehr DJ, Fasehun OA, Kwon NS, Gross SS, Gonzalez JA, Levi R, et al. Inhibition of macrophage and endothelial cell nitric oxide synthase by diphenyleneiodonium and its analogs. FASEB J. 1991;5:98–103. doi: 10.1096/fasebj.5.1.1703974. [DOI] [PubMed] [Google Scholar]
- Suzuki Y, Asano K, Shiraishi Y, Oguma T, Shiomi T, Fukunaga K, et al. Human bronchial smooth muscle cell proliferation via thromboxane A2 receptor. Prostaglandins Leukot Essent Fatty Acids. 2004;71:375–382. doi: 10.1016/j.plefa.2004.07.004. [DOI] [PubMed] [Google Scholar]
- Tew DG. Inhibition of cytochrome P450 reductase by the diphenyliodonium cation. Kinetic analysis and covalent modifications. Biochemistry. 1993;32:10209–10215. doi: 10.1021/bi00089a042. [DOI] [PubMed] [Google Scholar]
- Tolloczko B, Turkewitsch P, Choudry S, Bisotto S, Fixman ED, Martin JG. Src modulates serotonin-induced calcium signaling by regulating phosphatidylinositol 4,5-bisphosphate. Am J Physiol Lung Cell Mol Physiol. 2002;282:L1305–L1313. doi: 10.1152/ajplung.00304.2001. [DOI] [PubMed] [Google Scholar]
- Wijetunge S, Hughes AD. Activation of endogenous c-Src or a related tyrosine kinase by intracellular (pY)EEI peptide increases voltage-operated calcium channel currents in rabbit ear artery cells. FEBS Lett. 1996;399:63–66. doi: 10.1016/s0014-5793(96)01177-5. [DOI] [PubMed] [Google Scholar]
- Wijetunge S, Hughes AD. Src family tyrosine kinases mediate contraction of rat isolated tail arteries in response to a hyposmotic stimulus. J Hypertens. 2007;25:1871–1878. doi: 10.1097/HJH.0b013e328255e8f0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Worek F, Mast U, Kiderlen D, Diepold C, Eyer P. Improved determination of acetylcholinesterase activity in human whole blood. Clin Chim Acta. 1999;288:73–90. doi: 10.1016/s0009-8981(99)00144-8. [DOI] [PubMed] [Google Scholar]
- Worek F, Reiter G, Eyer P, Szinicz L. Reactivation kinetics of acetylcholinesterase from different species inhibited by highly toxic organophosphates. Arch Toxicol. 2002;76:523–529. doi: 10.1007/s00204-002-0375-1. [DOI] [PubMed] [Google Scholar]