

Abstract
Background and purpose:
Amitriptyline is often prescribed as a first-line treatment for neuropathic pain but its precise mode of analgesic action remains uncertain. Amitriptyline is known to inhibit voltage-dependent ion channels and also to act as an antagonist at ligand-gated ion channels, such as nicotinic acetylcholine receptors (nAChRs). In the present study, we tested the effect of amitriptyline on nicotinic responses of unmyelinated axons in isolated segments of human peripheral nerve. In particular, a comparison was made between the concentrations of amitriptyline necessary for inhibition of nAChRs and those required for inhibition of the compound C-fibre action potential.
Experimental approach:
Isolated axon fascicles were prepared from short segments of human sural nerve, and multiple measures of axonal excitability were recorded using computer-controlled threshold tracking software.
Key results:
Amitriptyline (EC50 2.6 µM) reduced the nicotine-induced increase in C-fibre excitability but only slightly altered the amplitude and latency to onset of the compound action potential. In contrast, tetrodotoxin produced a clear reduction in the amplitude and a prolongation of action potential onset latency but was without effect on the nicotine-induced increase in axonal excitability.
Conclusions and implications:
These data demonstrate that low concentrations of amitriptyline suppress the response of human peripheral C-type axons to nicotine by directly inhibiting nAChRs. Blockade of tetrodotoxin-sensitive, voltage-dependent sodium channels does not contribute to this effect. An inhibitory action of amitriptyline on nAChRs in unmyelinated nociceptive axons may be an important component of amitriptyline's therapeutic effect in the treatment of neuropathic pain.
Keywords: neuropathic pain, axonal excitability, nociceptive nerve fibres, neuronal nicotinic receptors
Introduction
Amitriptyline is often used in the treatment of neuropathic pain (Vadalouca et al., 2006; Saarto and Wiffen, 2007); however, its mode of analgesic action is uncertain. Effects in the CNS include an inhibitory effect on the re-uptake of 5-HT and noradrenaline that may reduce pain by enhancing synaptic inhibition in the spinal cord or brain stem (Sacerdote et al., 1987; Onghena and Van, 1992). Amitriptyline may also alleviate neuropathic pain via its action in the peripheral nervous system. For example, application of amitriptyline to the surface of the skin or by subcutaneous injection leads to elevated sensory thresholds to mechanical, heat and cold stimuli in human subjects (Duale et al., 2008) and produces prolonged cutaneous analgesia in rats (Khan et al., 2002). This effect on peripheral neurons is typically attributed to amitriptyline's inhibition of voltage-dependent sodium (Nau et al., 2000; Bräu et al., 2001; Wang et al., 2004; Dick et al., 2007; Leffler et al., 2007) and potassium channels (Galeotti et al., 1997; Punke and Friederich, 2007).
Amitriptyline is also an antagonist at ligand-gated ion channels (Rammes and Rupprecht, 2007). For example, inhibition of nicotinic acetylcholine receptors (nAChRs) by amitriptyline has been described (Connolly et al., 1992; Gumilar et al., 2003). Interestingly, it has been noted that this effect requires only sub-micromolar concentrations in contrast to the micromolar concentrations necessary to inhibit voltage-dependent sodium channels (Park et al., 1998). nAChRs are also expressed functionally in both the axons (Douglas and Ritchie, 1960; Keele and Armstrong, 1964; Tanelian, 1991; Steen and Reeh, 1993; Schmelz et al., 2003; Zhang et al., 2004; Moalem et al., 2005) and cell bodies (Liu et al., 1993; Flores et al., 1996; Liu et al., 1998; Genzen et al., 2001; Lips et al., 2002) of a sub-population of peripheral sensory neurons, some of which are nociceptors. The pharmacological profile and the functional activity of nAChRs in the axonal membrane of unmyelinated human nerve fibres have both been characterized (Lang et al., 2003). From that study, the rank order of potency for nicotinic agonists was epibatidine > 5-iodo-A-85380 > 1,1-dimethyl-4-phenylpiperazinium iodide > nicotine > cytosine > acetylcholine. Blockade of nAChRs in human axons could be achieved with mecamylamine and, albeit at higher doses, by application of methyllycacontine and dihydro-β-erythroidine. In the present study, we have examined the effect of amitriptyline on the responses to nicotine of unmyelinated axons in peripheral human nerve. In particular, a comparison was made between the concentrations necessary for inhibition of nAChRs and for changes in amplitude and latency of compound action potentials (CAPs).
Methods
Preparations
Approval for the experimental use of human tissue was granted by the Ethics Committee of the University of Munich, and patients were informed about the operation by an anaesthetist prior to the procedure. Segments of sural nerve were removed from patients already scheduled for sural nerve biopsy, and each patient gave their written consent to the removal of an additional portion of nerve for research purposes. Experiments were carried out on 18 isolated fascicles of human sural nerve from seven patients (all male) with a median age of 66 years and ranging from 44 to 84 years.
As described previously (Lang et al., 2003) individual nerve fascicles were carefully extracted from an isolated segment of sural nerve (ca. 15–25 mm long) by grasping them with jewellers forceps and gently pulling them free. Each end of the nerve fascicle was drawn into a glass suction electrode within the organ bath and embedded in Vaseline to establish both a mechanical and a high resistance electrical seal. The organ bath (volume 1 mL) was continuously superfused at a rate of 6–8 mL·min−1 with physiological solution of the following composition (in mM) NaCl 118, KCl 3.0, CaCl2 1.5, MgCl2 1.0, D-glucose 5.0, NaHCO3 25 and NaH2PO4 1.2, and was bubbled with 95% O2/5% CO2 to pH 7.4. The temperature of the solution perfusing the bath was held constant at 32°C.
Electrophysiological analysis
The nerve fascicle was stimulated extracellularly with a constant current stimulator (A395, WPI, Sarasota, FL, USA). A silver wire inside the suction electrode served as the anode and a second silver wire, wound around the suction pipette, served as the cathode. Extracellular signals were recorded over the sealing resistance of the second suction electrode using a differential amplifier (NPI, Tamm, Germany). The signal was typically amplified with a gain of 1000× and filtered, low pass 1.3 kHz and high pass 3 Hz. The distance between stimulating and recording electrodes varied for each fascicle and was in the range 3–6 mm. Axonal excitability was assessed using QTRAC software (© Institute of Neurology, London, UK). QTRAC is a flexible, stimulus-response data-acquisition programme, originally written for studies of human nerves in vivo (Bostock et al., 1998), but it is also suitable for electrophysiological recordings from isolated peripheral nerves. In the present study, QTRAC was used to record CAPs of C-fibres as well as to generate timed stimuli. The isolated fascicles were stimulated with constant current pulses of fixed duration (1 ms) and varying amplitude. Stimulus frequency was invariably 0.5 Hz. For threshold tracking experiments, the current amplitude was automatically adjusted to maintain the C-fibre CAP response at constant amplitude (40% of the maximum, defined as ‘threshold’).
Statistics
Data plotted in Figures 5 and 7 are shown as means ± SEM of n= number of observations. Significance of differences between means was assessed by paired Student's t-test (Figure 7). Curve fitting to determine values of EC50 was performed in IgorPro (Wavemetrics, Lake Oswego, OR, USA) which uses the Levenberg-Marquardt algorithm for least-squares minimization (Figure 5A).
Figure 5.
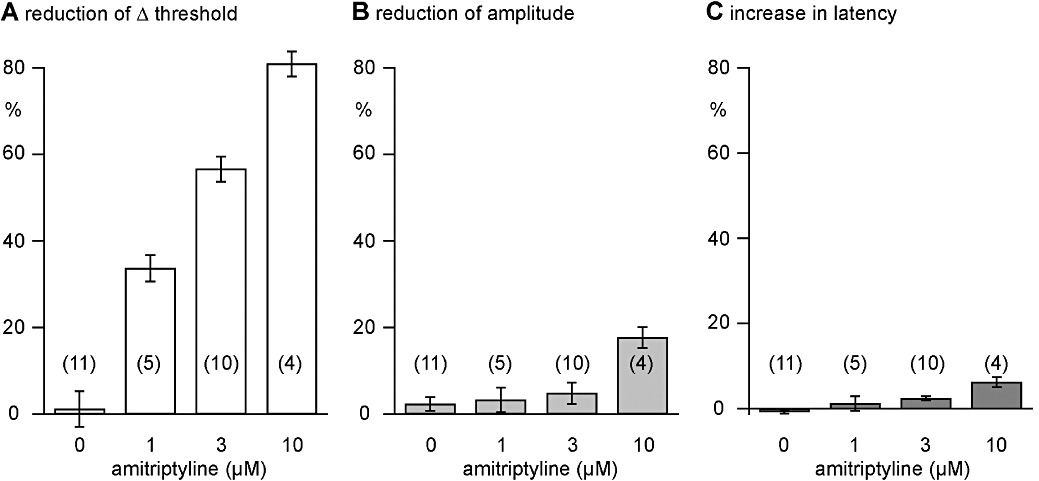
Quantitative analysis of the effects of amitriptyline on nicotine-induced (10 µM) increases in excitability and the amplitude and latency to onset of the C-fibre compound action potential (CAP) response to supra-maximal stimulation. In concentrations from 1 to 10 µM, amitriptyline reduced, concentration-dependently, the nicotine-induced (10 µM) increase in excitability in unmyelinated human axons. An EC50 for this effect of 2.6 ± 0.2 µM was calculated. In addition, in concentrations above 1 µM, amitriptyline concentration-dependently reduced the amplitude and increased the latency to half-maximum amplitude of the C-fibre CAP. However, these changes are much less prominent than the reduction of the nicotine-induced increase in axonal excitability observed in the presence of amitriptyline.
Figure 7.
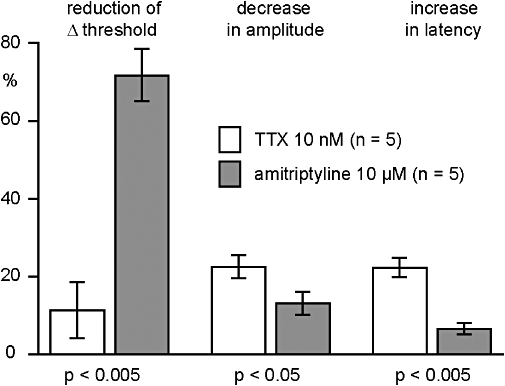
Quantitative analysis of the effects of amitriptyline (10 µM) and tetrodotoxin (TTX) (10 nM) on the C-fibre compound action potential response to electrical stimulation of human nerve fascicles. TTX (10 nM) reduced the amplitude and prolonged the latency to half-maximum amplitude of the C-fibre response to supra-maximal stimulation. These effects of TTX on the C-fibre response were significantly more pronounced than those of amitriptyline (10 µM). In contrast, amitriptyline (10 µM) produced a significantly stronger inhibition of the reduction in threshold produced by nicotine (10 µM).
Drugs
Nicotine hydrogen tartrate, tetrodotoxin (TTX) and amitriptyline hydrochloride were purchased from Sigma (Taufkirchen, Germany). The desired concentration was achieved by dilution of stock solutions on the day of each experiment.
Results
Effects of nicotine on axonal excitability
The effect of amitriptyline on unmyelinated axons in isolated fascicles of human sural nerve was tested by monitoring the compound C-fibre action potential response to electrical stimulation. A representative example of the CAP response is illustrated in Figure 1. An Aα/β peak corresponding to the activation of large-diameter myelinated axons occurred at short latency in response to low stimulus strengths (stimulus duration 0.1 ms). This peak is designated Aα/β because, while in the majority of people the sural nerve is exclusively sensory, in some individuals it may contain both cutaneous Aβ afferents as well as Aα axons comprising efferent motoneurons and Ia and Ib muscle afferents (Haupts and Amoiridis, 2007). At slightly higher stimulus intensities, small-diameter thinly myelinated axons were activated producing an Aδ peak at slightly longer latency. Further increases in stimulus intensity and duration (1 ms) activated unmyelinated axons and produced the C peak. In some human nerve fascicles, two classes of unmyelinated axon are discernible in the compound response, a lower threshold, more rapidly conducting class giving rise to the C1 peak and a higher threshold, more slowly conducting C2 peak. Quantitatively, the amplitude of the C-fibre compound potential varied between 0.1 and 1.5 mV.
Figure 1.
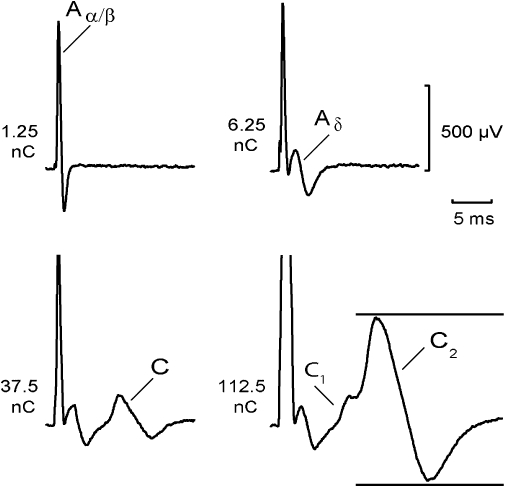
Components of the compound action potential (CAP) recorded from a segment of human sural nerve: an Aα/β peak corresponding to action potentials in large-diameter myelinated axons occurred at short latency in response to low stimulus strengths (12.5 µA, 0.1 ms). At slightly higher stimulus intensities (62.5 µA, 0.1 ms), small-diameter thinly myelinated axons were activated producing an Aδ peak at longer latency. Further increases in stimulus intensity (1 ms) activated two populations of unmyelinated axons. The fibre population contributing to the C1 peak had faster conduction velocities and lower electrical activation thresholds (37.5 µA, 1 ms) than the axonal population producing the C2 peak (112.5 µA, 1 ms). The distance between stimulus and recording electrode was 6 mm. Multiple measures of excitability were recorded by using the CAP (peak-to-peak) within the time window indicated by the horizontal bars.
The effect of nicotine on multiple measures of C-fibre excitability was then examined (Figure 2). Nerve fascicles were stimulated at a constant frequency of 0.5 Hz, and the following parameters (Figure 2A) were tested by means of a time window used to separate the C-fibre CAP from other components such as stimulus artefact and the A-fibre response: (i) the amplitude and latency of the maximal C-fibre CAP; (ii) the ‘threshold’ current necessary to maintain the amplitude of the C-fibre CAP at 40% of its maximum; and (iii) axonal electrical excitability 30 ms after a conditioning supra-maximal stimulus (post-spike excitability). Addition of nicotine (10 µM) to the bathing solution resulted in a slight decrease of the maximal C-fibre amplitude and an increase in axonal excitability, as indicated by the reduction in current required to maintain the test potential at 40% of the maximum (Figure 2B). In addition, nicotine strongly altered post-spike excitability (Figure 2B lowermost panel). The post-spike super-excitability of about 10% in the control solution was reversed to a post-spike sub-excitability of about 2–3% during application of nicotine. This change from super-excitability to sub-excitability induced by nicotine is consistent with membrane depolarization (Moalem-Taylor et al., 2007).
Figure 2.
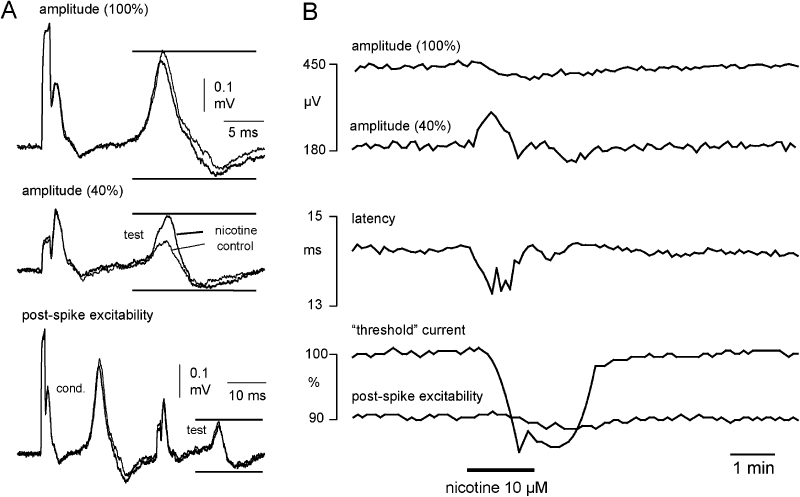
The effect of nicotine on unmyelinated axons in segments of human nerve is illustrated by representative examples of compound action potentials (CAPs) evoked by each of the three stimuli used in the stimulus paradigm to monitor excitability. To monitor the entire population (100%) of unmyelinated axons activated by electrical stimulation the peak-to-peak amplitude of the C-fibre CAP response to supra-maximal stimulation was monitored (A and B, upper trace). Amplitude was taken as the difference between the maximum and minimum excursions of the recorded signal within the time window indicated by upper and lower horizontal bars. The current (duration 1 ms) required to maintain a compound C-fibre action potential amplitude at 40% of the CAP evoked by supra-maximal stimulation (100%) was the second parameter monitored (A, middle trace). This parameter is used to monitor changes in the electrical activation threshold of the unmyelinated C-fibre population (‘threshold current’). To assess post-spike excitability, the current required to maintain a CAP amplitude at 40% of the supra-maximal response was monitored at a fixed delay of 30 ms after a preceding CAP of maximal amplitude had been evoked (A, lower trace). Each of these parameters was monitored sequentially at 2 s intervals during the recording period. (B) Upon application of nicotine (10 µM) the amplitude of the 40% CAP increases, that is, nicotine has an excitatory effect. The tracking software reduces the current ‘threshold’ (B, lower panel) to maintain the 40% amplitude. In addition, the normal post-spike super-excitability that follows an action potential is reversed to sub-excitability in the presence of nicotine, which is consistent with nicotine depolarizing the axons.
Amitriptyline reduces the effects of nicotine on axonal excitability
The effect of amitriptyline on several electrophysiological C-fibre parameters was also examined. First, we investigated whether amitriptyline modifies the increase in C-fibre excitability produced by bath application of nicotine (Figures 3 and 4). To this end, the effect of nicotine on axonal excitability was monitored using ‘threshold tracking’ as described above. Second, recordings were made of amitriptyline-induced changes in the amplitude and latency of the maximal C-fibre action potential (Figure 4B). This was examined because of the known blocking effect of amitriptyline on voltage-dependent sodium channels (see Introduction), a feature that is likely to bring about changes in the shape and latency of the compound C-fibre action potential.
Figure 3.
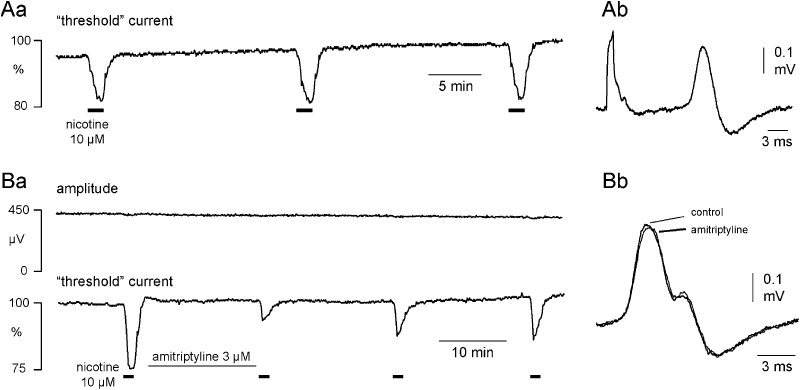
Amitriptyline reduces the magnitude of the nicotine effect on C-fibre excitability. Nicotine (10 µM) produces a robust increase in the electrical excitability of C-fibres as illustrated in panel Aa for three consecutive applications at 20 min intervals. There is no indication of tachyphylaxis. The inset to the right (Ab) shows an example of the C-fibre CAP recorded in response to supra-maximal stimulation. Amitriptyline (3 µM) substantially reduces the increase in excitability produced by nicotine (10 µM) as shown in the recording from a second isolated human nerve segment in B. At a concentration of 3 µM amitriptyline's action is restricted to an inhibition of the excitability increase brought about by nicotine (Ba), with only a small effect on the amplitude and latency to onset of the C-fibre response to supra-maximal stimulation (Bb, inset right).
Figure 4.
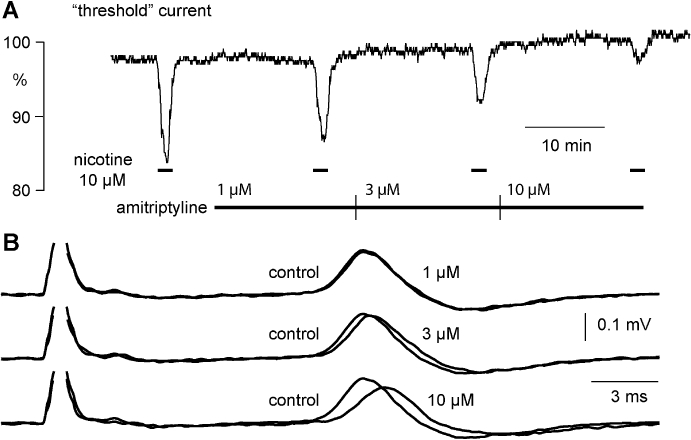
Amitriptyline affects the nicotine-induced increase in excitability as well as the amplitude and latency of the C-fibre CAP response to supra-maximal stimulation. (A) Amitriptyline (1–10 µM) inhibits concentration-dependently the increase in C-fibre excitability induced by nicotine (10 µM). In addition, amitriptyline (1–10 µM, B) produces a rather modest concentration-dependent decrease in the absolute amplitude (peak-to-peak) and an increase in the latency to half-maximum amplitude of the compound C-fibre action potential response to supra-maximal electrical stimulation.
In control experiments (n= 11), nicotine was applied for 90 s periods at intervals of 20 min, during which the bath was perfused with physiological solution (Figure 3A). We did not observe any tachyphylaxis of the axonal response to nicotine across repeat applications using such time intervals. In further experiments, the effect of amitriptyline on the axonal response to nicotine was examined, and a representative example is shown in Figure 3B. Amitriptyline (3 µM) reduced the nicotine-induced increase in axonal excitability by more than 50%. The population statistics for this effect are shown in Figure 5A, and from these data an EC50 for amitriptyline's inhibition of the response to nicotine of 2.6 ± 0.2 µM was calculated. The suppression of the nicotine response by amitriptyline was long lasting and apparent for up to 40 min following the beginning of amitriptyline washout (Figure 3Ba).
The effects of amitriptyline in concentrations ranging from 1 to 10 µM on the amplitude and latency of the compound C-type action potential and on the increase in the excitability of unmyelinated axons produced by bath application of nicotine are illustrated in Figure 4. Amitriptyline (1 µM) reduced the increase in excitability produced by bath application of nicotine without a clear effect on either the amplitude or latency of the compound C-fibre action potential response to electrical stimulation. At higher concentrations of amitriptyline (3–10 µM) the nicotine-induced increase in axonal excitability was further reduced (Figure 4A), and additionally the amplitude and latency of the C-type CAP were also marginally altered (Figure 4B). The quantitative analysis revealed differences between the lowest concentration of amitriptyline (1 µM) necessary for inhibition of the nicotine-induced changes in membrane threshold and that required to bring about changes in the CAP (Figure 5). Amitriptyline (1 µM) produced a clear reduction of the ‘nicotinic response’ (33.7 ± 3.0%; mean ± SEM, n= 5), while changes in the CAP at this concentration of amitriptyline were not significant (decrease in peak amplitude: 3.3 ± 2.8%; increase in latency: 1.2 ± 1.7%; mean ± SEM, n= 5).
The response of unmyelinated axons to nicotine is differentially affected by amitriptyline and TTX
In another series of experiments, the effects of amitriptyline (10 µM) on the C-fibre response to nicotine were compared with those of TTX (10 nM). A representative example is shown in Figure 6. Changes in axonal excitability following the addition of nicotine (10 µM) to the bathing solution were tested twice under control conditions (Figure 6B). Exposure of the nerve fascicle to TTX (10 nM) produced a clear decrease in the amplitude and an increase in the latency to onset of the compound C-fibre action potential (Figure 6A) but did not affect the nicotine-induced decrease in ‘threshold current’. The effects of TTX were reversible, and axonal excitability returned to the resting values within about 20 min of washout. In contrast, amitriptyline (10 µM) reduced only marginally the amplitude and latency of the compound C-fibre potential (Figure 6B), but it produced a substantial reduction of the ‘threshold current’ response to nicotine. The pooled data (five individual fascicles from three patients) for this effect is shown in Figure 7. Amitriptyline (10 µM) changed the shape of the compound C-fibre action potential considerably less than TTX (10 nM), but the reduction of the nicotine-induced increase in axonal excitability was much more pronounced than that of TTX.
Figure 6.
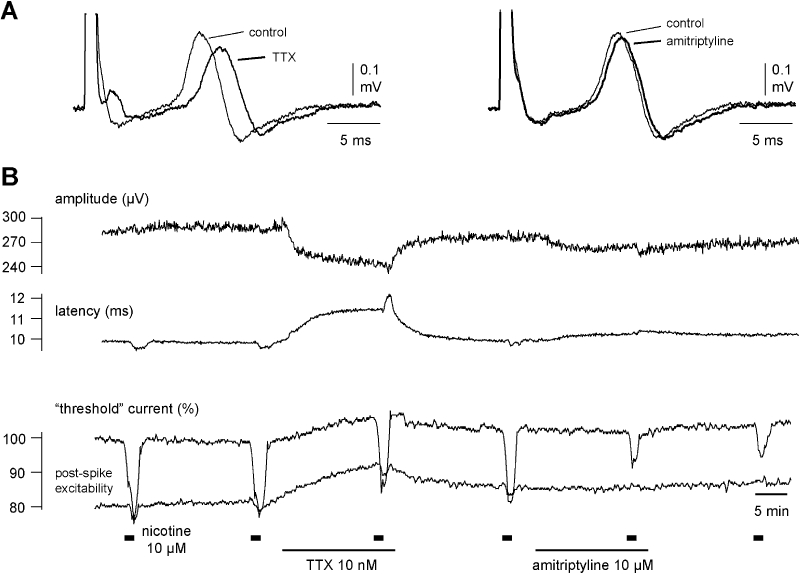
Amitriptyline (10 µM) but not tetrodotoxin (TTX) (10 nM) reduces the nicotine-induced increase in axonal excitability. The effect of both amitriptyline and TTX on the compound action potential (CAP) response to electrical stimulation of unmyelinated axons is shown for a single human nerve fascicle. In this recording nicotine (10 µM, 90 s, thick bars) was added to the bathing solution at regular intervals of 20 min. Over the 20 min period preceding the third application of nicotine, TTX (10 nM) was added to the bathing solution and during the interval between the fourth and fifth application of nicotine, amitriptyline (10 µM) was added to the solution perfusing the bath. TTX reduced the amplitude and prolonged the latency to half-maximum amplitude of the CAP response to supra-maximal stimulation and increased the current required to maintain a CAP response of 40% maximum (left panel A and B) but was without effect on the increase in excitability seen in the presence of nicotine (10 µM). Amitriptyline (10 µM) however considerably reduced the increase in excitability produced by nicotine (10 µM) but only marginally reduced the amplitude and prolonged the latency to half-maximum amplitude of the CAP response to supra-maximal stimulation. Amitriptyline (10 µM) was without effect on the current required to evoke a 40% maximum CAP.
Discussion
The key observation of the present study is an inhibition of nAChRs expressed in human unmyelinated axons by low concentrations of amitriptyline. Amitriptyline's effect on the response to nicotine of unmyelinated human axons resembles that previously reported for mecamylamine, a potent blocker of nAChRs (Lang et al., 2003). It has long been known that the depolarizing action of acetylcholine on unmyelinated axons can be blocked not only by specific antagonists of nAChRs but also by substances such as tetraethylammonium, local anaesthetics and morphine (Ritchie and Armett, 1963). It has been suggested, therefore, that all compounds with a quaternary or tertiary nitrogen atom in their structure are able to antagonize the action of acetylcholine on C-fibres (Ritchie and Armett, 1963). Amitriptyline seems to be another example consistent with this interpretation. However, amitriptyline also has effects on other ion channels (see Introduction). The small changes in latency and amplitude of the compound C-fibre action potential produced by amitriptyline may indicate that a reduction in the availability of voltage-dependent Na+ channels contributes to the amitriptyline-induced inhibition of the effects of nicotine. However, in the presence of TTX, pronounced changes in the shape of compound C-fibre action potential were seen without any concomitant effect on the response to nicotine. This indicates that amitriptyline has a direct action on nAChRs.
It seems possible that TTX-resistant sodium channels in unmyelinated peripheral axons could contribute to the observed interaction between the effects of nicotine and amitriptyline. Amitriptyline can inhibit the TTX-resistant voltage-dependent sodium channel (TTXr Nav) in rat dorsal root and trigeminal ganglion cells (Bräu et al., 2001; Hur et al., 2008). If this action of amitriptyline were to contribute to its observed blockade of axonal responses to nicotine, the action of nicotine would itself need to be in part mediated by Navs. While nicotine affects cell expressed TTXr Nav1.5 (hH1) currents (Liu et al., 2004), it does so at concentrations in the millimolar range, and with an inhibitory mode of action. Notwithstanding the differences in concentration, it is difficult to envisage how nicotine and amitriptyline, both of which would be expected to block TTXr NaV, might be able to interact to account for the observation here, namely that amitriptyline can suppress the excitatory action of nicotine on unmyelinated axons. Furthermore, the observation that amitriptyline blocks nAChR-mediated responses in skeletal muscle (Gumilar et al., 2003), a tissue in which TTX-resistant sodium channel isoforms are not expressed, suggests that TTXr Nav are not essential for amitriptyline-induced blockade of responses to nicotine. The alternative proposal, namely that amitriptyline is one of a large number of compounds with a chemical structure appropriate for blockade of nAChRs (Ritchie and Armett, 1963; Arias et al., 2006), seems considerably more reasonable to explain our findings.
Clinical relevance of concentrations used
The effect of nicotine on axonal excitability was reduced by concentrations of amitriptyline of around 1 µM (314 ng·mL−1), and this is consistent with a previous report illustrating the high sensitivity of nicotinic responses to amitriptyline (Park et al., 1998). Studies in human subjects suggest that plasma levels of amitriptyline after single or multiple oral doses range from 20 to 150 ng·mL−1 (ca. 70–540 nM; Schulz et al., 1985). It is therefore possible that nicotinic receptors on sensory axons are inhibited during treatment of neuropathic pain with amitriptyline. In fact, amitriptyline in moderate doses reduces the quantitative sudomotor axon reflex in human subjects (Low and Opfer-Gehrking, 1992), and nicotinic receptors on sympathetic axons are thought to contribute to this sweat response (Wada et al., 1952).
nAChRs and neuropathic pain
The findings of the present study indicate a possible contribution of nAChRs to the pathogenesis of neuropathic pain. There are previous reports that indicate such a connection: (i) The expression of nAChRs is altered following peripheral nerve lesions with nicotinic acetylcholine binding sites increasing on both sides of dorsal root ligation sites in rats (Gillberg and Askmark, 1991); (ii) an up-regulation of α5 subunit immunoreactivity in nerve fibres of the outer dorsal horn was observed after spinal nerve ligation (Vincler and Eisenach, 2004); and (iii) knock down of the α5 nAChR subunit is able to alleviate the mechanical allodynia usually accompanying spinal nerve ligation in rats (Vincler and Eisenach, 2005).
There are also pharmacological observations to support the involvement of nAChRs in the pathogenesis of neuropathic pain. Interestingly, anti-nociceptive effects of both nicotinic agonists (Marubio et al., 1999; Vincler, 2005; Meyer, 2006) and antagonists (Livett et al., 2006; Vincler et al., 2006) have been reported. The mechanism underlying the effect of nicotinic agonists is not entirely clear but is most probably attributable to an activation of α4 subunits of nAChRs on inhibitory neurons in supraspinal pain pathways (Marubio et al., 1999; Meyer, 2006). However, it is also possible that the anaesthetic property of nicotine can be due to inhibition of TTXr Navs without activation of nAChRs (Liu et al., 2004). The analgesic effects of antagonists seem plausible in situations where a direct, local excitatory effect of acetylcholine on afferent nociceptive neurons may be involved (Livett et al., 2006).
There are several possible sources of acetylcholine in the periphery. A likely candidate is lymphocytes because these cells release acetylcholine according to their degree of differentiation and activation (Rinner et al., 1998). Invasion of dorsal root ganglia by lymphocytes after peripheral nerve lesions has been reported (Hu and McLachlan, 2002; Kleinschnitz et al., 2006; Moalem and Tracey, 2006) and, as lymphocytes themselves express nAChRs (Kawashima and Fujii, 2003), amitriptyline may be able to exert a direct action on lymphocytes.
Together, the present data indicate that the functional activity of nicotinic receptors expressed on primary afferent nociceptive neurons is blocked by amitriptyline. This mode of action of amitriptyline may explain, at least in part, amitriptyline's therapeutic action in the treatment of neuropathic pain.
Acknowledgments
This work was supported by the Deutsche Forschungsgemeinschaft (GR 801/3-1). We would like to thank Christina Müller for expert technical assistance and the colleagues from the Department of Hand and Plastic Surgery (Head: Dr Stefan Deiler) who performed the sural nerve biopsies.
Glossary
Abbreviations:
- CAP
compound action potential
- nAChR
nicotinic acetylcholine receptor
- TTX
tetrodotoxin
- TTXr Nav
TTX-resistant voltage-dependent sodium channel
Conflict of interest
None.
References
- Arias HR, Bhumireddy P, Bouzat C. Molecular mechanisms and binding site locations for noncompetitive antagonists of nicotinic acetylcholine receptors. Int J Biochem Cell Biol. 2006;38:1254–1276. doi: 10.1016/j.biocel.2006.01.006. [DOI] [PubMed] [Google Scholar]
- Bostock H, Cikurel K, Burke D. Threshold tracking techniques in the study of human peripheral nerve. Muscle Nerve. 1998;21:137–158. doi: 10.1002/(sici)1097-4598(199802)21:2<137::aid-mus1>3.0.co;2-c. [DOI] [PubMed] [Google Scholar]
- Bräu ME, Dreimann M, Olschewski A, Vogel W, Hempelmann G. Effect of drugs used for neuropathic pain management on tetrodotoxin-resistant Na(+) currents in rat sensory neurons. Anesthesiology. 2001;94:137–144. doi: 10.1097/00000542-200101000-00024. [DOI] [PubMed] [Google Scholar]
- Connolly J, Boulter J, Heinemann SF. Alpha 4-2 beta 2 and other nicotinic acetylcholine receptor subtypes as targets of psychoactive and addictive drugs. Br J Pharmacol. 1992;105:657–666. doi: 10.1111/j.1476-5381.1992.tb09035.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dick IE, Brochu RM, Purohit Y, Kaczorowski GJ, Martin WJ, Priest BT. Sodium channel blockade may contribute to the analgesic efficacy of antidepressants. J Pain. 2007;8:315–324. doi: 10.1016/j.jpain.2006.10.001. [DOI] [PubMed] [Google Scholar]
- Douglas WW, Ritchie JM. The excitatory action of acetylcholine on cutaneous non-myelinated fibres. J Physiol (Lond) 1960;150:501–514. doi: 10.1113/jphysiol.1960.sp006401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duale C, Daveau J, Cardot JM, Boyer-Grand A, Schoeffler P, Dubray C. Cutaneous amitriptyline in human volunteers: differential effects on the components of sensory information. Anesthesiology. 2008;108:714–721. doi: 10.1097/ALN.0b013e3181672632. [DOI] [PubMed] [Google Scholar]
- Flores CM, DeCamp RM, Kilo S, Rogers SW, Hargreaves KM. Neuronal nicotinic receptor expression in sensory neurons of the rat trigeminal ganglion: demonstration of alpha3beta4, a novel subtype in the mammalian nervous system. J Neurosci. 1996;16:7892–7901. doi: 10.1523/JNEUROSCI.16-24-07892.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galeotti N, Ghelardini C, Capaccioli S, Quattrone A, Nicolin A, Bartolini A. Blockade of clomipramine and amitriptyline analgesia by an antisense oligonucleotide to mKv1.1, a mouse Shaker-like K+ channel. Eur J Pharmacol. 1997;330:15–25. doi: 10.1016/s0014-2999(97)10134-0. [DOI] [PubMed] [Google Scholar]
- Genzen JR, Van Cleve W, McGehee DS. Dorsal root ganglion neurons express multiple nicotinic acetylcholine receptor subtypes. J Neurophysiol. 2001;86:1773–1782. doi: 10.1152/jn.2001.86.4.1773. [DOI] [PubMed] [Google Scholar]
- Gillberg PG, Askmark H. Changes in cholinergic and opioid receptors in the rat spinal cord, dorsal root and sciatic nerve after ventral and dorsal root lesion. J Neural Transm Gen Sect. 1991;85:31–39. doi: 10.1007/BF01244655. [DOI] [PubMed] [Google Scholar]
- Gumilar F, Arias HR, Spitzmaul G, Bouzat C. Molecular mechanisms of inhibition of nicotinic acetylcholine receptors by tricyclic antidepressants. Neuropharmacology. 2003;45:964–976. doi: 10.1016/s0028-3908(03)00247-8. [DOI] [PubMed] [Google Scholar]
- Haupts MR, Amoiridis G. The sural nerve is not always a pure sensory nerve. Muscle Nerve. 2007;35:812. doi: 10.1002/mus.20767. [DOI] [PubMed] [Google Scholar]
- Hu P, McLachlan EM. Macrophage and lymphocyte invasion of dorsal root ganglia after peripheral nerve lesions in the rat. Neuroscience. 2002;112:23–38. doi: 10.1016/s0306-4522(02)00065-9. [DOI] [PubMed] [Google Scholar]
- Hur YK, Choi IS, Cho JH, Park EJ, Choi JK, Choi BJ, et al. Effects of carbamazepine and amitriptyline on tetrodotoxinresistant Na+ channels in immature rat trigeminal ganglion neurons. Arch Pharm Res. 2008;31:178–182. doi: 10.1007/s12272-001-1138-x. [DOI] [PubMed] [Google Scholar]
- Kawashima K, Fujii T. The lymphocytic cholinergic system and its contribution to the regulation of immune activity. Life Sci. 2003;74:675–696. doi: 10.1016/j.lfs.2003.09.037. [DOI] [PubMed] [Google Scholar]
- Keele CA, Armstrong D. Substances Producing Pain and Itch. London: Edward Arnold; 1964. [Google Scholar]
- Khan MA, Gerner P, Kuo WG. Amitriptyline for prolonged cutaneous analgesia in the rat. Anesthesiology. 2002;96:109–116. doi: 10.1097/00000542-200201000-00023. [DOI] [PubMed] [Google Scholar]
- Kleinschnitz C, Hofstetter HH, Meuth SG, Braeuninger S, Sommer C, Stoll G. T cell infiltration after chronic constriction injury of mouse sciatic nerve is associated with interleukin-17 expression. Exp Neurol. 2006;200:480–485. doi: 10.1016/j.expneurol.2006.03.014. [DOI] [PubMed] [Google Scholar]
- Lang PM, Burgstahler R, Sippel W, Irnich D, Schlotter-Weigel B, Grafe P. Characterization of neuronal nicotinic acetylcholine receptors in the membrane of unmyelinated human C-fiber axons by in vitro studies. J Neurophysiol. 2003;90:3295–3303. doi: 10.1152/jn.00512.2003. [DOI] [PubMed] [Google Scholar]
- Leffler A, Reiprich A, Mohapatra DP, Nau C. Use-dependent block by lidocaine but not amitriptyline is more pronounced in tetrodotoxin (TTX)-Resistant Nav1.8 than in TTX-sensitive Na+ channels. J Pharmacol Exp Ther. 2007;320:354–364. doi: 10.1124/jpet.106.109025. [DOI] [PubMed] [Google Scholar]
- Lips KS, Pfeil U, Kummer W. Coexpression of alpha 9 and alpha 10 nicotinic acetylcholine receptors in rat dorsal root ganglion neurons. Neuroscience. 2002;115:1–5. doi: 10.1016/s0306-4522(02)00274-9. [DOI] [PubMed] [Google Scholar]
- Liu L, Pugh W, Ma H, Simon SA. Identification of acetylcholine receptors in adult rat trigeminal ganglion neurons. Brain Res. 1993;617:37–42. doi: 10.1016/0006-8993(93)90609-q. [DOI] [PubMed] [Google Scholar]
- Liu L, Chang GQ, Jiao YQ, Simon SA. Neuronal nicotinic acetylcholine receptors in rat trigeminal ganglia. Brain Res. 1998;809:238–245. doi: 10.1016/s0006-8993(98)00862-2. [DOI] [PubMed] [Google Scholar]
- Liu L, Zhu W, Zhang ZS, Yang T, Grant A, Oxford G, et al. Nicotine inhibits voltage-dependent sodium channels and sensitizes vanillois receptors. J Neurophysiol. 2004;91:14782–11491. doi: 10.1152/jn.00922.2003. [DOI] [PubMed] [Google Scholar]
- Livett BG, Sandall DW, Keays D, Down J, Gayler KR, Satkunanathan N, et al. Therapeutic applications of conotoxins that target the neuronal nicotinic acetylcholine receptor. Toxicon. 2006;48:810–829. doi: 10.1016/j.toxicon.2006.07.023. [DOI] [PubMed] [Google Scholar]
- Low PA, Opfer-Gehrking TL. Differential effects of amitriptyline on sudomotor, cardiovagal, and adrenergic function in human subjects. Muscle Nerve. 1992;15:1340–1344. doi: 10.1002/mus.880151208. [DOI] [PubMed] [Google Scholar]
- Marubio LM, Mar Arroyo-Jimenez M, Cordero-Erausquin M, Lena C, Le Novere N, de Kerchove d'Exaerde A, et al. Reduced antinociception in mice lacking neuronal nicotinic receptor subunits. Nature. 1999;398:805–810. doi: 10.1038/19756. [DOI] [PubMed] [Google Scholar]
- Meyer MD. Neuronal nicotinic acetylcholine receptors as a target for the treatment of neuropathic pain. Drug Dev Res. 2006;67:355–359. [Google Scholar]
- Moalem G, Tracey DJ. Immune and inflammatory mechanisms in neuropathic pain. Brain Res Rev. 2006;51:240–264. doi: 10.1016/j.brainresrev.2005.11.004. [DOI] [PubMed] [Google Scholar]
- Moalem G, Grafe P, Tracey DJ. Chemical mediators enhance the excitability of unmyelinated sensory axons in normal and injured peripheral nerve of the rat. Neuroscience. 2005;134:1399–1411. doi: 10.1016/j.neuroscience.2005.05.046. [DOI] [PubMed] [Google Scholar]
- Moalem-Taylor G, Lang PM, Tracey DJ, Grafe P. Post-spike excitability indicates changes in membrane potential of isolated C-fibers. Muscle Nerve. 2007;36:172–182. doi: 10.1002/mus.20793. [DOI] [PubMed] [Google Scholar]
- Nau C, Seaver M, Wang SY, Wang GK. Block of human heart hH1 sodium channels by amitriptyline. J Pharmacol Exp Ther. 2000;292:1015–1023. [PubMed] [Google Scholar]
- Onghena P, Van HB. Antidepressant-induced analgesia in chronic non-malignant pain: a meta-analysis of 39 placebo-controlled studies. Pain. 1992;49:205–219. doi: 10.1016/0304-3959(92)90144-Z. [DOI] [PubMed] [Google Scholar]
- Park TJ, Shin SY, Suh BC, Suh EK, Lee IS, Kim YS, et al. Differential inhibition of catecholamine secretion by amitriptyline through blockage of nicotinic receptors, sodium channels, and calcium channels in bovine adrenal chromaffin cells. Synapse. 1998;29:248–256. doi: 10.1002/(SICI)1098-2396(199807)29:3<248::AID-SYN7>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- Punke MA, Friederich P. Amitriptyline is a potent blocker of human Kv1.1 and Kv7.2/7.3 channels. Anesth Analg. 2007;104:1256–1264. doi: 10.1213/01.ane.0000260310.63117.a2. [DOI] [PubMed] [Google Scholar]
- Rammes G, Rupprecht R. Modulation of ligand-gated ion channels by antidepressants and antipsychotics. Mol Neurobiol. 2007;35:160–174. doi: 10.1007/s12035-007-0006-1. [DOI] [PubMed] [Google Scholar]
- Rinner I, Kawashima K, Schauenstein K. Rat lymphocytes produce and secrete acetylcholine in dependence of differentiation and activation. J Neuroimmunol. 1998;81:31–37. doi: 10.1016/s0165-5728(97)00155-0. [DOI] [PubMed] [Google Scholar]
- Ritchie JM, Armett CJ. The role of acetylcholine in conduction in mammalian nonmyelinated nerve fibers. J Pharmacol Exp Ther. 1963;139:201–207. [PubMed] [Google Scholar]
- Saarto T, Wiffen PJ. Antidepressants for neuropathic pain. Cochrane Database Syst Rev. 2007;(4) doi: 10.1002/14651858.CD005454.pub2. CD005454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sacerdote P, Brini A, Mantegazza P, Panerai AE. A role for serotonin and beta-endorphin in the analgesia induced by some tricyclic antidepressant drugs. Pharmacol Biochem Behav. 1987;26:153–158. doi: 10.1016/0091-3057(87)90548-x. [DOI] [PubMed] [Google Scholar]
- Schmelz M, Schmidt R, Weidner C, Hilliges M, Torebjork ET, Handwerker HO. Chemical response pattern of different classes of C-nociceptors to pruritogens and algogens. J Neurophysiol. 2003;89:2441–2448. doi: 10.1152/jn.01139.2002. [DOI] [PubMed] [Google Scholar]
- Schulz P, Dick P, Blaschke TF, Hollister L. Discrepancies between pharmacokinetic studies of amitriptyline. Clin Pharmacokinet. 1985;10:257–268. doi: 10.2165/00003088-198510030-00005. [DOI] [PubMed] [Google Scholar]
- Steen KH, Reeh PW. Actions of cholinergic agonists and antagonists on sensory nerve endings in rat skin, in vitro. J Neurophysiol. 1993;70:397–405. doi: 10.1152/jn.1993.70.1.397. [DOI] [PubMed] [Google Scholar]
- Tanelian DL. Cholinergic activation of a population of corneal afferent nerves. Exp Brain Res. 1991;86:414–420. doi: 10.1007/BF00228966. [DOI] [PubMed] [Google Scholar]
- Vadalouca A, Siafaka I, Argyra E, Vrachnou E, Moka E. Therapeutic management of chronic neuropathic pain: an examination of pharmacologic treatment. Ann N Y Acad Sci. 2006;1088:164–186. doi: 10.1196/annals.1366.016. [DOI] [PubMed] [Google Scholar]
- Vincler M. Neuronal nicotinic receptors as targets for novel analgesics. Expert Opin Investig Drugs. 2005;14:1191–1198. doi: 10.1517/13543784.14.10.1191. [DOI] [PubMed] [Google Scholar]
- Vincler M, Eisenach JC. Plasticity of spinal nicotinic acetylcholine receptors following spinal nerve ligation. Neurosci Res. 2004;48:139–145. doi: 10.1016/j.neures.2003.10.007. [DOI] [PubMed] [Google Scholar]
- Vincler M, Wittenauer S, Parker R, Ellison M, Olivera BM, McIntosh JM. Molecular mechanism for analgesia involving specific antagonism of alpha9alpha10 nicotinic acetylcholine receptors. Proc Natl Acad Sci USA. 2006;103:17880–17884. doi: 10.1073/pnas.0608715103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vincler MA, Eisenach JC. Knock down of the alpha 5 nicotinic acetylcholine receptor in spinal nerve-ligated rats alleviates mechanical allodynia. Pharmacol Biochem Behav. 2005;80:135–143. doi: 10.1016/j.pbb.2004.10.011. [DOI] [PubMed] [Google Scholar]
- Wada M, Rai T, Takagaki T, Nakagawa T. Axon reflex mechanism in sweat responses to nicotine, acetylcholine and sodium chloride. J Appl Physiol. 1952;4:745–752. doi: 10.1152/jappl.1952.4.9.745. [DOI] [PubMed] [Google Scholar]
- Wang GK, Russell C, Wang SY. State-dependent block of voltage-gated Na+ channels by amitriptyline via the local anesthetic receptor and its implication for neuropathic pain. Pain. 2004;110:166–174. doi: 10.1016/j.pain.2004.03.018. [DOI] [PubMed] [Google Scholar]
- Zhang CL, Verbny Y, Malek SA, Stys PK, Chiu SY. Nicotinic acetylcholine receptors in mouse and rat optic nerves. J Neurophysiol. 2004;91:1025–1035. doi: 10.1152/jn.00769.2003. [DOI] [PubMed] [Google Scholar]