

Abstract
Background and purpose:
Our previous study showed that urocortin (Ucn1) exacerbates the hypercoagulable state and vasculitis in a rat model of sodium laurate-induced thromboangiitis obliterans. Furthermore, the inflammatory molecules COX-2 and ICAM-1 may participate in this effect. In the present study, the effects of Ucn1 on COX-2 and ICAM-1 expression in lipopolysaccharide (LPS)-induced rat aortic endothelial cells (RAECs) were investigated and the mechanisms involved explored.
Experimental approach:
RAECs were isolated from adult male Wistar rats, and identified at the first passage. Experiments were performed on cells, from primary culture, at passages 5–8. The expression of COX-2 and ICAM-1 at both mRNA and protein levels was determined by semi-quantitative RT-PCR and Western blot analysis. Levels of PGE2 and soluble ICAM-1 (sICAM-1) in culture medium were measured by enzyme-linked immunosorbent assay. Furthermore, the phosphorylation status of p38MAPK, ERK1/2, JNK, Akt and NF-κB was analysed by Western blot; nuclear translocation of NF-κB was observed by immunofluorescence.
Key results:
Ucn1 augmented LPS-induced expression of COX-2 and ICAM-1 in RAECs in a time- and concentration-dependent manner. Ucn1 increased PGE2 and sICAM-1 levels. These effects were abolished by the CRF2 receptor antagonist, antisauvagine-30, but not by the CRF1 receptor antagonist, NBI-27914. Moreover, Ucn2 activated p38MAPK and augmented NF-κB nuclear translocation and phosphorylation, whereas ERK1/2, JNK and Akt pathways were not involved in this process.
Conclusions and implications:
These findings suggest that Ucn1 exerts pro-inflammatory effects by augmenting LPS-induced expression of COX-2 and ICAM-1 in RAECs via CRF2 receptors and the activation of p38MAPK and NF-κB.
Keywords: urocortin, COX-2, ICAM-1, CRF2 receptor, rat aortic endothelial cells
Introduction
Vascular endothelial cells (ECs) play a pivotal role in modulating local and systemic inflammation. ECs express chemokines that initiate the activation and recruitment of circulating leukocytes at sites of tissue inflammation (Berman et al., 1993; Bannerman and Goldblum, 1997). Among these chemokines, COX-2 and ICAM-1 are two important modulators, with the former participating in a series of inflammatory diseases and the latter playing an important role in the adhesion process of inflammatory cells. The bacterial endotoxin, lipopolysaccharide (LPS), an essential component of the surface of Gram-negative bacteria (Raetz, 1990), has potent pro-inflammatory properties by acting on many cell types including ECs (Berman et al., 1993; Munshi et al., 2002). LPS-activated ECs may be changed with up-regulation of COX-2 and ICAM-1 expression (Chen et al., 2001b; Heo et al., 2008), pro-coagulant activity, enhanced endothelial permeability and abundant pro-inflammatory mediators' secretion (Berman et al., 1993; Bannerman and Goldblum, 1997; Bierhaus et al. 2000), which may lead to an imbalance in the immune system. Thus, finding a way to minimize COX-2 and ICAM-1 expression may have a dramatic impact on the treatment of inflammatory diseases.
Urocortin (Ucn), a 40 aa corticotrophin-releasing factor (CRF) family peptide that was first identified in rat midbrain (Vaughan et al., 1995), has been demonstrated to be widely expressed in peripheral tissues including the cardiovascular system, spleen, skin, lymphocytes, macrophages and mast cells (Fekete and Zorrilla, 2007). The effects of Ucn1 are mediated by two high-affinity receptors, the CRF1 receptor and the CRF2 receptor, with the former mainly distributed centrally and the latter primarily expressed peripherally (Fekete and Zorrilla, 2007). Besides its cardiovascular protective property (Okosi et al., 1998; Oki and Sasano, 2004; Yang et al., 2006), Ucn1 is now considered to be a potent immunomodulator which participates in various immune responses (Fekete and Zorrilla, 2007). Our previous studies have demonstrated that Ucn1 participates in the development of allergic asthma in the rat and can trigger mast cell infiltration and activation via CRF receptors (Wu et al., 2006; 2008;). Singh et al. (1999) found that Ucn1 is potent at inducing mast cell degranulation and triggering vascular permeability via CRF receptors. Moreover, our data showed that CRF plays a significant role in promoting the development of atherosclerosis (Wu et al., 2009). All of these findings suggest that immune-derived Ucn1 may participate in the pathophysiology of many inflammatory conditions as a pro-inflammatory mediator.
Previous studies have indicated that endogenous Ucn1 has an inhibitory role in the growth of ECs and blood vessels (Yang et al., 2008), and it may trigger ECs to change into a procoagulant state (Grignani and Maiolo, 2000) by stimulating interleukin-1b (IL-1b) and interleukin-6 (IL-6) secretion in vitro (Kohno et al., 2001). Recently, it was found that activation of CRF2 receptors suppresses vascularization (Bale et al., 2002; Hao et al., 2008). Thus, Ucn1 may have a dramatic impact on the normal function of ECs.
Kageyama et al. (2006) demonstrated that in rat aortic smooth muscle cells, Ucn1 induces the expression of COX-2 in a time- and concentration-dependent manner (Kageyama et al., 2006). Also, CRF has been shown to enhance the interferon-γ-stimulated expression of ICAM-1 on human skin keratinocytes (Quevedo et al., 2001). Our recent data demonstrated that Ucn1 promotes the development of vasculitis in the rat thromboangiitis obliterans (TAO) model (original article accepted by British Journal of Pharmacology). Because COX-2 and ICAM-1 are associated with the development of vasculitis (de Gaetano et al., 2003; Witkowska, 2005), our data also suggest that this effect of Ucn1 on vasculitis might be exerted through an effect on the expression of these two factors. The present study was performed to examine the effects of Ucn1 on COX-2 and ICAM-1 expression in LPS-induced rat aortic endothelial cells (RAECs) and explore the mechanisms involved. In the present study, it was first demonstrated that Ucn increased the expression of both COX-2 and ICAM-1 in a time- and concentration-dependent manner in LPS-activated RAECs. Furthermore, the p38MAPK and NF-κB pathways were involved in this effect, which was exerted via the CRF2 receptor.
Methods
Culture of RAECs
RAECs were isolated from male Wistar rats (Shanghai Laboratory Animal Center, Shanghai, China) according to the method described previously (McGuire and Orkin, 1987; Yang et al., 2006). The animal operation procedure was approved by the ethics review board of Nanjing Medical University. Cells were cultured in RPMI 1640 supplemented with 20% fetal bovine serum, 1% penicillin–streptomycin, at 37°C in a 95% air/5% CO2 incubator. Our previous studies were carried out with passages 5–8 RAECs, and it was found, using an F-VIII marker, that these cells maintain their properties (Yang et al., 2006). Thus, the present experiments were performed on cells, from primary culture, at passages 5–8. The identity of the RAECs was confirmed by immunofluorescence staining by the use of rabbit anti-rat factor VIII antibody and Cy-3 conjugated goat anti-rabbit IgG.
Immunofluorescence staining
RAECs were cultured on coverslips placed in tissue culture dishes. Following different treatments, the cells were washed with phosphate-buffered solution and fixed with fresh 4% paraformaldehyde for 30 min. Subsequently, the cells were permeabilized with 0.5% Triton X-100 for 15 min on ice and blocked in 5% BSA for 30 min at room temperature. After being blocked, the cells were incubated with factor VIII antibody (1:100) or NF-κB p65 antibody (1:25) overnight in a humid chamber at 4°C. Then, the cells were incubated with a secondary antibody conjugated to Cy-3 for 30 min in the dark. After being washed three times, the cells were mounted on a slide. The slides were visualized using a fluorescence microscope.
RNA isolation and semi-quantitative RT-PCR analysis
Total RNAs were extracted from RAECs, using TRIzol according to the manufacturer's protocol. For cDNA synthesis, Moloney murine leukaemia virus (MMLV) was applied as the reverse transcriptase. For PCR reaction, Taq DNA polymerase was used in the reaction system. Primers for COX-2 (Ohnaka et al., 2000), ICAM-1 (Taal et al., 2000) and GAPDH (Baigent and Lowry, 2000) were synthesized from published sequences as shown in Table 1. The products were visualized by electrophoresis in 2.0% agarose gel containing 0.5 µg·mL−1 ethidium bromide. Specific genes were verified by their predicted sizes. GAPDH was set as the internal control.
Table 1.
A summary of the RT-PCR primer sequences used to amplify GAPDH, COX-2 and ICAM-1
| Sequences | Product size (bp) | Annealing T (C) | ||
|---|---|---|---|---|
| GAPDH | Sense | TCCCAGAGCTGAACGGGAAGCTCACTG | 339 | 68.1 |
| Antisense | TGGAGGCCATGTAGGCCATGAGGTCCA | |||
| COX-2 | Sense | TTCACCAGACAGATTGCTGGC | 530 | 63.5 |
| Antisense | AGTCTGGAGTGGGAGGCACTTG | |||
| ICAM-1 | Sense | AGAAGGACTGCTTGGGGAA | 332 | 58.1 |
| Antisense | CCTCTGGCGGTAATAGGTG | |||
Western blot analysis
The protein samples were separated on a 10% sodium dodecyl sulphate–polyacrylamide gel and electrophoretically transferred to PVDF membranes in Tris–glycine transfer buffer. Then, membranes were blocked in 5% (w/v) instant non-fat dried milk for 2 h at room temperature, and incubated with primary antibodies corresponding to COX-2 (1:1000), ICAM-1 (1:500), β-actin (1:250), p38MAPK (1:1000), phospho-p38MAPK (1:1000), ERK1/2 (1:1000), phospho-ERK1/2 (1:1000), JNK (1:1000), phospho-JNK (1:1000), Akt (1:1000), phospho-Akt (Ser 473) (1:1000), NF-κB p65 (1:1000), phospho-NF- κB p65 (1:1000), respectively, at 4°C overnight. The membranes were subsequently washed with TBST [50 mM Tris–HCl (pH 7.5), 150 mM NaCl, 0.05% Tween 20] and incubated with secondary horseradish peroxidase-conjugated IgG for 2 h at room temperature. Immunoreactive proteins were visualized by LumiGLO (Cell Signaling Technology, Beverly, MA, USA) chemiluminescent reagent and peroxide. The light-emitting bands were detected with X-ray films.
Enzyme-linked immunosorbent assay (elisa)
Levels of PGE2 and soluble ICAM-1 (sICAM-1) in culture medium were determined by elisa assay using PGE2 and sICAM-1 kits according to the manufacturers' instructions. Kit standards and controls were performed for each assay completed. The final concentrations of PGE2 and sICAM-1 were calculated by converting the optical density reading using standard curves.
Statistical analysis
Data are expressed as means ± SEM. The significance for the difference among groups was analysed with SPSS 11.0 (Chicago, IL, USA) by one-way analysis of variance with Student–Newman–Keuls multiple comparison methods. Differences were considered to be statistically significant at a P value of <0.05.
Reagents
Rat urocortin1 (Ucn1), Ucn2, CRF1 receptor antagonist NBI-27914, CRF2 receptor antagonist antisauvagine-30, LPS, rabbit anti-rat factor VIII (a stable endothelial antigen) antibody and Cy-3 conjugated goat anti-rabbit IgG were purchased from Sigma (St. Louis, MO, USA). Polyclonal COX-2 antibody and monoclonal ICAM-1 antibody were obtained from Abcam (Cambridge, UK). PGE2 and sICAM-1 elisa kits were obtained from R&D (Minneapolis, MN, USA) and Boster (Wuhan, China) respectively. Specific antibodies to p38MAPK, phospho-p38MAPK, ERK1/2, phospho-ERK1/2, JNK, phospho-JNK, Akt, phospho-Akt (Ser 473), NF-κB p65, phospho-NF-κB p65, p38MAPK inhibitor SB203580, LumiGLO chemiluminescent reagent and peroxide were provided by Cell Signaling Technology. TRIzol and MMLV were obtained from Invitrogen (Carlsbad, CA, USA), Taq DNA polymerase from Promega (Madison, WI, USA) and the X-ray films from Kodak (Rochester, NY, USA). The other reagents used were derived from commercial sources. All drug/molecular target nomenclature conforms to British Journal of Pharmacology's Guide to Receptors and Channels (Alexander et al., 2008).
Results
Immunofluorescent identification of RAECs
After being isolated, RAECs were identified at the first passage. As shown in Figure 1, the cytoplasm was completely stained red with factor VIII-specific antibody and Cy-3 conjugated secondary antibody. Factor VIII-associated antigen is a cytological marker closely identified with ECs (Karasek, 1989); the cells we obtained were undoubtedly RAECs.
Figure 1.

Immunofluorescent identification of rat aortic endothelial cells. Cells cultured on coverslips were incubated with rabbit anti-rat factor VIII (a stable endothelial antigen) antibody and then incubated with Cy-3 conjugated secondary antibody; the nuclei were stained with DAPI. In the negative control, factor VIII antibody was omitted (magnification: 200×).
Ucn augmented LPS-induced COX-2 and ICAM-1 expression in RAECs
As depicted in Figure 2, after exposure of RAECs to 10 µg·mL−1 LPS, 10−7 M Ucn (Ucn1) induced COX-2 and ICAM-1 expression in a time-dependent manner, with the mRNA and protein levels reaching their peaks after 4 and 8 h respectively. With increased time, their expression was decreased. Hence, in the following experiments, the time points of 4 and 8 h were selected to determine the mRNA and protein levels respectively. Ucn1 augmented LPS-induced COX-2 and ICAM-1 expression in a concentration-dependent manner; a marked effect was observed with 10−9 M Ucn1, which did not increase at higher concentrations (Figure 3).
Figure 2.
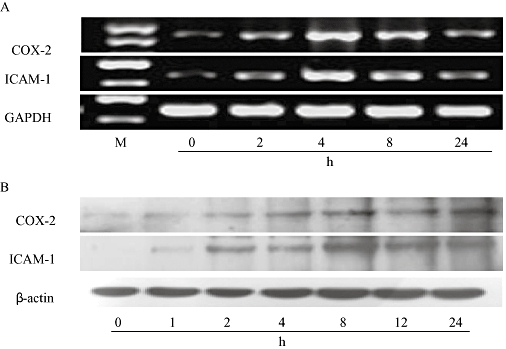
COX-2 and ICAM-1 expression induced by Ucn1 occurred in a time-dependent manner in lipopolysaccharide (LPS)-activated rat aortic endothelial cells. In the presence of LPS (10 µg·mL−1), cells were pretreated with Ucn1 (10−7 M) and incubated for the durations shown. (A) COX-2 and ICAM-1 mRNA expression induced by Ucn1 reached their peak at the time point of 4 h. (B) Ucn1 promoted COX-2 and ICAM-1 protein expression with significant effects at the time point of 8 h. Similar results were obtained from more than three independent cultures, and a representative experiment is shown.
Figure 3.
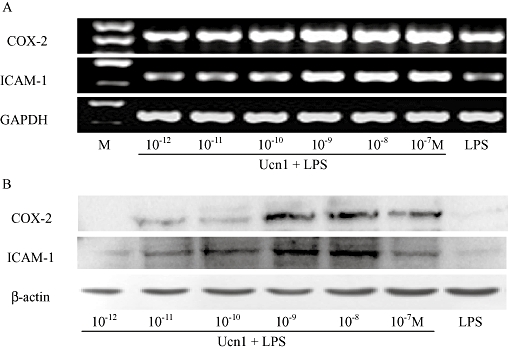
COX-2 and ICAM-1 expression induced by Ucn1 was in a concentration-dependent manner in lipopolysaccharide (LPS)-activated rat aortic endothelial cells. On exposure to LPS (10 µg·mL−1), cells were pretreated with Ucn1 at concentrations ranging from 10−12 to 10−7 M for 30 min. (A) Cells were incubated for 4 h, and COX-2 and ICAM-1 mRNA expression was determined by RT-PCR. (B) Cells were incubated for 8 h, and COX-2 and ICAM-1 protein level was measured by Western blot. Experiments were done from more than three independent cultures, and a representative experiment is shown.
COX-2 and ICAM-1 expression was mediated by the CRF2 receptor
To illustrate the involvement of CRF receptors in Ucn1-induced augmentation of COX-2 and ICAM-1 expression in LPS-activated RAECs, cells were pretreated with Ucn1 alone or together with CRF receptor antagonists (CRF1 receptor antagonist, NBI-27914, or CRF2 receptor antagonist, antisauvagine-30). Ucn1 pretreatment augmented LPS-induced COX-2 and ICAM-1 mRNA expression to levels 1.36- and 1.40-fold observed after LPS pretreatment alone, respectively; antisauvagine-30 reversed this augmentation (Figure 4IA,B). A similar result was observed on COX-2 and ICAM-1 protein expression (Figure 4IC,D). However, NBI-27914 had no significant effect on COX-2 and ICAM-1 expression.
Figure 4.


Effect of Ucn1 on COX-2 and ICAM-1 expression in rat aortic endothelial cells. Cells were treated with vehicle, Ucn1 (10−9 M) alone or together with NBI-27914 (10−8 M) or antisauvegine-30 (10−8 M), or NS-398 (10−5 M) in the presence of lipopolysaccharide (LPS) (10 µg·mL−1) as indicated. (I) Effect of Ucn1 on COX-2 and ICAM-1 expression. Cells were incubated for 4 h (mRNA) or 8 h (protein) to determine COX-2 and ICAM-1 levels by RT-PCR (IA and B) or Western blot (IC and D). (II) Effect of Ucn1 on PGE2 and soluble ICAM-1 (sICAM-1) production measured by enzyme-linked immunosorbent assay. Cells were incubated for 24 h, and the culture supernatant was obtained to determine PGE2 (IIA) or sICAM-1 (IIB) levels. Data given are the means ± SEM of values taken from three independent cultures. *P < 0.05, versus LPS group; #P < 0.05, versus Ucn1 + LPS group. Similar results were obtained from more than three independent cultures, and a representative experiment is shown.
The concentrations of PGE2, the main metabolite of COX-2, and sICAM-1 in the supernatant of the culture were measured by elisa. Similar results were observed for both PGE2 and sICAM-1 in that levels were increased by the application of Ucn1, and pretreatment with antisauvagine-30 abolished this increase (Figure 4IIA,B).
Interestingly, it was found that the two molecules, COX-2 and ICAM-1, changed in a similar manner. To explore their possible relationship, NS-398, a selective inhibitor of COX-2, was used. As shown in Figure 4IIA, in the presence of both Ucn1 and LPS, PGE2 production was significantly decreased after NS-398 pretreatment. Furthermore, Ucn1-induced elevation of ICAM-1 was dramatically reduced on blockade of COX-2 in LPS-activated RAECs (Figure 4IIB). These observations indicate that Ucn1-induced ICAM-1 expression was partially mediated by COX-2.
Ucn2-induced p38MAPK and NF-κB phosphorylation in LPS-activated RAECs
Mitogen-activated protein kinases (MAPKs) and Akt pathways have been implicated in the activation of the pro-inflammatory process (Bhat et al., 1998; Ojaniemi et al., 2003), and have been shown to be key signalling intermediates downstream of CRF2 receptor activation (Dermitzaki et al., 2002; Sananbenesi et al., 2003; Karteris et al., 2004; Moss et al., 2007; Markovic et al., 2008). To investigate the role of these two pathways in the effect of CRF2 receptor on LPS-induced COX-2 and ICAM-1 expression, the cells were pretreated with Ucn2, a selective CRF2 receptor agonist (Fekete and Zorrilla, 2007). Subsequently, the phosphorylation status of p38MAPK, ERK1/2, JNK and Akt were analysed by Western blot.
As depicted in Figure 5A, in the presence of 10 µg·mL−1 LPS, 10−9 M Ucn2 induced a transient phosphorylation of p38MAPK with peak activation at 15 min. Furthermore, LPS-induced significant phosphorylation of p38MAPK and Ucn2 application dramatically augmented this phosphorylation (Figure 5B,C). This phosphorylation of p38MAPK was completely blocked in the presence of 10−5 M SB203580, a p38MAPK inhibitor. However, the phosphorylation of ERK1/2, JNK and Akt did not appear to be altered by Ucn2 treatment (Figure 5D–F).
Figure 5.

Effect of Ucn2 on mitogen-activated protein kinases (MAPKs) and Akt in lipopolysaccharide (LPS)-induced activation of rat aortic endothelial cells. Cells were treated with vehicle or Ucn2 (10−9 M) in the presence of LPS (10 µg·mL−1). (A) For p38MAPK determination, cells were treated with Ucn2 (10−9 M) and incubated for the durations shown. Phospho-p38MAPK was obviously observed at the time point of 15 min. The p38MAPK inhibitor SB203580 (10−5 M) was used to investigate the effect of p38MAPK phosphorylation (B and C). (D and E) For ERK1/2 and JNK measurement, cells were incubated for 15 min, and phospho-ERK1/2, JNK were analysed by Western blot. (F) Effect of Ucn2 on Akt phosphorylation. Results given are the means ± SEM of values taken from three independent cultures. *P < 0.05, versus LPS group. Similar results were obtained from more than three independent cultures, and a representative experiment is shown.
NF-κB is known to participate in the inflammatory process (Ghosh and Hayden, 2008). To determine whether it is involved in Ucn2-induced inflammation, nuclear translocation of NF-κB was observed by immunofluorescence. As shown in Figure 6A, NF-κB was visualized primarily in the cytoplasm in untreated control cells, while LPS treatment for 1 h induced NF-κB translocation from the cytoplasm into the nucleus. What is more important, compared with LPS treatment, Ucn2 noticeably augmented NF-κB nuclear translocation as after Ucn2 treatment, most NF-κB was visualized in the nucleus of the cells. Furthermore, the phosphorylation status of NF-κB was determined by Western blot analysis. As shown in Figure 6B,C, LPS pretreatment significantly increased NF-κB phosphorylation; Ucn2 application further enhanced the phosphorylation of NF-κB. Combined with the observation in Figure 6A, we came to the conclusion that Ucn2 can increase LPS-induced phosphorylation of NF-κB in RAECs.
Figure 6.

Effect of Ucn2 on the NF-κB pathway in lipopolysaccharide (LPS)-activated rat aortic endothelial cells. (A) Effect of Ucn2 on nuclear translocation of NF-κB. Cells cultured on coverslips were incubated with specific NF-κB p65 antibody and then incubated with Cy-3 conjugated secondary antibody; the nucleus was stained with DAPI. A negative control was obtained by omitting NF-κB p65 antibody. (Magnification: 200×). (B and C) Effect of Ucn2 on NF-κB phosphorylation determined by Western blot. Results are presented as the means ± SEM of values taken from three independent cultures. *P < 0.05, versus LPS group. Similar results were obtained from more than three independent cultures, and a representative experiment is shown.
Discussion and conclusions
Ucn (Ucn1), a CRF family peptide (Vaughan et al., 1995), has been demonstrated to stimulate the release of pro-inflammatory mediators under inflammatory conditions (Kohno et al., 2001; Saruta et al., 2004). In this study, we found that Ucn1 could increase LPS-induced COX-2 and ICAM-1expression in RAECs via the CRF2 receptor. Furthermore, p38MAPK and NF-κB pathways participated in this process.
It has been well established that, in addition to the indirect anti-inflammatory effect via ACTH/cortisol (Elenkov and Chrousos, 1999; Elenkov et al., 1999), the CRF family peptides play a direct pro-inflammatory role in the regulation of the inflammatory process (Elenkov and Chrousos, 1999; Elenkov et al., 1999). Moreover, the pro-inflammatory effect of CRF family peptides is partially attributed to their stimulation of immune cells, such as mast cells and macrophages (Theoharides et al., 1995; 1998; Agelaki et al., 2002; Tsatsanis et al., 2007). It has been demonstrated that these immune cells express CRF1 and CRF2 receptors, and can synthesize CRF family peptides (Theoharides et al., 1998; Baigent, 2001). CRF and Ucn1 stimulate the production and release of pro-inflammatory mediators in mast cells (Theoharides et al., 1995; 1998;), such as IL-1, IL-6 and TNF-α. Moreover, CRF intensifies the response of macrophages to bacterial LPS by augmenting their synthesis of the pro-inflammatory cytokines, TNF-α and IL-6, at the mRNA level (Agelaki et al., 2002).
Vascular ECs play a pivotal role in modulating local and systemic inflammation. LPS has potent pro-inflammatory properties and acts on many cell types including ECs (Berman et al., 1993; Munshi et al., 2002). There is much evidence indicating that COX-2, the rate-limiting enzyme in the metabolism of arachidonic acid, is involved in inflammatory diseases (Dubois et al., 1998; Cipollone and Fazia, 2006); ICAM-1, as an important cellular adhesion molecule, has also been shown to have a key role in modulating peripheral inflammatory disease, such as artherosclerosis and TAO (Halacheva et al., 2002; Witkowska, 2005). Several studies have illustrated that LPS-activated ECs demonstrate an up-regulation of COX-2 and ICAM-1 expression (Chen et al., 2001b; Heo et al., 2008). However, the function of Ucn in RAECs and its potential role in modulating COX-2 and ICAM-1 expression have rarely been illustrated.
In the present study, we found that Ucn1 exerted a pro-inflammatory effect by augmenting LPS-induced COX-2 and ICAM-1 expression in RAECs in a time- and concentration-dependent manner via the CRF2 receptor. Indeed, Ucn1 up-regulated the COX-2 and ICAM-1 mRNA expression to reach a maximum at 4 h, and the protein levels peaked at 8 h. This effect was via CRF2 receptors as it could be abolished by the CRF2 receptor antagonist, antisauvagine-30, but not by the CRF1 receptor antagonist, NBI-27914. The above effects were further confirmed by measuring the levels of PGE2 (the main metabolite of COX-2) and sICAM-1 in the culture supernatant of LPS-activated RAECs. The present data are consistent with those from previous studies. For example, it has been demonstrated that CRF-related peptides induce COX-2 expression and PG production in human placental trophoblasts and macrophages (Tsatsanis et al., 2007; Gao et al., 2008). Kageyama et al. (2006) showed that Ucn1 and -2 induced the expression of COX-2 in a time- and dose-dependent manner via CRF2 receptors in rat aortic smooth muscle cells. Previously, it was found that CRF could enhance the interferon-γ-stimulated expression of ICAM-1 on human skin keratinocytes (Quevedo et al., 2001). Taken together, our results demonstrate, for the first time, that Ucn can induce COX-2 and ICAM-1 expression in RAECs via the CRF2 receptor.
Our previous study showed that the application of Ucn1 induced COX-2 and ICAM-1 expression via the CRF1 receptor in the rat TAO model. This discrepancy with the present results might be attributed to the different distribution of CRF receptor types. In RAECs, the dominant receptor type is the CRF2 receptor (Fekete and Zorrilla, 2007). Therefore, the effects of Ucn1 on RAECs are mainly implemented via the CRF2 receptor. Kageyama et al. (2006) showed that Ucn1 and-2 could induce the expression of COX-2 via the CRF2 receptor in rat aortic smooth muscle cells. However, in the rat TAO model, the level of CRF1 receptors was elevated, while the expression of CRF2 receptors was not significantly changed. This might be attributed to both the infiltration of immune cells bearing CRF1 receptors and an effect of inflammatory mediators on CRF1 receptor expression. On the one hand, the pro-inflammatory action of CRF peptides is partially the result of their effects on immune cells (such as mast cells, macrophages and lymphocytes) (Bamberger et al., 1998; Theoharides et al., 1998; Agelaki et al., 2002), which can synthesize CRF peptides and express their receptors (Theoharides et al., 1998; Baigent, 2001). On the other hand, inflammatory mediators could induce CRF1 receptor expression (Inada et al., 2009).
The MAPKs pathway is known to play a critical role in cytokine production (Bhat et al., 1998; Means et al., 2000), and Akt is involved in LPS–TLR4-mediated cytokine expression in macrophages and microglia (Jones et al., 2001; Ojaniemi et al., 2003; Kim et al. 2004). Recent studies have shown MAPKs, Akt and NF-κB pathways are key downstream signalling intermediates of CRF2 receptor activation (Dermitzaki et al., 2002; Sananbenesi et al., 2003; Karteris et al., 2004; Moss et al., 2007; Markovic et al., 2008). In order to further clarify the mechanisms of the effect of Ucn on the expression of COX-2 and ICAM-1 in LPS-induced RAECs activation, the intracellular cell signalling pathways involved were investigated. Because the modulating effect was via the CRF2 receptor, Ucn2, which binds with high affinity to the CRF2 receptor (Fekete and Zorrilla, 2007), was used. We found that Ucn2 activated the p38MAPK pathway, and SB203580, the highly specific inhibitor of p38MAPK, abolished this effect. In contrast to previous studies showing that the ERK1/2 and Akt pathways positively regulate CRF peptide-induced expression of pro-inflammatory genes (Kim et al., 2004; Moss et al., 2007), our present study did not provide significant evidence that ERK1/2, JNK and Akt pathways are involved in the effects of Ucn2 on LPS-induced activation of RAECs. These differences may be attributed to the use of different cell types. In addition to the activation of the p38MAPK pathway, Ucn2 induced nuclear translocation and phosphorylation of NF-κB, which is consistent with previous findings (Moss et al., 2007). Taken together, by showing the positive effect of Ucn2 on LPS-induced p38MAPK and NF-κB activation in RAECs, the present study revealed part of the mechanism of the CRF2 receptor in inflammation.
Interestingly, in the present study, we found that the change in the levels of COX-2 and ICAM-1 showed a similar trend. To explore their possible relationship, NS-398, the selective COX-2 inhibitor, was used. It was found that NS-398 reduced the expression of sICAM-1 (Figure 4IA,B). This indicates that ICAM-1 expression is partially mediated by COX-2. Previous studies have shown that COX-2 and ICAM-1 are expressed in a wide variety of cell types, such as HUVECs and mouse brain ECs (Seok et al., 2006; Zhao et al., 2008). Aspirin, the non-selective COX-2 inhibitor, significantly suppressed COX-2 and ICAM-1 expression induced by ox-LDL (Zhao et al., 2008). This is consistent with our present findings. Furthermore, COX-2 and ICAM-1 are now thought to be regulated by NF-κB, via the expression of inflammatory genes induced by the activation of NF-κB (Chen, 2006). In human alveolar epithelial cells, TNF-α-induced ICAM-1 expression was mediated through activation of IKKβ, phosphorylation of IκBα at serine and the subsequent activation of NF-κB (Rahman et al., 1999; Huang et al., 2003a), and COX-2 expression is elevated after activation of IKKα/β and NF-κB (Chen et al., 2000; Huang et al., 2003b). Degradation of IκB or blockade of IκB phosphorylation has been shown to suppress TNF-α-induced expression of COX-2 and ICAM-1 (Chen et al., 1995; 2001a;). These findings combined with the present results suggest that an intrinsic relationship exists between the two molecules in that ICAM-1 expression is partially mediated by COX-2, and NF-κB is involved in this process.
In conclusion, this is the first time that Ucn has been shown to increase LPS-induced COX-2 and ICAM-1 expression, in RAECs, in a time- and concentration-dependent manner. Furthermore, this effect is mediated via the CRF2 receptor via a mechanism involving both the p38MAPK and NF-κB pathways.
Acknowledgments
This work was supported by a key project from the Natural Scientific Foundation of Jiangsu Province (no. BK2006727), a project from New Century Excellent Talents (no. NCET-06-0507) and a project funded by the Summit of the ‘Six Great Talents’ of Jiangsu Province (06-C-017).
Glossary
Abbreviations:
- CRF
corticotrophin-releasing factor
- CRF1 receptor
corticotrophin-releasing factor type 1 receptor
- CRF2 receptor
corticotrophin-releasing factor type 2 receptor
- ECs
endothelial cells
- RAECs
rat aortic endothelial cells
- TAO
thromboangiitis obliterans
- Ucn
urocortin
Conflicts of interest
None.
References
- Agelaki S, Tsatsanis C, Gravanis A, Margioris AN. Corticotropin-releasing hormone augments proinflammatory cytokine production from macrophages in vitro and in lipopolysaccharide-induced endotoxin shock in mice. Infect Immun. 2002;70:6068–6074. doi: 10.1128/IAI.70.11.6068-6074.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SP, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC), 3rd edition. Br J Pharmacol. 2008;153(Suppl. 2):S1–S209. doi: 10.1038/sj.bjp.0707746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baigent SM. Peripheral corticotropin-releasing hormone and urocortin in the control of the immune response. Peptides. 2001;22:809–820. doi: 10.1016/s0196-9781(01)00395-3. [DOI] [PubMed] [Google Scholar]
- Baigent SM, Lowry PJ. mRNA expression profiles for corticotrophin-releasing factor (CRF), urocortin, CRF receptors and CRF-binding protein in peripheral rat tissues. J Mol Endocrinol. 2000;25:43–52. doi: 10.1677/jme.0.0250043. [DOI] [PubMed] [Google Scholar]
- Bale TL, Giordano FJ, Hickey RP, Huang Y, Nath AK, Peterson KL, et al. Corticotropin-releasing factor receptor 2 is a tonic suppressor of vascularization. Proc Natl Acad Sci USA. 2002;99:7734–7739. doi: 10.1073/pnas.102187099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bamberger CM, Wald M, Bamberger AM, Ergün S, Beil FU, Schulte HM. Human lymphocytes produce urocortin, but not corticotropin-releasing hormone. J Clin Endocrinol Metab. 1998;83:708–711. doi: 10.1210/jcem.83.2.4693. [DOI] [PubMed] [Google Scholar]
- Bannerman DD, Goldblum SE. Endotoxin induces endothelial barrier dysfunction through protein tyrosine phosphorylation. Am J Physiol. 1997;273:L217–L226. doi: 10.1152/ajplung.1997.273.1.L217. [DOI] [PubMed] [Google Scholar]
- Berman RS, Frew JD, Martin W. Endotoxin-induced arterial endothelial barrier dysfunction assessed by an in vitro model. Br J Pharmacol. 1993;110:1282–1284. doi: 10.1111/j.1476-5381.1993.tb13956.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhat NR, Zhang P, Lee JC, Hogan EL. Extracellular signal-regulated kinase and p38 subgroups of mitogen-activated protein kinases regulate inducible nitric oxide synthase and tumor necrosis factor-alpha gene expression in endotoxin-stimulated primary glial cultures. J Neurosci. 1998;18:1633–1641. doi: 10.1523/JNEUROSCI.18-05-01633.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bierhaus A, Chen J, Liliensiek B, Nawroth PP. LPS and cytokine-activated endothelium. Semin Thromb Hemost. 2000;26:571–587. doi: 10.1055/s-2000-13214. [DOI] [PubMed] [Google Scholar]
- Chen CC. Signal transduction pathways of inflammatory gene expressions and therapeutic implications. Curr Pharm Des. 2006;12:3497–3508. doi: 10.2174/138161206778343028. [DOI] [PubMed] [Google Scholar]
- Chen CC, Rosenbloom CL, Anderson DC, Manning AM. Selective inhibition of E-selectin, vascular cell adhesion molecule-1, and intercellular adhesion molecule-1 expression by inhibitors of I kappa B-alpha phosphorylation. J Immunol. 1995;155:3538–3545. [PubMed] [Google Scholar]
- Chen CC, Sun YT, Chen JJ, Chiu KT. TNF-alpha-induced cyclooxygenase-2 expression in human lung epithelial cells: involvement of the phospholipase C-gamma 2, protein kinase Calpha, tyrosine kinase, NF-kappa B-inducing kinase, and I-kappa B kinase 1/2 pathway. J Immunol. 2000;165:2719–2728. doi: 10.4049/jimmunol.165.5.2719. [DOI] [PubMed] [Google Scholar]
- Chen CC, Chiu KT, Chan ST, Chern JW. Conjugated polyhydroxybenzene derivatives block tumor necrosis factor-alpha-mediated nuclear factor-kappaB activation and cyclooxygenase-2 gene transcription by targeting IkappaB kinase activity. Mol Pharmacol. 2001a;60:1439–1448. doi: 10.1124/mol.60.6.1439. [DOI] [PubMed] [Google Scholar]
- Chen JX, Berry LC, Jr, Christman BW, Tanner M, Myers PR, Meyrick BO. NO regulates LPS-stimulated cyclooxygenase gene expression and activity in pulmonary artery endothelium. Am J Physiol Lung Cell Mol Physiol. 2001b;280:L450–L457. doi: 10.1152/ajplung.2001.280.3.L450. [DOI] [PubMed] [Google Scholar]
- Cipollone F, Fazia ML. COX-2 and atherosclerosis. J Cardiovasc Pharmacol. 2006;47:S26–S36. doi: 10.1097/00005344-200605001-00006. [DOI] [PubMed] [Google Scholar]
- Dermitzaki E, Tsatsanis C, Gravanis A, Margioris AN. Corticotropin-releasing hormone (CRH) induces Fas ligand production and apoptosis in PC12 cells via activation of p38 MAPK. J Biol Chem. 2002;277:12280–12287. doi: 10.1074/jbc.M111236200. [DOI] [PubMed] [Google Scholar]
- Dubois RN, Abramson SB, Crofford L, Gupta RA, Simon LS, Van De Putte LB, et al. Cyclooxygenase in biology and disease. FASEB J. 1998;12:1063–1073. [PubMed] [Google Scholar]
- Elenkov IJ, Chrousos GP. Stress hormones, Th1/Th2 patterns, pro/anti-inflammatory cytokines and susceptibility to disease. Trends Endocrinol Metab. 1999;10:359–368. doi: 10.1016/s1043-2760(99)00188-5. [DOI] [PubMed] [Google Scholar]
- Elenkov IJ, Webster EL, Torpy DJ, Chrousos GP. Stress, corticotropin-releasing hormone, glucocorticoids, and the immune/inflammatory response: acute and chronic effects. Ann N Y Acad Sci. 1999;876:1–11. doi: 10.1111/j.1749-6632.1999.tb07618.x. [DOI] [PubMed] [Google Scholar]
- Fekete EM, Zorrilla EP. Physiology, pharmacology, and therapeutic relevance of urocortins in mammals: ancient CRF paralogs. Front Neuroendocrinol. 2007;28:1–27. doi: 10.1016/j.yfrne.2006.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Gaetano G, Donati MB, Cerletti C. Prevention of thrombosis and vascular inflammation: benefits and limitations of selective or combined COX-1, COX-2 and 5-LOX inhibitors. Trends Pharmacol Sci. 2003;24:245–252. doi: 10.1016/S0165-6147(03)00077-4. [DOI] [PubMed] [Google Scholar]
- Gao L, Lu C, Xu C, Tao Y, Cong B, Ni X. Differential regulation of prostaglandin production mediated by CRH receptor type 1 and type 2 in cultured human placental trophoblasts. Endocrinology. 2008;149:2866–2876. doi: 10.1210/en.2007-1377. [DOI] [PubMed] [Google Scholar]
- Ghosh S, Hayden MS. New regulators of NF-kappaB in inflammation. Nat Rev Immunol. 2008;8:837–848. doi: 10.1038/nri2423. [DOI] [PubMed] [Google Scholar]
- Grignani G, Maiolo A. Cytokines and hemostasis. Haematologica. 2000;85:967–972. [PubMed] [Google Scholar]
- Halacheva K, Gulubova MV, Manolova I, Petkov D. Expression of ICAM-1, VCAM-1, E-selectin and TNF-alpha on the endothelium of femoral and iliac arteries in thromboangiitis obliterans. Acta Histochem. 2002;104:177–184. doi: 10.1078/0065-1281-00621. [DOI] [PubMed] [Google Scholar]
- Hao Z, Huang Y, Cleman J, Jovin IS, Vale WW, Bale TL, et al. Urocortin2 inhibits tumor growth via effects on vascularization and cell proliferation. Proc Natl Acad Sci USA. 2008;105:3939–3944. doi: 10.1073/pnas.0712366105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heo SK, Yun HJ, Noh EK, Park WH, Park SD. LPS induces inflammatory responses in human aortic vascular smooth muscle cells via Toll-like receptor 4 expression and nitric oxide production. Immunol Lett. 2008;120:57–64. doi: 10.1016/j.imlet.2008.07.002. [DOI] [PubMed] [Google Scholar]
- Huang WC, Chen JJ, Chen CC. c-Src-dependent tyrosine phosphorylation of IKKbeta is involved in tumor necrosis factor-alpha induced intercellular adhesion molecule-1 expression. J Biol Chem. 2003a;278:9944–9952. doi: 10.1074/jbc.m208521200. [DOI] [PubMed] [Google Scholar]
- Huang WC, Chen JJ, Inoue H, Chen CC. Tyrosine phosphorylation of I-kappa B kinase alpha/beta by protein kinase C-dependent c-Src activation is involved in TNF-alpha-induced cyclooxygenase-2 expression. J Immunol. 2003b;170:4767–4775. doi: 10.4049/jimmunol.170.9.4767. [DOI] [PubMed] [Google Scholar]
- Inada Y, Ikeda K, Tojo K, Sakamoto M, Takada Y, Tajima N. Possible involvement of corticotropin-releasing factor receptor signaling on vascular inflammation. Peptides. 2009;30:365–372. doi: 10.1016/j.peptides.2008.10.015. [DOI] [PubMed] [Google Scholar]
- Jones BW, Heldwein KA, Means TK, Saukkonen JJ, Fenton MJ. Differential roles of Toll-like receptors in the elicitation of proinflammatory responses by macrophages. Ann Rheum Dis. 2001;60:iii6–iii12. doi: 10.1136/ard.60.90003.iii6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kageyama K, Hanada K, Nigawara T, Moriyama T, Terui K, Sakihara S, et al. Urocortin induces interleukin-6 gene expression via cyclooxygenase-2 activity in aortic smooth muscle cells. Endocrinology. 2006;147:4454–4462. doi: 10.1210/en.2006-0008. [DOI] [PubMed] [Google Scholar]
- Karasek MA. Microvascular endothelial cell culture. J Invest Dermatol. 1989;93:S33–S38. doi: 10.1111/1523-1747.ep12580906. [DOI] [PubMed] [Google Scholar]
- Karteris E, Hillhouse EW, Grammatopoulos D. Urocortin II is expressed in human pregnant myometrial cells and regulates myosin light chain phosphorylation: pote potential role of the type-2 corticotropin-releasing hormone receptor in the control of myometrial contractility. Endocrinology. 2004;145:890–900. doi: 10.1210/en.2003-1210. [DOI] [PubMed] [Google Scholar]
- Kim WK, Hwang SY, Oh ES, Piao HZ, Kim KW, Han IO. TGF-beta1 represses activation and resultant death of microglia via inhibition of phosphatidylinositol 3-kinase activity. J Immunol. 2004;172:7015–7023. doi: 10.4049/jimmunol.172.11.7015. [DOI] [PubMed] [Google Scholar]
- Kohno M, Kawahito Y, Tsubouchi Y, Hashiramoto A, Yamada R, Inoue KI, et al. Urocortin expression in synovium of patients with rheumatoid arthritis and osteoarthritis: relation to inflammatory activity. J Clin Endocrinol Metab. 2001;86:4344–4352. doi: 10.1210/jcem.86.9.7827. [DOI] [PubMed] [Google Scholar]
- McGuire PG, Orkin RW. Isolation of rat aortic endothelial cells by primary explant techniques and their phenotypic modulation by defined substrata. Lab Invest. 1987;57:94–105. [PubMed] [Google Scholar]
- Markovic D, Punn A, Lehnert H, Grammatopoulos DK. Intracellular mechanisms regulating corticotropin-releasing hormone receptor-2beta endocytosis and interaction with extracellularly regulated kinase 1/2 and p38 mitogen-activated protein kinase signaling cascades. Mol Endocrinol. 2008;22:689–706. doi: 10.1210/me.2007-0136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Means TK, Pavlovich RP, Roca D, Vermeulen MW, Fenton MJ. Activation of TNF-alpha transcription utilizes distinct MAP kinase pathways in different macrophage populations. J Leukoc Biol. 2000;67:885–893. doi: 10.1002/jlb.67.6.885. [DOI] [PubMed] [Google Scholar]
- Moss AC, Anton P, Savidge T, Newman P, Cheifetz AS, Gay J, et al. Urocortin II mediates pro-inflammatory effects in human colonocytes via corticotropin-releasing hormone receptor 2alpha. Gut. 2007;56:1210–1217. doi: 10.1136/gut.2006.110668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munshi N, Fernandis AZ, Cherla RP, Park IW, Ganju RK. Lipopolysaccharide-induced apoptosis of endothelial cells and its inhibition by vascular endothelial growth factor. J Immunol. 2002;168:5860–5866. doi: 10.4049/jimmunol.168.11.5860. [DOI] [PubMed] [Google Scholar]
- Ohnaka K, Numaguchi K, Yamakawa T, Inagami T. Induction of cyclooxygenase-2 by angiotensin II in cultured rat vascular smooth muscle cells. Hypertension. 2000;35:68–75. doi: 10.1161/01.hyp.35.1.68. [DOI] [PubMed] [Google Scholar]
- Ojaniemi M, Glumoff V, Harju K, Liljeroos M, Vuori K, Hallman M. Phosphatidylinositol 3-kinase is involved in Toll-like receptor 4-mediated cytokine expression in mouse macrophages. Eur J Immunol. 2003;33:597–605. doi: 10.1002/eji.200323376. [DOI] [PubMed] [Google Scholar]
- Okosi A, Brar BK, Chan M, D'Souza L, Smith E, Stephanou A, et al. Expression and protective effects of urocortin in cardiac myocytes. Neuropeptides. 1998;32:167–171. doi: 10.1016/s0143-4179(98)90033-6. [DOI] [PubMed] [Google Scholar]
- Oki Y, Sasano H. Localization and physiological roles of urocortin. Peptides. 2004;25:1745–1749. doi: 10.1016/j.peptides.2004.06.023. [DOI] [PubMed] [Google Scholar]
- Quevedo ME, Slominski A, Pinto W, Wei E, Wortsman J. Pleiotropic effects of corticotropin releasing hormone on normal human skin keratinocytes. In Vitro Cell Dev Biol Anim. 2001;37:50–54. doi: 10.1290/1071-2690(2001)037<0050:peocrh>2.0.co;2. [DOI] [PubMed] [Google Scholar]
- Raetz CR. Biochemistry of endotoxins. Annu Rev Biochem. 1990;59:129–170. doi: 10.1146/annurev.bi.59.070190.001021. [DOI] [PubMed] [Google Scholar]
- Rahman A, Bando M, Kefer J, Anwar KN, Malik AB. Protein kinase C-activated oxidant generation in endothelial cells signals intercellular adhesion molecule-1 gene transcription. Mol Pharmacol. 1999;55:575–583. [PubMed] [Google Scholar]
- Sananbenesi F, Fischer A, Schrick C, Spiess J, Radulovic J. Mitogen-activated protein kinase signaling in the hippocampus and its modulation by corticotropin-releasing factor receptor 2: a possible link between stress and fear memory. J Neurosci. 2003;23:11436–11443. doi: 10.1523/JNEUROSCI.23-36-11436.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saruta M, Takahashi K, Suzuki T, Torii A, Kawakami M, Sasano H. Urocortin 1 in colonic mucosa in patients with ulcerative colitis. J Clin Endocrinol Metab. 2004;89:5352–5361. doi: 10.1210/jc.2004-0195. [DOI] [PubMed] [Google Scholar]
- Seok SM, Park DH, Kim YC, Moon CH, Jung YS, Baik EJ, et al. COX-2 is associated with cadmium-induced ICAM-1 expression in cerebrovascular endothelial cells. Toxicol Lett. 2006;165:212–220. doi: 10.1016/j.toxlet.2006.04.007. [DOI] [PubMed] [Google Scholar]
- Singh LK, Boucher W, Pang X, Letourneau R, Seretakis D, Green M, et al. Potent mast cell degranulation and vascular permeability triggered by urocortin through activation of corticotropin-releasing hormone receptors. J Pharmacol Exp Ther. 1999;288:1349–1356. [PubMed] [Google Scholar]
- Taal MW, Zandi-Nejad K, Weening B, Shahsafaei A, Kato S, Lee KW, et al. Proinflammatory gene expression and macrophage recruitment in the rat remnant kidney. Kidney Int. 2000;58:1664–1676. doi: 10.1111/j.1523-1755.2000.00327.x. [DOI] [PubMed] [Google Scholar]
- Theoharides TC, Spanos C, Pang X, Alferes L, Ligris K, Letourneau R, et al. Stress-induced intracranial mast cell degranulation: a corticotropin-releasing hormone-mediated effect. Endocrinology. 1995;136:5745–5750. doi: 10.1210/endo.136.12.7588332. [DOI] [PubMed] [Google Scholar]
- Theoharides TC, Singh LK, Boucher W, Pang X, Letourneau R, Webster E, et al. Corticotropin-releasing hormone induces skin mast cell degranulation and increased vascular permeability, a possible explanation for its proinflammatory effects. Endocrinology. 1998;139:403–413. doi: 10.1210/endo.139.1.5660. [DOI] [PubMed] [Google Scholar]
- Tsatsanis C, Androulidaki A, Dermitzaki E, Gravanis A, Margioris AN. Corticotropin releasing factor receptor 1 (CRF1) and CRF2 agonists exert an anti-inflammatory effect during the early phase of inflammation suppressing LPS-induced TNF-alpha release from macrophages via induction of COX-2 and PGE2. J Cell Physiol. 2007;210:774–783. doi: 10.1002/jcp.20900. [DOI] [PubMed] [Google Scholar]
- Vaughan J, Donaldson C, Bittencourt J, Perrin MH, Lewis K, Sutton S, et al. Urocortin, a mammalian neuropeptide related to fish urotensin I and to corticotropin-releasing factor. Nature. 1995;378:287–292. doi: 10.1038/378287a0. [DOI] [PubMed] [Google Scholar]
- Witkowska AM. Soluble ICAM-1: a marker of vascular inflammation and lifestyle. Cytokine. 2005;31:127–134. doi: 10.1016/j.cyto.2005.04.007. [DOI] [PubMed] [Google Scholar]
- Wu Y, Xu Y, Zhou H, Tao J, Li S. Expression of urocortin in rat lung and its effect on pulmonary vascular permeability. J Endocrinol. 2006;189:167–178. doi: 10.1677/joe.1.06607. [DOI] [PubMed] [Google Scholar]
- Wu Y, Hu J, Zhang R, Zhou C, Xu Y, Guan X, et al. Enhanced intracellular calcium induced by urocortin is involved in degranulation of rat lung mast cells. Cell Physiol Biochem. 2008;21:173–182. doi: 10.1159/000113759. [DOI] [PubMed] [Google Scholar]
- Wu Y, Zhang R, Zhou C, Xu Y, Guan X, Hu J, et al. Enhanced expression of vascular cell adhesion molecule-1 by corticotrophin-releasing hormone contributes to progression of atherosclerosis in LDL receptor-deficient mice. Atherosclerosis. 2009;203:360–370. doi: 10.1016/j.atherosclerosis.2008.05.059. [DOI] [PubMed] [Google Scholar]
- Yang C, Xu Y, Mendez T, Wang F, Yang Q, Li S. Effects of intravenous urocortin on angiotensin-converting enzyme in rats. Vascul Pharmacol. 2006;44:238–246. doi: 10.1016/j.vph.2006.01.001. [DOI] [PubMed] [Google Scholar]
- Yang C, Xu Y, Li S. Urocortin: a beneficial or detrimental agent to endothelium? Biochem Biophys Res Commun. 2008;371:345–349. doi: 10.1016/j.bbrc.2008.01.157. [DOI] [PubMed] [Google Scholar]
- Zhao J, Qi R, Li R, Wu W, Gao X, Bao L, et al. Protective effects of aspirin against oxidized LDL-induced inflammatory protein expression in human endothelial cells. J Cardiovasc Pharmacol. 2008;51:32–37. doi: 10.1097/FJC.0b013e318159ebaf. [DOI] [PubMed] [Google Scholar]