

Abstract
Background and purpose:
The lipid phosphatase known as SH2 domain-containing inositol 5′-phosphatase 2 (SHIP2) plays an important role in the regulation of the intracellular insulin signalling pathway. Recent studies have suggested that inhibition of SHIP2 could produce significant benefits in treatment of type 2 diabetes. However, there were no small molecule SHIP2 inhibitors and we, therefore, aimed to identify this type of compound.
Experimental approach:
The phosphatase assay with malachite green was used for high-throughput screening. The pharmacological profiles of suitable compounds were further characterized in phosphatase assays, cellular assays and oral administration in normal and diabetic (db/db) mice.
Key results:
During high-throughput screening, AS1949490 was identified as a potent SHIP2 inhibitor (IC50= 0.62 µM for SHIP2). This compound was also selective for SHIP2 relative to other intracellular phosphatases. In L6 myotubes, AS1949490 increased the phosphorylation of Akt, glucose consumption and glucose uptake. In FAO hepatocytes, AS1949490 suppressed gluconeogenesis. Acute administration of AS1949490 inhibited the expression of gluconeogenic genes in the livers of normal mice. Chronic treatment of diabetic db/db mice with AS1949490 significantly lowered the plasma glucose level and improved glucose intolerance. These in vivo effects were based in part on the activation of intracellular insulin signalling pathways in the liver.
Conclusions and implications:
This is the first report of a small molecule inhibitor of SHIP2. This compound will help to elucidate the physiological functions of SHIP2 and its involvement in various diseases, such as type 2 diabetes.
Keywords: SHIP2, AS1949490, small-molecule inhibitor, type 2 diabetes, insulin signalling
Introduction
Type 2 diabetes mellitus is becoming an increasingly widespread global health issue (Amos et al., 1997). Although the prevalence of this disease has grown drastically, the increase in treatment options has lagged behind. In addition, the therapeutic approaches currently available are often limited by undesirable side effects associated with long-term treatment (Wagman and Nuss, 2001; Martens et al., 2002). Therefore, the discovery of safer and more efficient novel treatments for type 2 diabetes is both important and necessary.
It is well known that disordered insulin signalling leads to type 2 diabetes (Leng et al., 2004). Thus, one of the most interesting research areas for the discovery of new drug targets in type 2 diabetes is the insulin signalling pathway. The intracellular protein, SH2 domain-containing inositol 5′-phosphatase 2 (SHIP2), which negatively regulates insulin signalling by hydrolyzing phosphatidylinositol-3,4,5-trisphosphate, is one such new drug target (Baumgartener, 2003; Lazar and Saltiel, 2006; Sasaoka et al., 2006). In vitro, the overexpression of the wild or dominant negative type of SHIP2 resulted in the reduction or enhancement of insulin signalling respectively (Sasaoka et al., 2001; Wada et al., 2001). The transgenic overexpression of SHIP2 in a model of diabetes in rodents was shown to impair glucose tolerance and insulin sensitivity (Kagawa et al., 2008). The liver-specific expression of dominant negative SHIP2 by adenovirus-mediated gene transfer improved the symptoms of type 2 diabetes in a diabetic model (Fukui et al., 2005; Grempler et al., 2007). A deficiency in the SHIP2 gene resulted in resistance to diet-induced obesity and insulin resistance (Sleeman et al., 2005).
Considering all these results, alteration of SHIP2 activity might be a new valuable therapeutic approach for type 2 diabetes with disordered insulin signalling (Lazar and Saltiel, 2006; Sasaoka et al., 2006). However, little progress has been made in identifying small molecule inhibitors of SHIP2. In the present study, we discovered for the first time, a potent, selective, small molecule SHIP2 inhibitor, AS1949490 (Figure 1A). This compound activated intracellular insulin signalling and decreased both fasting and post-prandial blood glucose in db/db diabetic mice.
Figure 1.

(A) Chemical structure of AS1949490, (B) Representative dose-dependent inhibition of recombinant human SH2 domain-containing inositol 5′-phosphatase 2 activity with AS1949490. Signal output was converted to percent activation. The graph shown is representative of three experiments.
Methods
Expression and purification of recombinant phosphatases
Human SHIP2 (residues 419–732; NP_001558), human SHIP1 (residues 399–714; NP_005532), mouse SHIP2 (residues 421–733; AAF28187), human synaptojanin (residues 492–856, XM_009729) catalytic domains and full-length human myotubularin (U46024) were cloned from human or mouse cDNA using polymerase chain reaction (PCR). These phosphatases were expressed in Escherichia coli with an 6His tag and purified using immobilized metal affinity chromatograpy, as previously described (Pesesse et al., 1998; Chi et al., 2004). For the purification of human phosphatase and tensin homolog (PTEN), truncations and deletions were introduced (deleted residues: 2–6, 286–309 and 354–403; NP_000305) to remove unstructured regions, as previously reported (Lee et al., 1999). This deleted PTEN was expressed with a GST-tag at the N-terminal in Sf-9 cells, and then purified with glutathione sepharose. Purified enzymes were suspended in lysis buffer (25 mM Tris-HCl pH 7.4, 300 mM NaCl) containing 50% glycerol and complete protease inhibitor cocktail (Roche, Mannheim, Germany).
Malachite green phosphate assay
The catalytic activity of purified phosphatase was determined using D-myo-inositol-1,3,4,5-tetrakisphosphate [Ins(1,3,4,5)P4] (Cayman, MI, USA) previously reported as a substrate for SHIP2 (Chi et al., 2004) or D-myo-phosphatidylinositol-3,4,5-trisphosphate (PtdIns(3,4,5)P3) (Echelon, UT, USA). Enzyme protein (100 ng) in reaction buffer (10 mM HEPES pH 7.25, 6 mM MgCl2, 0.1% CHAPS, 250 mM sucrose and 0.25 mM EDTA) was mixed with either an aliquot of Ins(1,3,4,5)P4 for SHIP2 and SHIP1 or an aliquot of PtdIns(3,4,5)P3 for PTEN, synaptojanin, and myotubularin. The final concentration of the substrates was 100 µM for Ins(1,3,4,5)P4 or 250 µM for PtdIns(3,4,5)P3 in a final volume of 20 µL reaction mixture.
A sample (20 µL) of this solution was placed in each well of 384 well plates, after which the plates were incubated at room temperature for 20 min. Malachite green reagent (Biomol, PA, USA) was added to each well, and then the plates were incubated for another 25 min at room temperature. Absorbance at 630 nm was measured using a Safire microplate reader (TECAN, Mannedorf, Switzerland).
Cell-based enzyme-linked immunosorbent assay (ELISA)
L6 myoblasts were seeded on 96 well plates. The confluent cells were cultured for 5 or 6 days in Dulbecco's Modified Eagle's Medium (DMEM) containing 2% horse serum to promote the differentiation to myotubes. The L6 myotubes were serum-starved for 16 h in DMEM, and then treated with AS1949490 for 15 min before stimulation with insulin or fetal bovine serum (FBS) for 10 min. The cells were immediately fixed with 4% paraformaldehyde for 15 min. After being washed with Tris-buffered saline containing 0.1% Tween-20 (TBS-T), the cells were permeabilized with 0.1% Triton X-100 for 10 min and blocked for 1 h in Blocking One solution (Nacalai Tesque, Kyoto, Japan). The cells were incubated with Ser473 phosphorylated specific Akt antibody (BIOMOL, PA, USA) for 2 h at room temperature. The primary antibody was labelled with horseradish peroxidase-conjugated second antibody (Cell Signaling Technology, MA, USA) and incubated with tetramethylbenzidine substrate. The absorbance was measured at 450 nm.
Glucose uptake and glucose consumption assays
The L6 myotubes were cultured for 48 h under differentiating conditions (2% horse serum in DMEM without phenol red) with AS1949490 in the presence (for glucose consumption assay) or absence (for glucose uptake assay) of insulin. Glucose uptake and glucose consumption were then measured as previously described (Somwar et al., 1998; Shimokawa et al., 2000).
Gluconeogenesis assay
Rat FAO hepatoma-derived cells were grown in DMEM containing 10% FBS. The FAO cells were seeded on 96 well plates and treated with AS1949490 and insulin for 24 h. After being washed with phosphate-buffered saline (PBS), glucose production buffer (glucose-free DMEM pH 7.4 containing 20 mM sodium pyruvate without phenol red) was added to each well. After 6 h, the glucose concentration in the medium was measured using Glucose CII-Test reagent (Wako, Osaka, Japan).
Expression analysis of mRNA
Samples of liver tissue were pulverized in liquid nitrogen and the total RNA was extracted using an Isogen kit (Nippon Gene, Tokyo, Japan) according to the manufacturer's instructions. cDNAs were synthesized as described previously (Matsumoto et al., 2000). mRNA for glucose-6 phosphatase (G6Pase) and phosphoenolpyruvate carboxykinase (PEPCK) were quantified using reverse transcription-polymerase chain reaction (RT-PCR) with the SYBR green method and a PRISM 7900 Sequence Detector according to the manufacturer's instructions (Perkin-Elmer Applied Biosystems, CA, USA). Mouse 18S ribosomal RNA (18S rRNA) was measured as an internal control. The primers for G6Pase were as follows: forward (TTAAAGAGACTGTGGGCATCAAT), reverse (ATCCACTTGAAGACGAGGTTG); and for PEPCK: forward (GGTATTGAACTGACAGACTCGC), reverse (CACAGATATGCCCATCCGA). Primers for 18S rRNA were purchased from Perkin-Elmer Applied Biosystems (4319413E).
Expression analysis of protein
The cell lysates or liver tissue samples were homogenized in lysis buffer (50 mM Tris-HCl pH 7.5, 150 mM NaCl, 1% NP40, 0.1% SDS, Complete Protease Inhibitor Cocktail). Equal amounts of protein lysates were separated on SDS-polyacrylamide gel, transferred to a PVDF membrane, and then incubated with the primary antibody [Ser473 phosphorylated specific Akt antibody (BIOMOL), anti-Akt antibody, phospho-specific glycogen synthase kinase (GSK)3β antibody, or anti-GSK3β antibody (Cell Signaling Technology)]. The primary antibody was detected with a horseradish peroxidase-conjugated second antibody and enhanced using ECL Western Blotting Systems (GE Healthcare Bio-Sciences KK, Tokyo, Japan). The chemiluminescence intensity was subsequently detected and quantified using a quantitative digital imaging system (VersaDoc, BIO-RAD, CA, USA).
In vivo anti-diabetic effect of AS1949490
All animal care and experimental procedures were approved by the Animal Ethical Committee of Astellas Pharma Inc. Six week old male ICR mice (Japan SLC, Inc., Shizuoka, Japan) and 6 week old male C57BL/KsJ Jcl-dbm +db/+db mice (CLEA Japan, Inc., Tokyo, Japan) were used in the study. The animals were cared for under standard maintenance conditions [controlled temperature, humidity, and light (12-h light-dark cycle)] and allowed free access to laboratory chow and water while in their home cage. In the chronic treatment study, the mice were given 300 mg·kg−1 of AS1949490 suspended in 0.5% methylcellulose twice daily by oral gavage, after which the blood or plasma glucose level was measured. Plasma insulin levels were determined using an insulin ELISA kit (Morinaga Bioscience Laboratory, Kanagawa, Japan).
Oral glucose tolerance test (OGTT)
Blood samples were collected from db/db mice treated long term with either the vehicle or AS1949490, after which they were fasted overnight. After 30 min, the blood glucose levels were measured again, and then glucose solution (2 g·kg−1) was orally administered. At 0.5, 1, 2 and 3 h after glucose loading, blood glucose levels were then measured.
Statistical analysis
Data are shown as the means ± standard error. Statistical and data analysis were conducted using the SAS 8.2 software package (SAS Institute Japan, Ltd., Tokyo, Japan). The IC50 and Ki values were calculated using regression analysis. The statistical significance of the difference between two groups was determined using the Student's t-test. The significance of differences between multiple groups was assessed using Dunnett's multiple range test. P values less than <0.05 were considered significant.
Materials
3-[(4-chlorobenzyl)oxy]-N-[(1S)-1-phenylethyl]-2-thiophenecarboxamide (AS1949490) was synthesized by Astellas Pharma Inc. (Ibaraki, Japan) following the synthetic scheme outlined below.
 |
Methyl 3-[(4-chlorobenzyl)oxy]-2-thiophenecarboxylate: (compound 1)
To a mixture of methyl 3-hydroxy-2-thiphenecarboxylate (10 g, 63 mM) and K2CO3 (13.1 g, 94.8 mM) in 2-butanone (100 mL) was added a solution of 4-chlorobenzyl chloride (12.2 g, 75.9 mM) in 2-butanone (50 mL) and refluxed overnight. The reaction mixture was poured into a mixture of water and EtOAc and separated. The organic layer was washed with 1N-HCl and brine. The organic layer was dried over anhydrous MgSO4 and concentrated in vacuo. The residue was recrystallized from MeOH to give pale yellow crystals (15.8 g, 88%). 1H-NMR (300 MHz, CDCl3): 3.86 (3H, s), 5.21 (2H, s), 6.81 (1H, d, J = 5.7 Hz), 7.32–7.45 (5H, m).
3-[(4-Chlorobenzyl)oxy]-2-thiophenecarboxylic acid: (compound 2)
To a solution of (1) (23.5 g, 83.1 mM) in THF (150 mL) and MeOH (200 mL) was added 2N-NaOH (125 mL, 0.25 mM) and stirred for 4 h at 50°C. The reaction mixture was concentrated in vacuo. To the residue was added water (200 mL), then was added conc. HCl dropwise (22.5 mL). The precipitates were collected by suction. The precipitates were recrystallized from EtOAc-hexane to give colorless crystals (19.3 g, 86%). 1H-NMR (300 MHz, DMSO-d6): 5.26 (2H, s), 6.87 (1H, d, J = 5.5 Hz), 7.34–7.43 (4H, m), 7.52 (1H, d, J = 5.5 Hz).
3-[(4-chlorobenzyl)oxy]-N-[(1S)-1-phenylethyl]-2-thiophenecarboxamide: (AS 1949490)
A mixture of (2) (6.0 g, 22.3 mmol), HOBt (3.47 g, 25.7 mmol), WSCD·HCl (4.92 g, 25.7 mM) and (1S)-1-phenylethylamine (3.13 mL, 24.6 mM) in DMF (60 mL) was stirred overnight at room temperature. The reaction mixture was poured into mixture of EtOAc and water. The organic layer was separated and washed with sat.NH4Cl, sat.NaHCO3 and brine successively. The organic layer was dried over anhydrous Na2SO4 and concentrated in vacuo. The residue was purified by column chromatography (silica gel, EtOAc-hexane). The crude product was recrystallized from EtOH to give colourless crystals (7.33 g, 88%). 1H-NMR (300 MHz, DMSO-d6): 1.36 (3H, d, J = 6.9 Hz), 4.98–5.09 (1H, m), 5.32 (2H, s), 7.16–7.30 (6H, m), 7.44–7.53 (4H, m), 7.61 (1H, d, J = 7.9 Hz), 7.74 (1H, d, J = 6.0 Hz).
Results
Identification of a potent selective small molecule SHIP2 inhibitor
Our in-house chemical library was subjected to high-throughput screening, which resulted in the identification of AS1949490 as a SHIP2 inhibitor. AS1949490 showed potent inhibitory activity against human and mouse SHIP2 in a concentration-dependent manner, with IC50 values of 0.62 and 0.34 µM respectively (Figure 1B and Table 1). To identify the kinetic patterns of SHIP2 inhibition by AS1949490, we performed kinetic inhibition analyses. In this study, the Km for Ins(1,3,4,5)P4 as substrate for SHIP2 was 44 ± 10 mM and Lineweaver–Burke analysis (Figure S1) indicated that AS1949490 was a competitive inhibitor, with a Ki value estimated to be 0.44 µM for human SHIP2. To confirm the selectivity of AS1949490 for SHIP2, the inhibitory effect of AS1949490 was also assessed for SHIP1, which has an overall structure similar to that of SHIP2. The Km for Ins(1,3,4,5)P4 with human SHIP1 enzyme was 45 ± 16 mM. AS1949490 was not as potent as an inhibitor of human SHIP1 activity, with Ki and IC50 values (Table 1) approximately 30 times higher than we found for SHIP2. Additionally, we examined the effects of this compound on other types of phosphatases, PTEN, synaptojanin and myotubularin. AS1949490 did not inhibit PTEN, synaptojanin and myotublarin phosphatase activity, even when added at concentrations up to 50 µM (Table 1). These results demonstrate that AS1949490 is a selective SHIP2 inhibitor.
Table 1.
In vitro affinity of AS1949490 for SHIP2, SHIP1, PTEN, synaptojanin and myotubularin
| IC50 (µM) | Ki (µM) | |
|---|---|---|
| Human SHIP2 | 0.62 ± 0.02 | 0.44 ± 0.19 |
| Human SHIP1 | 13 ± 2 | 11 ± 1.4 |
| Mouse SHIP2 | 0.34 ± 0.10 | N.T. |
| Human PTEN | >50 | N.T. |
| Human synaptojanin | >50 | N.T. |
| Human myotubularin | >50 | N.T. |
The values are the means ± standard error of three independent experiments.
N.T., not tested; PTEN, phosphatase and tensin homologue; SHIP, SH2 domain-containing inositol 5′-phosphatase.
AS1949490 increases the phosphorylation of Akt
Inhibition of SHIP2 activity increases the phosphorylation of various insulin signalling-related molecules, including Akt, a key enzyme in the regulation of insulin signalling (Sasaoka et al., 2004). The development of a quantitative cell-based assay was necessary to efficiently determine the functional character of AS1949490. Therefore, we established a cell-based ELISA to detect the phosphorylation of Akt. First, we confirmed that the phospho-specific Akt antibody specifically recognized a 65 kDa band corresponding to the Akt phosphorylated on Ser473 while being treated with 1 nM insulin using the Western blot method (Figure 2A). Next, we experimented with a cell-based ELISA in a 96-well plate format using this antibody. As shown in Figure 2B,C, the quantitative phosphorylation of Akt, dependent on insulin or FBS treatment, could be detected in L6 myotubes, using this assay method. The results of this cell-based ELISA indicated that treatment of L6 myotubes with AS1949490 dose-dependently increased insulin-induced phosphorylation of Akt (Figure 2C). There were no associated changes in the overall level of Akt protein following treatment with this compound (data not shown).
Figure 2.
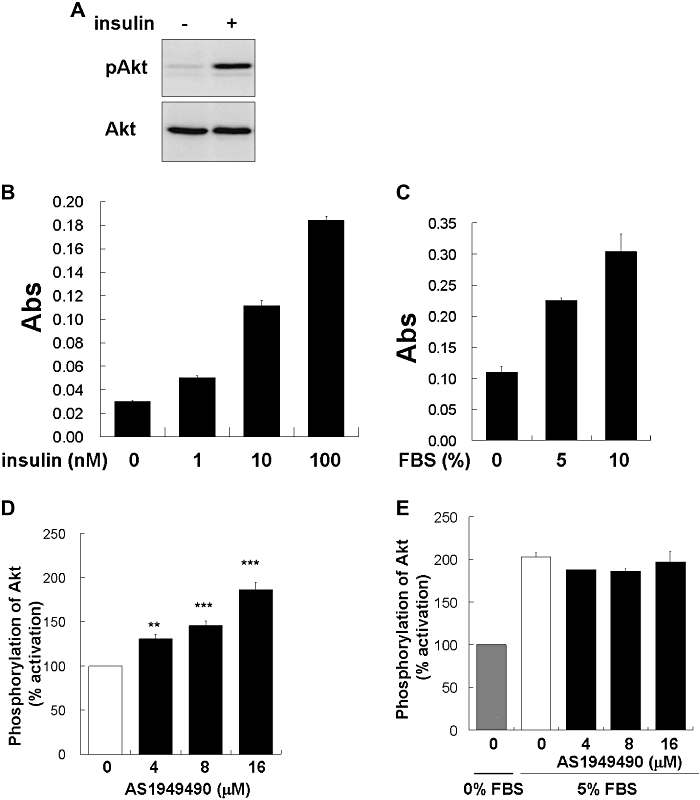
Effect of AS1949490 on insulin-induced Akt phosphorylation in L6 myotubes. (A) Protein lysates from L6 myotubes were immunoblotted with anti-phosphorylated Akt antibody or anti-Akt antibody. L6 myotubes were incubated for 15 min both with (D,E) and without (B,C) AS1949490 treatment before being stimulated with insulin at indicated concentration (B) and 1 nM (D) or fetal bovine serum (FBS) at indicated concentrations (C,E) for 10 min. Signal output was converted to percent activation (D,E). The values are the means ± standard error of three independent experiments. Asterisks indicate significant differences: **P < 0.01, ***P < 0.001 versus 0 nM AS1949490 treatment.
Previous reports have shown that SHIP2 is specifically stimulated by insulin (Sasaoka et al., 2004). Therefore, the change in the phosphorylation of Akt was examined via stimulation by FBS as a growth factor. AS1949490 caused no alteration in the FBS-induced phosphorylation of Akt in L6 myotubes (Figure 2D). These findings suggest that SHIP2 inhibition by AS1949490 can enhance the phosphorylation of Akt, related specifically to insulin signalling.
AS1949490 activates glucose metabolism
It is well known that the enhancement of insulin signalling by the activation of Akt leads to activation of glucose metabolism (Ueki et al., 1998; Hanada et al., 2004). We measured the rate of glucose consumption in L6 myotubes treated with AS1949490 and found that when L6 myotubes were treated with several concentrations of AS1949490 and 1 nM insulin for 48 h, the consumption of glucose in the medium increased concentration dependently (Figure 3A). The uptake of glucose, which is an important indicator of glucose metabolism, was then examined (Ueki et al., 1998). Treatment with AS1949490 for 48 h profoundly stimulated glucose uptake activity in L6 myotubes (Figure 3B). These results indicate that AS1949490 can stimulate the activation of glucose metabolism.
Figure 3.
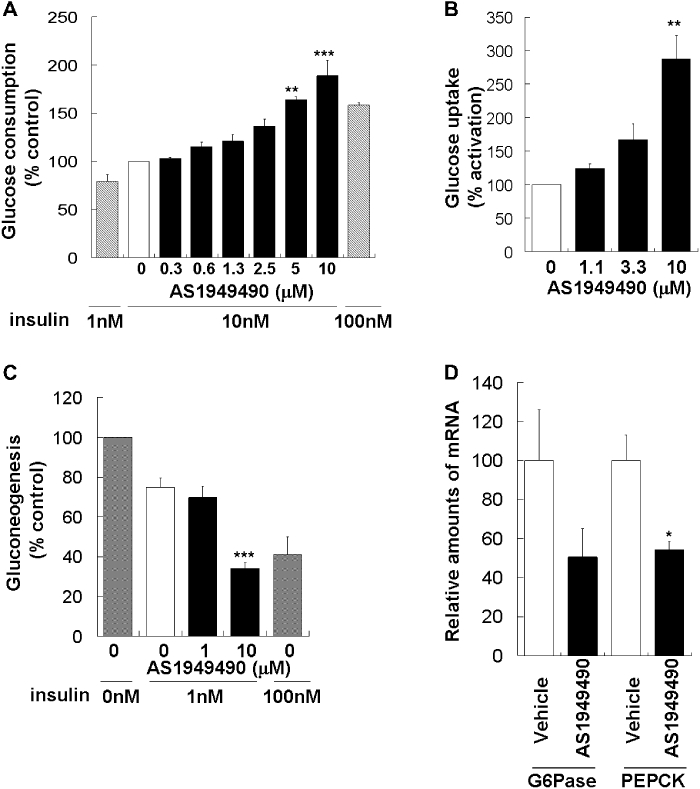
Effect of AS1949490 on (A) glucose consumption and (B) glucose uptake in L6 myotubes, (C) gluconeogenesis in FAO cells and (D) hepatic gene expression in ICR mice. L6 myotubes were incubated with AS1949490 for 48 h under conditions conducive to differentiation. (A) Glucose consumption and (B) glucose uptake were then measured. The values are the means ± standard error (SE) of three independent experiments. FAO cells were treated with AS19494940 and insulin for 24 h. (C) The glucose concentration in the medium was measured after incubation for 6 h in gluconeogenesis buffer. The values are the means ± SE of four independent experiments. AS19494940 was administered to normal ICR mice at a dose of 300 mg·kg−1, p.o. after overnight fasting (n= 4 per group). After 8 h, the total RNA was isolated from the liver. (D) G6Pase and PEPCK mRNA levels were measured using real-time quantitative polymerase chain reaction. The values are the means ± SE (n= 4). Asterisks indicate significant differences: *P < 0.05, **P < 0.01, ***P < 0.001 versus 0 nM treatment with AS1949490 or vehicle.
AS1949490 suppresses gluconeogenesis and the expression of related genes
In the liver, insulin signalling regulates gluconeogenesis negatively (L'age et al., 1968; Barthel and Schmoll, 2003). Therefore, we tested the impact of AS1949490 on gluconeogenesis. Cultured hepatocyte FAO cells were treated with AS1949490 and 1 nM insulin for 24 h. Treatment with AS1949490 significantly decreased the level of insulin-induced gluconeogenesis (Figure 3C).
The regulation of gluconeogenesis by insulin is known to be due to the suppression of the expression of related genes, such as those for PEPCK and G6Pase (Granner et al., 1983; Barthel and Schmoll, 2003). To confirm that AS1949490 also can suppress the expression levels of these genes in vivo, we gave AS1949490 acutely to normal mice. Oral administration of AS1949490 to ICR mice resulted in an approximately 50% reduction of both PEPCK and G6Pase mRNA levels in the liver (Figure 3D). These results indicate that AS1949490 can regulate gluconeogenesis both in vitro and in vivo.
Effects of AS1949490 in diabetic mice
We investigated the anti-hyperglycaemic effect of AS1949490 in db/db diabetic mice. AS1949490 was administered to diabetic db/db mice twice daily p.o. for 7 or 10 days. This treatment significantly decreased plasma glucose (23% reduction, relative to vehicle), without affecting body weight, insulin levels or food intake (Figure 4A–C, and data not shown). In the 10 day study, the effect of AS1949490 on glucose homeostasis using the OGTT in db/db mice was examined. AS1949490 treatment significantly reduced both fasting blood glucose (37% reduction, relative to vehicle; time =−30 min) and the area under the blood glucose concentration time curve (AUC) (Figure 5A,B).
Figure 4.
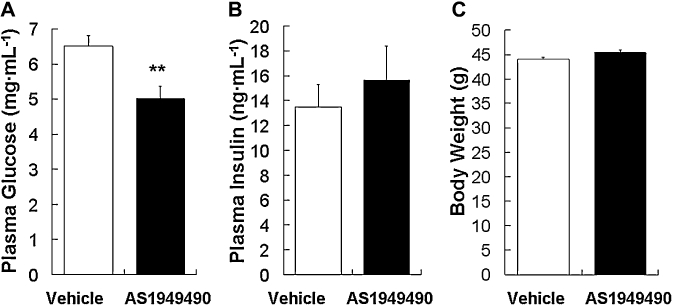
Effect of chronic treatment with AS1949490 on plasma glucose and insulin levels, and body weight in db/db mice. AS1949490 was orally administered to db/db mice twice daily for 7 days at a dose of 300 mg·kg−1. Plasma glucose levels (A), plasma insulin levels (B) and body weight (C) were measured at the start of the light cycle. The values are the means ± standard error (n= 8). Asterisks indicate significant differences: **P < 0.01 versus vehicle.
Figure 5.

Effect of chronic treatment with AS1949490 on (A,B) blood glucose during oral glucose tolerance tests (OGTT), and (C,D) phosphorylation of GSK3β in db/db mice. AS1949490 was given orally to db/db mice twice daily for 10 days at a dose of 300 mg·kg−1. Food was withheld for 16 h after the final drug dosing, and then glucose solution (2 g·kg−1) was loaded. (A) Blood glucose levels were measured at the indicated times before and after glucose loading (at time 0). (B) AUC levels during hours 0–2 of the OGTT were calculated for each group after subtracting the baseline values (time 0). The livers of the mice treated with AS1949490 were homogenized. (C) The liver protein lysates were immunoblotted with anti-phosphorylated GSK3β antibody or anti-GSK3β antibody. (D) The densitometric quantification of pGSK3β was normalized using the total GSK3β levels. The values of (A) and (B) are the means ± standard error (n= 8). Asterisks indicate significant differences: *P < 0.05, **P < 0.01 versus vehicle. The values shown in (D) are the means ± standard deviation for two animals.
AS1949490 activates intracellular insulin signalling in the liver
To confirm whether these anti-diabetic effects occurred due to enhancement of insulin signalling, we examined this signalling in the liver after chronic administration of AS1949490. The phosphorylation levels of GSK3β were measured as an indicator of insulin signalling activity (Cross et al., 1995; Summers et al., 1999). Chronic administration of AS1949490 significantly increased the phosphorylation of GSK3β in the liver without changing the overall levels of GSK3β protein (Figure 5C,D).
Discussion
The intracellular phosphatase, SHIP2, appears to be an important negative regulator of insulin signalling (Pesesse et al., 1997; Ishihara et al., 1999; Sleeman et al., 2005). Several previous studies have shown that the inhibition of SHIP2 has therapeutic potential as a treatment for type 2 diabetes (Clement et al., 2001; Fukui et al., 2005; Sleeman et al., 2005; Buettner et al., 2007; Grempler et al., 2007). However, these studies were limited by the genetics, for example, loss of function approaches such as knock out, dominant negative expression and antisense strategies. The effect of pharmacological inhibition of SHIP2 on the regulation of insulin signaling and glucose metabolism was unknown. Thus, it was necessary to find a small molecule SHIP2 inhibitor that could be used for studying the physiological and pathophysiological functions of SHIP2. In this study, AS1949490 showed potent and competitive inhibition of SHIP2 (IC50= 0.62 µM, Ki= 0.44 µM). In contrast, the inhibition of SHIP1, which is very similar to SHIP2, was significantly less effective (IC50= 12 µM, Ki= 11 µM). In addition, AS1949490 had no effect on the phosphatase activity of PTEN, synaptojanin and myotubularin, which are other types of intracellular phosphatase. These results indicate that AS1949490 is a potent, selective and competitive SHIP2 inhibitor.
Inhibition of SHIP2 leads to the activation of intracellular insulin signalling, such as the phosphorylation of Akt and the enhancement of glucose uptake (Sasaoka et al., 2001; 2004; Wada et al., 2001). Treatment with AS1949490 increased the insulin-induced phosphorylation of Akt. Peripheral tissues contain two Akt isoforms: Akt1 and Akt2 (Hanada et al., 2004), the latter being more important for insulin signalling (Cho et al., 2001; Garofalo et al., 2003). Insulin treatment mainly stimulates the phosphorylation of Akt2 (Calera et al., 1998), while growth factor stimulation activates Akt1 (Irie et al., 2005; Kim et al., 2008). Interestingly, Sasaoka et al. (2004) showed that SHIP2 is also specifically stimulated by insulin, and this is associated with the subtype-specific phosphorylation of Akt2. Indeed, AS1949490 failed to stimulate Akt phosphorylation induced by serum stimulation, which agrees with that report. Moreover, the addition of AS1949490 to cultured myotubes and hepatocytes also induced the activation of glucose metabolism and the reduction of gluconeogenesis, which is known to be downstream to the insulin signalling cascade (L'age et al., 1968; Granner et al., 1983; Czech, 1995). These in vitro studies agree well with previous reports using siRNA and the dominant negative inhibition of SHIP2 (Fukui et al., 2005; Buettner et al., 2007; Grempler et al., 2007). These results from cellular functional assays suggest that AS1949490 inhibited SHIP2 in cultured cells and activated metabolic responses to insulin. In this cellular characterization study, however, we focused on a few important key molecules and insulin signalling pathway events. Therefore, to directly and definitively demonstrate AS1949490's specificity for SHIP2, an additional study would have to explore whether AS1949490 could interfere with other molecules involved in insulin signalling, especially molecules upstream to Akt such as the insulin receptor, phosphoinositide 3-kinase, and, at the cellular level, phosphoinositide-3 phosphate.
To evaluate the in vivo effects of AS1949490 in this study, we used db/db mice, which provide a well-established model presenting key signs of diabetes: obesity, hyperglycaemia, insulin resistance and glucose intolerance (Kobayashi et al., 2000; Shao et al., 2000). In addition, increased SHIP2 protein production has been previously reported in the peripheral tissues, such as muscle and fat, of db/db mice (Hori et al., 2002). Chronic administration of AS1949490 markedly improved hyperglycaemia (non-fasting glucose: 23% reduction; fasting glucose: 37% reduction) and glucose intolerance, without affecting body weight or food intake in db/db mice. These results are consistent with previous reports indicating that hepatic overexpression of the dominant-negative SHIP2 mutant reduces the fasting blood glucose level and improves glucose tolerance in db/db mice (Fukui et al., 2005). These findings are also compatible with the results of an earlier study using KKAy mice (Grempler et al., 2007), in which liver-specific dominant-negative inhibition of SHIP2 reduced the non-fasting blood glucose level and improved glucose tolerance.
Pharmacokinetic analyses of exposure to AS1949490 revealed that, while serum concentrations were low, the concentration of AS1949490 was highest in the liver, (data not shown), which was consistent with the inhibition of hepatic SHIP2. Therefore, AS1949490 may function mainly in the liver. We observed that a single oral dose of AS1949490 inhibited the expression of gluconeogenic genes such as G6Pase and PEPCK in the liver. This agrees with the reduction of G6Pase and PEPCK gene expression in the liver observed with the SHIP2 dominant negative mutant (Fukui et al., 2005; Grempler et al., 2007). In addition, chronic treatment with AS1949490 induced the phosphorylation of GSK3β in the liver. GSK3β is downstream to insulin signalling, and is phosphorylated and inactivated by Akt. GSK3β is important to the metabolic action of insulin; for example, it plays a key role in glycogen synthesis (Cross et al., 1995). The negative regulation of GSK3β by SHIP2 has been well documented in previous reports (Wada et al., 2001; Sasaoka et al., 2004). These findings indicate that AS1949490 can inhibit SHIP2 in the liver, which suggests that the improvement of type 2 diabetes observed with AS1949490 treatment could be partially attributed to this effect. However, the possibility that AS1949490 has actions in other tissues, such as muscle or fat, which have been reported to be SHIP2 target tissues, cannot be ruled out. Therefore, additional studies are needed in a variety of target tissues to elucidate the contribution of each tissue to the overall improvement in glucose metabolism. In our experiments, in spite of a relatively high dose, no toxic effects such as hypophagia and body weight reduction were observed. However, this does not preclude the possibility that unknown targets of AS1949490 contribute to the anti-diabetic effects. To elucidate these issues, additional studies profiling AS1949490 in more detail (as previously mentioned) are necessary, as is the discovery of additional, more potent types of selective SHIP2 inhibitors.
In summary, the first novel, potent, selective small molecule SHIP2 inhibitor, AS1949490, has been identified. The full biological characterization of AS1949490 will be of great importance for the understanding of the physiological and pathophysiological functions of SHIP2.
Acknowledgments
We would like to thank Drs Masao Kato, Masayuki Shibasaki, Hiroshi Kayakiri, Shigeki Sato, Tatsuya Niimi and Hiroyuki Moritomo, as well as Ms Aya Takehana (Astellas Pharma Inc.) for their helpful advice and experimental support.
Glossary
Abbreviations:
- G6Pase
glucose-6 phosphatase
- GSK3β
glycogen synthase kinase 3β
- Ins(1,3,4,5)P4
D-myo-Inositol-1,3,4,5-tetrakisphosphate
- OGTT
oral glucose tolerance test
- PEPCK
phosphoenolpyruvate carboxykinase
- PIP3
phosphatidylinositol-3,4,5-trisphosphate
- PtdIns(3,4,5)P3
D-myo-phosphatidylinositol-3,4,5-trisphosphate
- PTEN
phosphatase and tensin homologue
- SHIP
SH2 domain-containing inositol 5′-phosphatase
Conflict of interest
None.
Supporting information
Additional Supporting Information may be found in the online version of this article:
Figure S1 (A) Inhibitory effect of AS1949490 on the catalytic activity of SHIP2 and SHIP1. AS1949490 inhibition constant and its mode of inhibition for SHIP2 were determined by varying the ins(1,3,4,5)P4 concentration at fixed levels of AS1949490, 0 nM (circle), 630 nM (triangle), 1300 nM (square). AS1949490 is a competitive inhibitor with respect to ins(1,3,4,5)P4 with a Ki value of 0.44 ± 0.19 μM for SHIP2. (B) AS1949490 inhibition constant and its mode of inhibition for SHIP1 were determined by varying the ins(1,3,4,5)P4 concentration at fixed levels of AS1949490, 0 nM (circle), 5 μM (triangle), 20 μM (square), 40 μM (diamond). AS1949490 is a competitive inhibitor with respect to ins(1,3,4,5)P4 with a Ki value of 11 ± 1.4 μM for SHIP1.
Please note: Wiley-Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
References
- Amos AF, McCarty DJ, Zimmet P. The rising global burden of diabetes and its complications: estimates and projections to the year 2010. Diabet Med. 1997;14:S1–S85. [PubMed] [Google Scholar]
- Barthel A, Schmoll D. Novel concepts in insulin regulation of hepatic gluconeogenesis. Am J Physiol Endocrinol Metab. 2003;285:E685–E692. doi: 10.1152/ajpendo.00253.2003. [DOI] [PubMed] [Google Scholar]
- Baumgartener JW. SHIP2: an emerging target for the treatment of type 2 diabetes mellitus. Curr Drug Targets Immune Endocr Metabol Disord. 2003;3:291–298. doi: 10.2174/1568008033340144. [DOI] [PubMed] [Google Scholar]
- Buettner R, Ottinger I, Gerhardt-Salbert C, Wrede CE, Scholmerich J, Bollheimer LC. Antisense oligonucleotides against the lipid phosphatase SHIP2 improve muscle insulin sensitivity in a dietary rat model of the metabolic syndrome. Am J Physiol Endocrinol Metab. 2007;292:E1871–E1878. doi: 10.1152/ajpendo.00263.2006. [DOI] [PubMed] [Google Scholar]
- Calera MR, Martinez C, Liu H, Jack AK, Birnbaum MJ, Pilch PF. Insulin increases the association of Akt-2 with Glut4-containing vesicles. J Biol Chem. 1998;273:7201–7204. doi: 10.1074/jbc.273.13.7201. [DOI] [PubMed] [Google Scholar]
- Chi Y, Zhou B, Wang WQ, Chung SK, Kwon YU, Ahn YH, et al. Comparative mechanistic and substrate specificity study of inositol polyphosphate 5-phosphatase Schizosaccharomyces pombe synaptojanin and SHIP2. J Biol Chem. 2004;279:44987–44995. doi: 10.1074/jbc.M406416200. [DOI] [PubMed] [Google Scholar]
- Cho H, Mu J, Kim JK, Thorvaldsen JL, Chu Q, Crenshaw EB, et al. Insulin resistance and a diabetes mellitus-like syndrome in mice lacking the protein kinase Akt2 (PKBbeta) Science. 2001;292:1728–1731. doi: 10.1126/science.292.5522.1728. [DOI] [PubMed] [Google Scholar]
- Clement S, Krause U, Desmedt F, Tanti JF, Behrends J, Pesesse X, et al. The lipid phosphatase SHIP2 controls insulin sensitivity. Nature. 2001;409:92–97. doi: 10.1038/35051094. [DOI] [PubMed] [Google Scholar]
- Cross DAE, Alessi DR, Cohen P, Andjelkovich M, Hemmings BA. Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature. 1995;378:785–789. doi: 10.1038/378785a0. [DOI] [PubMed] [Google Scholar]
- Czech MP. Molecular actions of insulin on glucose transport. Annu Rev Nutr. 1995;15:441–471. doi: 10.1146/annurev.nu.15.070195.002301. [DOI] [PubMed] [Google Scholar]
- Fukui K, Wada T, Kagawa S, Nagira K, Ikubo M, Ishihara H, et al. Impact of the liver-specific expression of SHIP2 (SH2-containing inositol 5′-phosphatase 2) on insulin signaling and glucose metabolism in mice. Diabetes. 2005;54:1958–1967. doi: 10.2337/diabetes.54.7.1958. [DOI] [PubMed] [Google Scholar]
- Garofalo RS, Orena SJ, Rafidi K, Torchia AJ, Stock JL, Hildebrandt AL, et al. Severe diabetes, age-dependent loss of adipose tissue, and mild growth deficiency in mice lacking Akt2/PKB beta. J Clin Invest. 2003;112:197–208. doi: 10.1172/JCI16885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Granner D, Andreone T, Sasaki K, Beale E. Inhibition of transcription of the phosphoenolpyruvate carboxykinase gene by insulin. Nature. 1983;305:549–551. doi: 10.1038/305549a0. [DOI] [PubMed] [Google Scholar]
- Grempler R, Zibrova D, Schoelch C, Van Marle A, Rippmann JF, Redemann N. Normalization of prandial blood glucose and improvement of glucose tolerance by liver-specific inhibition of SH2 domain containing inositol phosphatase 2 (SHIP2) in diabetic KKAy mice: SHIP2 inhibition causes insulin-mimetic effects on glycogen metabolism, gluconeogenesis, and glycolysis. Diabetes. 2007;56:2235–2241. doi: 10.2337/db06-1660. [DOI] [PubMed] [Google Scholar]
- Hanada M, Feng J, Hemmings BA. Structure, regulation and function of PKB/AKT – a major therapeutic target. Biochim Biophys Acta. 2004;1697:3–16. doi: 10.1016/j.bbapap.2003.11.009. [DOI] [PubMed] [Google Scholar]
- Hori H, Sasaoka T, Ishihara H, Wada T, Murakami S, Ishiki M, et al. Association of SH2-containing inositol phosphatase 2 with the insulin resistance of diabetic db/db mice. Diabetes. 2002;51:2387–2394. doi: 10.2337/diabetes.51.8.2387. [DOI] [PubMed] [Google Scholar]
- Irie HY, Pearline RV, Grueneberg D, Hsia M, Ravichandran P, Kothari N, et al. Distinct roles of Akt1 and Akt2 in regulating cell migration and epithelial-mesenchymal transition. J Cell Biol. 2005;171:1023–1034. doi: 10.1083/jcb.200505087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishihara H, Sasaoka T, Hori H, Wada T, Hirai H, Haruta T, et al. Molecular cloning of rat SH2-containing inositol phosphatase 2 (SHIP2) and its role in the regulation of insulin signaling. Biochem Biophys Res Commun. 1999;260:265–272. doi: 10.1006/bbrc.1999.0888. [DOI] [PubMed] [Google Scholar]
- Kagawa S, Soeda Y, Ishihara H, Oya T, Sasahara M, Yaguchi S, et al. Impact of transgenic overexpression of SH2-containing inositol 5′-phosphatase 2 on glucose metabolism and insulin signaling in mice. Endocrinology. 2008;149:642–650. doi: 10.1210/en.2007-0820. [DOI] [PubMed] [Google Scholar]
- Kim EK, Tucker DF, Yun SJ, Do KH, Kim MS, Kim JH, et al. Linker region of Akt1/protein kinase B[alpha] mediates platelet-derived growth factor-induced translocation and cell migration. Cell Signal. 2008;20:2030–2037. doi: 10.1016/j.cellsig.2008.07.012. [DOI] [PubMed] [Google Scholar]
- Kobayashi K, Forte TM, Taniguchi S, Ishida BY, Oka K, Chan L. The db/db mouse, a model for diabetic dyslipidemia: molecular characterization and effects of western diet feeding. Metabolism. 2000;49:22–31. doi: 10.1016/s0026-0495(00)90588-2. [DOI] [PubMed] [Google Scholar]
- L'age M, Henning HV, Ohly B, Seubert W. On the role of insulin in the control of gluconeogenesis. Biochem Biophys Res Commun. 1968;19:241–246. doi: 10.1016/0006-291x(68)90736-5. [DOI] [PubMed] [Google Scholar]
- Lazar Df, Saltiel AR. Lipid phosphatases as drug discovery targets for type 2 diabetes. Nat Rev Drug Discov. 2006;5:333–342. doi: 10.1038/nrd2007. [DOI] [PubMed] [Google Scholar]
- Lee JO, Yang H, Georgescu MM, Di Cristofano A, Maehama T, Shi Y, et al. Crystal structure of the PTEN tumor suppressor: implications for its phosphoinositide phosphatase activity and membrane association. Cell. 1999;99:323–334. doi: 10.1016/s0092-8674(00)81663-3. [DOI] [PubMed] [Google Scholar]
- Leng Y, Karlsson HK, Zierath JR. Insulin signaling defects in type 2 diabetes. Rev Endocr Metab Disord. 2004;5:111–117. doi: 10.1023/B:REMD.0000021432.84588.f6. [DOI] [PubMed] [Google Scholar]
- Martens FM, Visseren FL, Lemay J, De Koning EJ, Rabelink TJ. Metabolic and additional vascular effects of thiazolidinediones. Drugs. 2002;62:1463–1480. doi: 10.2165/00003495-200262100-00004. [DOI] [PubMed] [Google Scholar]
- Matsumoto M, Kamohara M, Sugimoto T, Hidaka K, Takasaki J, Saito T, et al. The novel G-protein coupled receptor SALPR shares sequence similarity with somatostatin and angiotensin receptors. Gene. 2000;248:183–189. doi: 10.1016/s0378-1119(00)00123-2. [DOI] [PubMed] [Google Scholar]
- Pesesse X, Deleu S, De Smedt F, Drayer L, Erneux C. Identification of a second SH2-domain-containing protein closely related to the phosphatidylinositol polyphosphate 5-phosphatase SHIP. Biochem Biophys Res Commun. 1997;239:697–700. doi: 10.1006/bbrc.1997.7538. [DOI] [PubMed] [Google Scholar]
- Pesesse X, Moreau C, Drayer AL, Woscholski RI, Parker P, Erneux C. The SH2 domain containing inositol 5-phosphatase SHIP2 displays phosphatidylinositol 3,4,5-trisphosphate and inositol 1,3,4,5-tetrakisphosphate 5-phosphatase activity. FEBS Lett. 1998;437:301–303. doi: 10.1016/s0014-5793(98)01255-1. [DOI] [PubMed] [Google Scholar]
- Sasaoka T, Hori H, Wada T, Ishiki M, Haruta T, Ishihara H, et al. SH2-containing inositol phosphatase 2 negatively regulates insulin-induced glycogen synthesis in L6 myotubes. Diabetologia. 2001;44:1258–1267. doi: 10.1007/s001250100645. [DOI] [PubMed] [Google Scholar]
- Sasaoka T, Wada T, Fukui K, Murakami S, Ishihara H, Suzuki R, et al. SH2-containing inositol phosphatase 2 predominantly regulates Akt2, and not Akt1, phosphorylation at the plasma membrane in response to insulin in 3T3-L1 adipocytes. J Biol Chem. 2004;279:14835–14843. doi: 10.1074/jbc.M311534200. [DOI] [PubMed] [Google Scholar]
- Sasaoka T, Wada T, Tsuneki H. Lipid phosphatases as a possible therapeutic target in cases of type 2 diabetes and obesity. Pharmacol Ther. 2006;112:799–809. doi: 10.1016/j.pharmthera.2006.06.001. [DOI] [PubMed] [Google Scholar]
- Shao J, Yamashita H, Qiao L, Friedman JE. Decreased Akt kinase activity and insulin resistance in C57BL/KsJ-Leprdb/db mice. J Endocrinol. 2000;167:107–115. doi: 10.1677/joe.0.1670107. [DOI] [PubMed] [Google Scholar]
- Shimokawa T, Kagami M, Kato M, Kurosaki E, Shibasaki M, Katoh M. Effect of YM-126414 on glucose uptake and redistribution of glucose transporter isotype 4 in muscle cells. Eur J Pharmacol. 2000;410:1–5. doi: 10.1016/s0014-2999(00)00880-3. [DOI] [PubMed] [Google Scholar]
- Sleeman MW, Wortley KE, Lai KMV, Gowen LC, Kintner J, Kline WO, et al. Absence of the lipid phosphatase SHIP2 confers resistance to dietary obesity. Nat Med. 2005;11:199–205. doi: 10.1038/nm1178. [DOI] [PubMed] [Google Scholar]
- Somwar R, Sweeney G, Ramlal T, Klip A. Stimulation of glucose and amino acid transport and activation of the insulin signaling pathways by insulin lispro in L6 skeletal muscle cells. Clin Ther. 1998;20:125–140. doi: 10.1016/s0149-2918(98)80040-4. [DOI] [PubMed] [Google Scholar]
- Summers SA, Kao AW, Kohn AD, Backus GS, Roth RA, Pessin JE, et al. The role of glycogen synthase kinase 3beta in insulin-stimulated glucose metabolism. J Biol Chem. 1999;274:17934–17940. doi: 10.1074/jbc.274.25.17934. [DOI] [PubMed] [Google Scholar]
- Ueki K, Yamamoto-Honda R, Kaburagi Y, Yamauchi T, Tobe K, Burgering BMT, et al. Potential role of protein kinase B in insulin-induced glucose transport, glycogen synthesis, and protein synthesis. J Biol Chem. 1998;273:5315–5322. doi: 10.1074/jbc.273.9.5315. [DOI] [PubMed] [Google Scholar]
- Wada T, Sasaoka T, Funaki M, Hori H, Murakami S, Ishiki M, et al. Overexpression of SH2-containing inositol phosphatase 2 results in negative regulation of insulin-induced metabolic actions in 3T3-L1 adipocytes via its 5′-phosphatase catalytic activity. Mol Cell Biol. 2001;21:1633–1646. doi: 10.1128/MCB.21.5.1633-1646.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagman AS, Nuss JM. Current therapies and emerging targets for the treatment of diabetes. Curr Pharm Des. 2001;7:417–450. doi: 10.2174/1381612013397915. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.