

Abstract
Background and purpose:
Cell-to-cell interactions between mast cells and activated T cells are increasingly recognized as a possible mechanism in the aetiology of allergic or non-allergic inflammatory disorders. To determine the anti-allergic effect of fisetin, we examined the ability of fisetin to suppress activation of the human mast cell line, HMC-1, induced by activated Jurkat T cell membranes.
Experimental approach:
HMC-1 cells were incubated with or without fisetin for 15 min and then co-cultured with Jurkat T cell membranes activated by phorbol-12-myristate 13-acetate for 16 h. We determined gene expression in activated HMC-1 cells by DNA microarray and quantitative reverse transcription (RT)-PCR analysis. We also examined activation of the transcription factor NF-κB and MAP kinases (MAPKs) in activated HMC-1 cells.
Key results:
Fisetin suppresses cell spreading and gene expression in HMC-1 cells stimulated by activated T cell membranes. Additionally, we show that these stimulated HMC-1 cells expressed granzyme B. The stimulatory interaction also induced activation of NF-κB and MAPKs; these activations were suppressed by fisetin. Fisetin also reduced the amount of cell surface antigen CD40 and intercellular adhesion molecule-1 (ICAM-1) on activated HMC-1 cells.
Conclusions and implications:
Fisetin suppressed activation of HMC-1 cells by activated T cell membranes by interfering with cell-to-cell interaction and inhibiting the activity of NF-κB and MAPKs and thereby suppressing gene expression. Fisetin may protect against the progression of inflammatory diseases by limiting interactions between mast cells and activated T cells.
Keywords: inflammation, fisetin, human mast cell line (HMC-1), mast cells, T cells, cell-to-cell interaction, granzyme B, CD40, intercellular adhesion molecule 1 (ICAM-1), NF-κB
Introduction
Mast cells play a major role in the development of allergy such as asthma and pollenosis. The high-affinity IgE receptor FcεRI mediates release of chemical mediators, such as histamine, prostaglandins, tryptase, cytokines and chemokines, and this release is a primary function of mast cells in allergic responses (Mekori and Metcalfe, 2000; Gilfillan and Tkaczyk, 2006). In addition, mast cell activation by cell-to-cell interaction with T cells has also attracted attention as a possible mechanism of T cell-mediated cutaneous delayed-type hypersensitivity, of remodelling in asthma and other inflammatory responses.
Morphological studies have shown that mast cells are particularly numerous and situated close to T cells in inflamed tissues (Friedman and Kaliner, 1985; Smith and Weis, 1996; Church and Levi-Schaffer, 1997). Nakae et al. (2005) showed that T cells in the dermis of mice with cutaneous delayed-type hypersensitivity, an allergic inflammatory response mediated by T cells, appeared to be in contact with the resident mast cells. Studies on mast cell-deficient mice showed that mast cells are involved in T cell-mediated inflammatory responses such as encephalomyelitis, cutaneous delayed-type hypersensitivity and airway inflammation and remodelling in asthma (Biedermann et al., 2000; Secor et al., 2000; Nakae et al., 2005; 2007; Okayama et al., 2007).
Activated T cells and fractionated T cell membranes have been shown to induce mast cell activation, triggering degranulation and the release of tumour necrosis factor-α (TNFα), interleukin (IL)-8 and other cytokines (Krishnaswamy et al., 1997; Bhattacharyya et al., 1998; Baram et al., 2001; Salamon et al., 2005; 2008;). Activated T cell membranes, but not IgE cross-linking, strongly induced the expressions of amphiregulin and activin A in LAD 2 human mast cells; both proteins have been associated with irreversible tissue remodelling by inducing either proliferation of airway smooth muscle cells or mucin expression in airway epithelial cells (Okayama et al., 2007; Salamon et al., 2008). Thus, interactions between mast cells and activated T cells may increase the risk of inflammatory disorders.
Flavonoids show a variety of biological activities, such as anti-inflammatory, antioxidant, antibacterial and anti-allergic effects (Middleton et al., 2000; Williams and Grayer, 2004; Melgarejo et al., 2007). Fisetin (3,7,3′,4′-tetrahydroxyflavone) (Figure 1), present in vegetables and fruits such as onions (5 mg·kg−1), strawberries (160 mg·kg−1), apples (27 mg·kg−1) and the Japanese fruit Kaki (persimmon) (11 mg·kg−1) (Arai et al., 2000), strongly suppresses degranulation and IL-4 and IL-13 production in RBL-2H3 rat basophilic cells stimulated by IgE cross-linking (Morimoto et al., 2003; Hirano et al., 2004; Kawai et al., 2007; Park et al., 2008). Park et al. (2007; 2008;) showed that in HMC-1 human mast cells stimulated by phorbol-12-myristate 13-acetate (PMA) plus calcium ionophore A23187, fisetin suppressed gene expression and decreased production of TNFα, IL-1β, IL-4, IL-6 and IL-8 by inhibiting the phosphorylation of MAP kinases (MAPKs) and activation of NF-κB (Park et al., 2007). Thus fisetin was proposed to suppress the IgE or PMA plus calcium ionophore-mediated activation of basophils or mast cells. However, the ability of fisetin to suppress mast cell activation by activated T cells has not yet been described.
Figure 1.
The chemical structure of fisetin.
In the present study, we show that fisetin strongly inhibits the activation of mast cells by activated T cell membranes. To investigate how interaction with T cell membranes activates mast cells and how fisetin suppresses this effect, we used a DNA microarray to examine gene expression in HMC-1 cells stimulated by activated T cell membranes. Here, we first show that HMC-1 cells can be induced to express granzyme B, an important inflammatory mediator, by activated T cell membranes. We also found that interaction with activated T cell membranes induced the phosphorylation of IκBα and subsequent activation of NF-κB in HMC-1 cells. Finally, we demonstrate that fisetin can inhibit the induction of IκBα phosphorylation and NF-κB activation in HMC-1 cells stimulated by activated T cell membranes.
Methods
Cell lines and culture
Jurkat T lymphoma cells (ATCC TIB-152) were purchased from Dainippon Pharmaceutical Co. (Osaka, Japan) and cultured in RPMI 1640 (Invitrogen Japan, Tokyo, Japan) supplemented with 10% fetal calf serum (FCS; JRH Bioscience, Inc., Aurora, OH, USA). HMC-1 human mast cells (a kind gift from Dr JH Butterfield, Mayo Clinic, Rochester, MN, USA) were cultured in Iscove's modified Dulbecco's medium (IMDM; Invitrogen Japan K.K., Tokyo, Japan) supplemented with 10% defined iron-supplemented bovine calf serum (HyClone, Logan, UT) and 1.2 mM α-thioglycerol (Sigma-Aldrich Japan) (Butterfield et al., 1988). LAD 2 human mast cells (a kind gift from Dr Kirshenbaum, National Institutes of Health, Bethesda, MD) were cultured in StemPro-34 SFM serum-free complete medium (Invitrogen Japan K.K.) supplemented with 100 ng·mL−1 stem cell factor (SCF), 2 mM glutamine, 100 IU·mL−1 penicillin and 100 µg·mL−1 streptomycin (Kirshenbaum et al., 2003). The cells were maintained at 37°C in 5% CO2 in a humidified incubator.
Preparation of resting and activated T cell membranes
Resting or activated (by PMA) T cell membranes were obtained as previously described (Baram et al., 2001). Briefly, Jurkat T cells (1 × 106 cells·mL−1) were incubated with 50 ng·mL−1 PMA for 1 h at 37°C. The cells were then washed three times with phosphate-buffered saline (PBS) and resuspended at 1 × 107cells·mL−1 in cold TKMS buffer [50 mM Tris-HCl (pH 7.4), 25 mM KCl, 5 mM MgCl, 0.25 mM sucrose and 1 mM PMSF]. The cells were incubated on ice for 20 min, and then homogenized using an ultrasonic homogenizer (Branson Sonifier 250; Branson, Danbury, CT, USA). Cell lysates were centrifuged at 800×g for 5 min at 4°C. The supernatants containing T cell membranes were centrifuged at 100 000×g for 60 min at 4°C using a Beckman SW50.1 rotor (Beckman, Tokyo, Japan), and the resultant pellets were suspended at 1 × 105 cell membranes per microlitre in PBS buffer and stored at −80°C. The resting or activated T cell membranes in all experiments were added at a ratio of 1:1 to HMC-1 cells (1 × 106 cells·mL−1).
Analysis of β-hexosaminidase release
Degranulation of mast cells was determined by measuring β-hexosaminidase release. Cells were seeded on a 48-well plate at a density of 2 × 105 cells per well. LAD 2 cells were incubated overnight with 1 µg·mL−1 chimeric human IgE anti-4-hydroxy-3-nitrophenylacetate (NP; hapten) (Serotec, Oxford, UK). The cells were then washed once with modified Tyrode's (MT) buffer [137 mM NaCl, 2.7 mM KCl, 1.8 mM CaCl2, 1 mM MgCl2, 5.6 mM glucose, 20 mM HEPES, 0.1% bovine serum albumin (BSA) and pH 7.3] and resuspended in MT buffer. IgE-sensitized LAD 2 cells were incubated with or without fisetin for 15 min. Then 100 ng·mL−1 NP-BSA (Biosearch Technologies, Novato, CA, USA) was added to the medium, and the cells were further incubated for 30 min. Alternatively, HMC-1 cells or LAD 2 cells were incubated with or without fisetin in MT buffer for 15 min, and then T cell membranes were added to the medium, and the cells were further incubated for 30 min. Supernatants were then collected, and total cell lysates were obtained by addition of 0.1% Triton X-100 in MT buffer. Supernatants and total cell lysates were incubated with 3.3 mM p-nitrophenyl-2-acetaamido-2-deoxy-β-D-glucopyranoside in 100 mM citrate buffer (pH 4.5) for 25 min at 37°C. The reaction was stopped by adding 2 M glycine (pH 10.4). Supernatants were incubated with 2 M glycine for 25 min at 37°C, and then with 3.3 mM p-nitrophenyl-2-acetaamido-2-deoxy-β-D-glucopyranoside in 100 mM citrate buffer (pH 4.5) for the blank reaction (blank supernatant). Absorbance at 405 nm was determined using a Wallac ARVO SX 1420 multilabel counter (Perkin Elmer Life Sciences, Tokyo, Japan). The rate of β-hexosaminidase release was calculated as β-hexosaminidase release rate (%) = 100 × {(supernatant − blank supernatant)/[(supernatant − blank supernatant) + total cell lysate]}.
Analysis of HMC-1 cell spreading
HMC-1 cells were seeded on a 24-well plate (1.25 × 105 cells per well), incubated for 1 h in IMDM, then with or without fisetin for 15 min, and finally co-cultured with T cell membranes for 16 h. Fisetin was present throughout the 16 h incubation period with T cell membranes.
Morphological changes in the cells were observed under a phase-contrast microscope (Eclipse TS100; Nikon, Tokyo, Japan) at 200× original magnification. Spreading of HMC-1 cells was calculated as the proportion (%) of spreading cells in the total cell population; photomicrographs from three separate experiments were scored, without knowledge of the treatments. Approximately 200 cells were counted from each photomicrograph.
Cell viability assay
HMC-1 cells were incubated with or without fisetin for 15 min and then co-cultured with T cell membranes for 20 h. Fisetin was present throughout the 20 h incubation period with T cell membranes. Cell viability was determined using a Premix WST-1 Cell Proliferation Assay System (TaKaRa, Tokyo, Japan) according to the manufacturer's instructions.
DNA microarray analysis
HMC-1 cells were incubated with or without fisetin for 15 min, and then co-cultured with T cell membranes for 16 h. Fisetin was present throughout the 16 h incubation period with T cell membranes. A DNA microarray analysis was performed using the Allergy Chip Genopal ARIH-GX that we developed previously (Mitsubishi Rayon, Tokyo, Japan) (Kobori et al., 2008). Briefly, total RNA was extracted from HMC-1 cells using an RNeasy Mini Kit (Qiagen KK, Tokyo, Japan) according to the manufacturer's instructions. Total RNA (5 µg) was reverse transcribed into cDNA, which was then transcribed into biotin-labelled amplified RNA using the MessageAmp II-Biotin Enhanced kit (Applied Biosystems Japan, Tokyo, Japan) according to the manufacturer's instructions. The biotin-labelled RNA was fragmented for 7.5 min at 70°C using Fragmentation Reagents (Applied Biosystems Japan, Tokyo, Japan). The fragmented amplified RNA was incubated in TNT buffer [0.12 M Tris-HCl (pH 7.5), 0.12 M NaCl, 0.05% Tween-20] for 2 min at 94°C, kept at 37°C, and then hybridized to the Allergy Chip Genopal ARIH-GX for 16 h at 65°C in a hybridization chamber in the dark. After hybridization, the Chip was washed twice in 15 mL TNT buffer for 20 min at 65°C, and once in 15 mL TN buffer [0.12 M Tris-HCl (pH 7.5), 0.12 M NaCl] for 10 min at 65°C. Biotin-labelled amplified RNA on the Chip was then labelled with streptavidin-Cy5 (GE Healthcare BioScience, Tokyo, Japan) for 30 min at room temperature in the dark. The chip was washed four times in 6 mL TNT buffer for 5 min at room temperature using a rotator (MTR-103; AS ONE, Osaka, Japan) and stored in 6 mL TN buffer at 4°C in the dark. Data were collected using a DNA microarray reader with multibeam excitation technology (Yokogawa Electric, Tokyo, Japan).
Quantitative real-time reverse transcription (RT)-PCR analysis
HMC-1 cells were prepared in the same manner as for the DNA microarray analysis. Total RNA was extracted from HMC-1 cells using an RNeasy Mini Kit (Qiagen KK) according to the manufacturer's instructions. Total RNA (1 µg) was reverse-transcribed into cDNA using SuperScript II reverse transcriptase and random primer oligonucleotides (Invitrogen Japan) in a 20 µL final volume, according to the manufacturer's instructions. Quantitative RT-PCR was performed with an ABI Prism 7000 sequence detection system (Applied Biosystems Japan, Chiba, Japan) in a total volume of 25 µL in the presence of 12.5 µL of SYBR Green Realtime PCR Master Mix (TOYOBO, Osaka, Japan), 1 µL of 10 µM specific sets primer and 2.5 µL cDNA solution under the following conditions: one cycle for 1 min at 95°C; 40 cycles for 15 s at 95°C, 15 s at 65°C and 40 s at 72°C. The following primers were chosen from the list in the Universal Probe Library Assay Design Center (Roche Diagnostics, Basel, Switzerland): granzyme B forward (agatgcaaccaatcctgctt), reverse (catgtcccccgatgatct); perforin forward (ccgcttctctatacgggattc), reverse (gcagcagcaggagaaggat); CD40 forward (ggtctcacctcgctatggtt), reverse (cagtgggtggttctggatg); TNFα forward (cagcctcttctccttcctgat), reverse (gccagagggctgattagaga); IL-1β forward (ctgtcctgcgtgttgaaaga), reverse (ttgggtaatttttgggatctaca); glyceraldehyde-3-phosphate dehydrogenase (GAPDH) forward (agccacatcgctcagacac), reverse (gcccaatacgaccaaatcc). The relative amount of each transcript was normalized against the amount of GAPDH transcript in the same cDNA. ICAM-1 and GAPDH were detected using a High-Capacity cDNA Archive Kit (Applied Biosystems Japan, Tokyo, Japan) and a TaqMan Universal Master Mix (Applied Biosystems Japan) under the following by conditions: one cycle for 2 min at 50°C and 10 min at 95°C; 40 cycles for 15 s at 95°C and 1 min at 60°C. The relative amount of ICAM-1 was normalized against the amount of GAPDH transcript in the same cDNA.
elisa
HMC-1 cells were prepared by the same manner as DNA microarray analysis. elisa for granzyme B and perforin was performed using the appropriate elisa kit according to the manufacturer's instructions (Mabtech AB, Nacka Strand, Sweden). Briefly, a microtiter plate was coated overnight with anti-granzyme B mAb GB10 (2 µg·mL−1) or anti-perforin mAb Pf-80/164 (4 µg·mL−1) at 4°C. Samples or standards diluted in incubation buffer (PBS with 0.05% Tween-20 and 0.1% BSA) were added to the microtiter plate and incubated for 2 h at room temperature. Granzyme B or perforin were then labelled with anti-granzyme B mAb GB11-biotin (1 µg·mL−1) or anti-perforin mAb Pf-344-biotin (1 µg·mL−1), respectively, for 1 h at room temperature and detected with streptavidin-horseradish peroxidase (HRP) and TMB-E substrate solution (Moss, Pasadena, MD, USA). Data were collected using a Wallac ARVO SX 1420 multilabel counter (Perkin Elmer Life Sciences, Tokyo, Japan).
Western blot analysis
HMC-1 cells were incubated with or without fisetin for 15 min, and then co-cultured with T cell membranes for the appropriate period. Fisetin was present throughout the incubation period with T cell membranes. Cells were lysed in the sample buffer [10 mM Tris-HCl (pH 6.8), 1% SDS, 1% 2-mercaptoethanol and 10% glycerol] and denatured for 5 min at 95°C. Denatured protein samples were separated by 12% SDS-polyacrylamide gel electrophoresis and electrophoretically transferred to a polyvinylidene difluoride membrane (Immobilon PVDF; Millipore, Bedford, MA, USA). Proteins were detected using specific antibodies and an ECL Plus detection system (GE Healthcare Bio-Sciences KK, Tokyo, Japan).
Measurement of NF-κB-DNA binding activity
HMC-1 cells were incubated with or without fisetin for 15 min, and then co-cultured with T cell membranes for 4 h. Fisetin was present throughout the 4 h incubation period with T cell membranes. Nuclear extracts of HMC-1 cells were prepared using a Nuclear Extract Kit (Active Motif, Carlsbad, CA, USA) according to the manufacturer's instructions and stored at −80°C. NF-κB-DNA binding activity was measured with a TransAM NF-κB p65 Chemi Kit (Active Motif, Carlsbad, CA, USA) according to the manufacturer's instructions. Briefly, cell nuclear extract was added to a cell plate on which an oligonucleotide containing a p65-binding region was immobilized. Binding of the NF-κB p65 to the oligonucleotide was detected by elisa using chemiluminescence.
Flow cytometry analysis
HMC-1 cells were prepared in the same manner as for the DNA microarray analysis. Cells were washed twice in 2% FCS/PBS buffer and suspended in 1 mL of 1 µg·mL−1 fluorescein (FITC)-conjugated specific antibody in 2% FCS/PBS buffer. After 30 min incubation on ice, cells were centrifuged at 800×g for 5 min, washed twice in 2% FCS/PBS buffer, and then transferred to a 5 mL polystyrene round-bottom tube (BD Biosciences, Franklin Lakes, NJ, USA). Fluorescently labelled cells were detected with a BD FACSCanto II Flow Cytometer (BD Biosciences). Mouse IgG2a–FITC-conjugated antibody was used as the isotype control.
Statistical analysis
Statistical analyses were performed using GraphPad Prism5 software (GraphPad Software, San Diego, CA, USA). Data are shown as means ± standard error. Significant differences between groups were determined by repeated measures anova followed by Tukey's post hoc test (Lew, 2007). A P-value of less than 0.05 was considered statistically significant.
Materials
Phorbol-12-myristate 13-acetate was purchased from Sigma-Aldrich Japan (Tokyo, Japan). Fisetin was purchased from Extrasynthese (Genay Cedex, France) and dissolved in dimethyl sulphoxide at a final concentration of 10 mM. BAY11-7082 was purchased from Alexis Biochemicals (San Diego, CA, USA). Anti-intercellular adhesion molecule-1 (ICAM-1), anti-GAPDH, FITC-conjugated anti-CD40 and normal mouse IgG2a isotype control antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Antibodies against IκBα and phospho-IκBα were purchased from Cell Signaling Technology (Danvers, MA, USA). A MAPK Family Antibody Sampler Kit [extracellular signal-regulated kinase (ERK)1/2, phospho-ERK1/2, p38 and phospho-p38] was purchased from Cell Signaling Technology.
Results
Fisetin suppresses degranulation in LAD 2 cells stimulated by IgE cross-linking
Fisetin has been reported to inhibit degranulation of basophils stimulated by IgE cross-linking. Therefore, we investigated whether fisetin suppressed degranulation of LAD 2 human mast cells stimulated by IgE/antigen. IgE-sensitized LAD 2 cells were incubated with or without fisetin for 15 min and then stimulated with the antigen NP-BSA for 30 min. Fisetin was present during the stimulation with NP-BSA. Degranulation of LAD 2 cells was measured using the rate of β-hexosaminidase release. As shown in Figure 2, IgE cross-linking (IgE/antigen) increased β-hexosaminidase release from LAD 2 cells and incubation of these cells with either 25 or 50 µM fisetin abolished β-hexosaminidase release, on antigen challenge (Figure 2).
Figure 2.
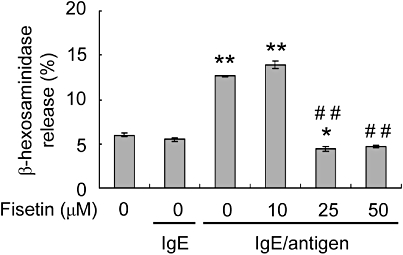
The effect of fisetin on β-hexosaminidase release from LAD 2 cells stimulated by IgE cross-linking. Cells were incubated overnight with 1 µg·mL−1 chimeric human IgE anti-NP (4-hydroxy-3-nitrophenylacetate). IgE sensitized cells were incubated with or without fisetin for 15 min; 100 ng·mL−1 of the antigen (NP-bovine serum albumin) was then added and the cells incubated for 30 min. The rate of β-hexosaminidase release was determined using an enzymatic assay. Data are expressed as mean ± standard error from three different experiments. *P < 0.05, **P < 0.01, significant difference from LAD 2 cells alone; and ##P < 0.01, significant difference from IgE/antigen.
We also investigated degranulation of HMC-1 and LAD 2 human mast cells that had been stimulated by PMA (50 ng·mL−1)-activated Jurkat T cell membranes. Activated T cell membranes did not induce β-hexosaminidase release from either HMC-1 or LAD 2 cells after 30 min stimulation (data not shown).
Fisetin suppresses morphological changes in HMC-1 cells stimulated by activated T cell membranes
The interaction of mast cells with CD3- or PMA-activated T cells induces the formation of heterotypic aggregates after 16–24 h incubation (Bhattacharyya et al., 1998). We examined the morphology of HMC-1 cells stimulated by resting or PMA (50 ng·mL−1)-activated Jurkat T cell membranes. HMC-1 cells incubated alone or with resting T cell membranes showed no morphological changes after 16 h incubation (Figure 3A,B). By contrast, cell spreading and adhesion were observed in HMC-1 cells incubated with PMA-activated T cell membranes for 16 h (Figure 3A,B). Cell spreading was observed within 6 h and peaked after 16 h of adding PMA-activated T cell membranes (data not shown). The effect of fisetin on the morphological changes induced in HMC-1 cells by activated T cell membranes was determined by pre-incubating the cells with 1, 10, 25 or 50 µM fisetin for 15 min followed by co-culture with PMA-activated T cell membranes for 16 h. We found that fisetin suppressed cell spreading in a dose-dependent manner (Figure 3A,B). The proportions of spreading cells in cultures treated with 1, 10, 25 and 50 µM fisetin decreased concentration-dependently (Figure 3B). The reduction in the rate of spreading was significant at 25 and 50 µM but not at 1 and 10 µM fisetin (Figure 3A,B). Fisetin, over this concentration range of 10–50 µM, did not affect the viability of HMC-1 cells stimulated with T cell membranes (Figure 3C). These results suggest that fisetin inhibited the activation of the mast cells by PMA-activated T cell membranes but did not adversely affect cell viability.
Figure 3.
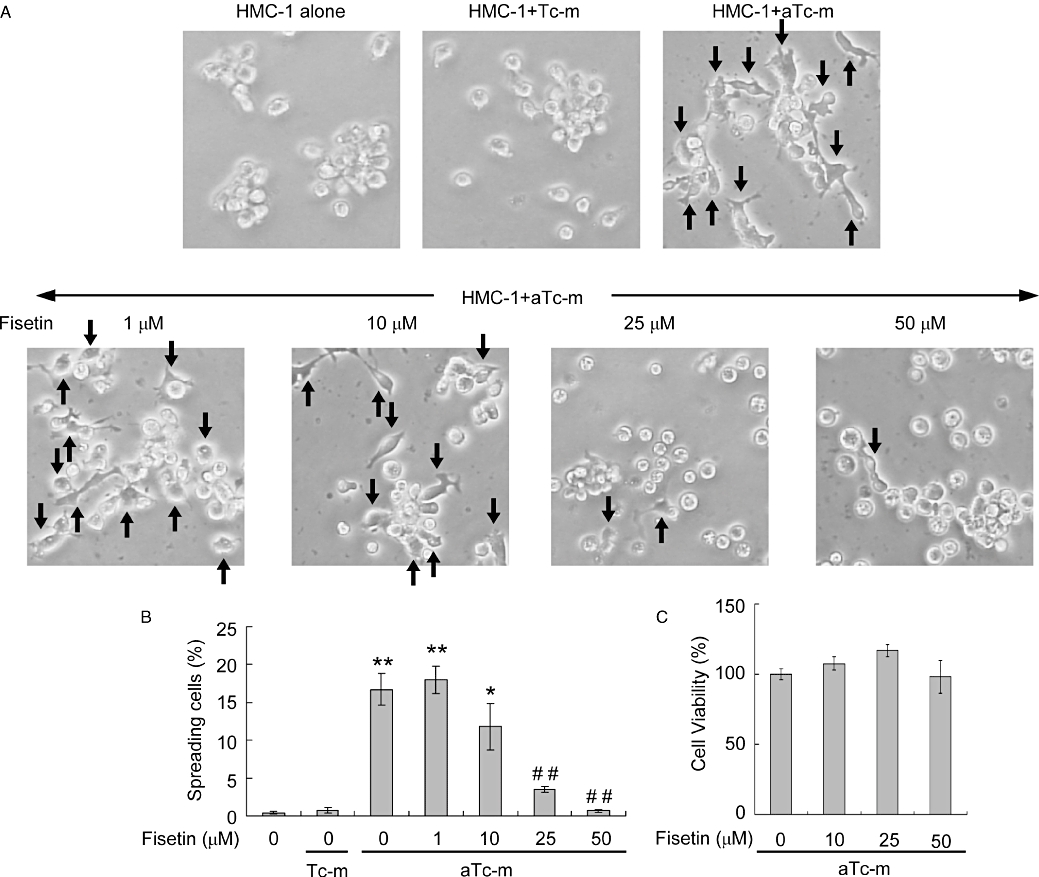
The effect of fisetin on spreading and viability of HMC-1 cells stimulated by activated-T cell membranes. (A) Cells were pre-incubated with or without the indicated concentration of fisetin for 15 min. Resting (Tc-m) or phorbol-12-myristate 13-acetate-activated T cell membranes (aTc-m) were then added and the cells incubated for 16 h. HMC-1 cell morphology was observed by phase-contrast microscopy (200× original magnification). The arrows indicate spreading cells. (B) Summary data of the proportions (%) of spreading cells under the various culture conditions shown in the micrographs above. (C) HMC-1 cells were treated as in (A) except for incubation for 20 h, at which time cell viability was analysed. The data are expressed as mean ± standard error from three different experiments. *P < 0.05, **P < 0.01, significant difference from HMC-1 cells alone; ##significant difference from aTc-m (P < 0.01).
Fisetin suppresses induced gene expression in HMC-1 cells stimulated by activated T cell membranes
Activated T cell membranes induce the expression of genes for inflammatory cytokines, such as TNFα and IL-8, in mast cells. We performed a microarray analysis of the expression of genes related to inflammation, immunity and housekeeping in HMC-1 cells that had been co-cultured with PMA-activated T cell membranes for 16 h. Genes that showed a greater than threefold increase in expression compared with controls are listed in Figure 4. Gene expression patterns in HMC-1 cells co-cultured with resting T cell membranes were essentially unchanged. However, in the presence of PMA-activated T cell membranes, 34 genes, including those for chemokines, ILs and cell surface membrane proteins, showed significantly increased expression in the HMC-1 cells (Figure 4). We then pre-incubated HMC-1 cells with fisetin for 15 min, and then incubated the cells for 16 h, with addition of PMA-activated T cell membranes. The elevated levels of gene expression observed in the HMC-1 cells in the presence of PMA-activated T cell membranes were substantially reduced by treating the cultures with 10, 25 or 50 µM fisetin (Figure 4). Indeed, many of the genes that were induced by PMA-activated T cell membranes had levels of expression similar to those of control cells after culture in 10, 25 or 50 µM fisetin (Figure 4).
Figure 4.
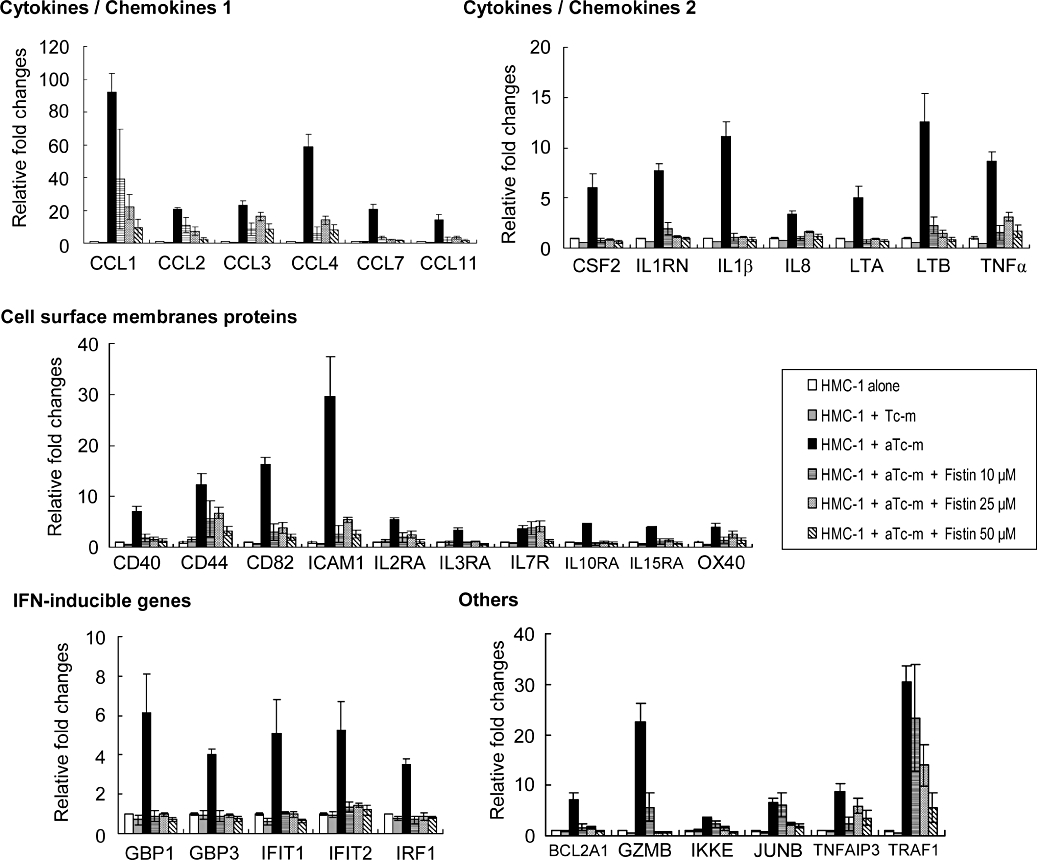
The effect of fisetin on gene expression in HMC-1 cells induced by PMA-activated T cell membranes. Cells were pre-incubated with or without the indicated concentration of fisetin for 15 min. Resting (Tc-m) or PMA-activated T cell membranes (aTc-m) were then added and the cells incubated for a further 16 h. Gene expression in the cells was determined using a DNA microarray (Genopal ARIH-GX, Mitsubishi Rayon). The degree of change (fold) was calculated compared with control cells. Data are expressed as mean ± standard error from three different experiments. CCL, CC chemokine ligand; CSF2, colony stimulating factor 2; HMC-1, HMC-1 cells; ICAM-1, intercellular adhesion molecule-1; IFN, interferon; IL, interleukin; PMA, phorbol-12-myristate 13-acetate; TNFα, tumour necrosis factor-α.
Fisetin suppresses the expression of granzyme B and perforin in HMC-1 cells stimulated by activated T cell membranes
Granzyme A, granzyme B and perforin are expressed by cytotoxic lymphocytes (Heusel et al., 1994; Lowin et al., 1994). A recent study suggested that perforin- or granzyme A-independent granzyme B may be a novel mediator of allergic inflammation (Tschopp et al., 2006). As the DNA microarray analysis performed here indicated that PMA-activated T cell membranes induced granzyme A-independent expression of granzyme B in HMC-1 cells (Figure 4), we examined mRNA expression and protein production in these cells by quantitative RT-PCR and elisa respectively. The quantitative RT-PCR analysis indicated that expression of granzyme B was strongly induced in HMC-1 cells stimulated by PMA-activated T cell membranes (Figure 5A). Fisetin, at a concentration of 10, 25 or 50 µM, significantly reduced this elevated granzyme B expression (Figure 5A). The increase in expression of RNA in the stimulated HMC-1 cells was matched by a significant increase in secretion of protein; again, treatment with fisetin significantly reduced secretion of granzyme B (Figure 5B).
Figure 5.
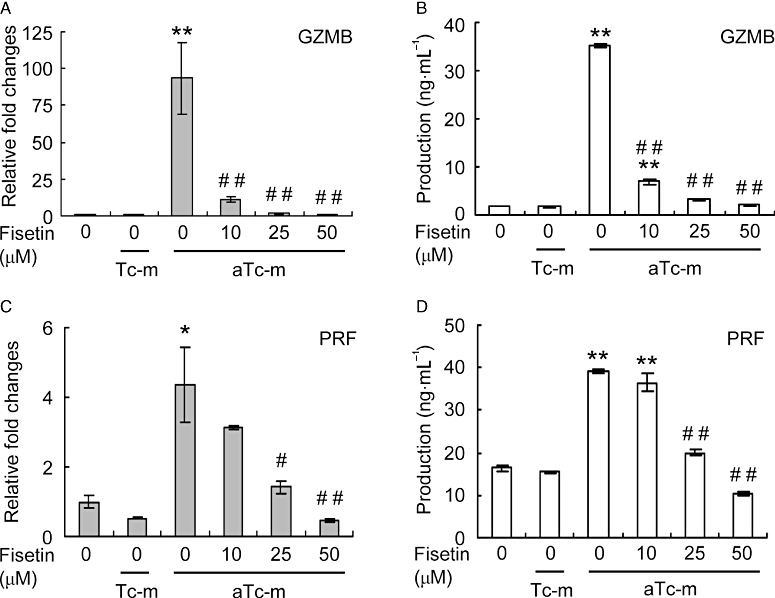
The effect of fisetin on expression of granzyme B (GZMB) and perforin (PRF) in HMC-1 cells stimulated by activated T cell membranes. Cells were pre-incubated with or without the indicated concentration of fisetin for 15 min. Resting (Tc-m) or phorbol-12-myristate 13-acetate-activated T cell membranes (aTc-m) were then added and the cells incubated for a further 16 h. (A,C) Gene expression levels were determined by real-time RT-PCR, normalized against glyceraldehyde-3-phosphate dehydrogenase and plotted relative to those in cells treated with aTc-m alone. (B,D) Production of granzyme B and perforin proteins was determined by elisa of culture supernatants. Data are expressed as mean ± standard error from three different experiments. *P < 0.05, **P < 0.01, significant difference from HMC-1 cells alone; and #P < 0.05, ##P < 0.01, significant difference from aTc-m.
Perforin is required for cytolysis, while the granzymes efficiently induce death of target cells (Heusel et al., 1994; Lowin et al., 1994). In a similar fashion as for granzyme B, we determined perforin mRNA expression and protein production in HMC-1 cells stimulated with PMA-activated T cell membranes. We found that expression and secretion of perforin were significantly increased in the stimulated HMC-1 cells (Figure 5C,D) and that fisetin significantly reduced this elevation in expression and secretion (Figure 5C,D).
Fisetin suppresses the expression of the cell surface membrane proteins ICAM-1 and CD40 on HMC-1 cells stimulated by activated T cell membranes
The DNA microarray analysis showed that expression of the cell surface membrane proteins ICAM-1, CD40, CD44 and CD82, and of IL receptors were increased in HMC-1 cells stimulated by PMA-activated T cell membranes and that fisetin suppressed this increased expression (Figure 4). Cell surface membrane proteins are associated with the co-stimulation and adhesion of leukocyte cells. As lymphocyte function-associated antigen-1 (LFA-1), the ligand of ICAM-1, and CD40 ligand (CD40L) are abundantly expressed on activated T cells (Borrow et al., 1996; Bennett et al., 1998; Ridge et al., 1998; Schoenberger et al., 1998), we examined the levels of mRNA and protein expression of ICAM-1 and CD40 on HMC-1 cells stimulated by activated T cell membranes. Quantitative RT-PCR analysis showed that the levels of expression of ICAM-1 and CD40 were elevated in stimulated HMC-1 cells and that this increased expression was significantly reduced by fisetin in a dose-dependent manner (Figure 6A,B). Similarly, the level of ICAM-1 protein increased in stimulated HMC-1 cells and could be reduced by fisetin (Figure 6C). In contrast, the level of CD40 protein, which is constitutively expressed on HMC-1 cells, was not increased on stimulated cells (Figure 6D). Nevertheless, the level of CD40 protein was slightly reduced by treatment with 50 µM fisetin (Figure 6D).
Figure 6.
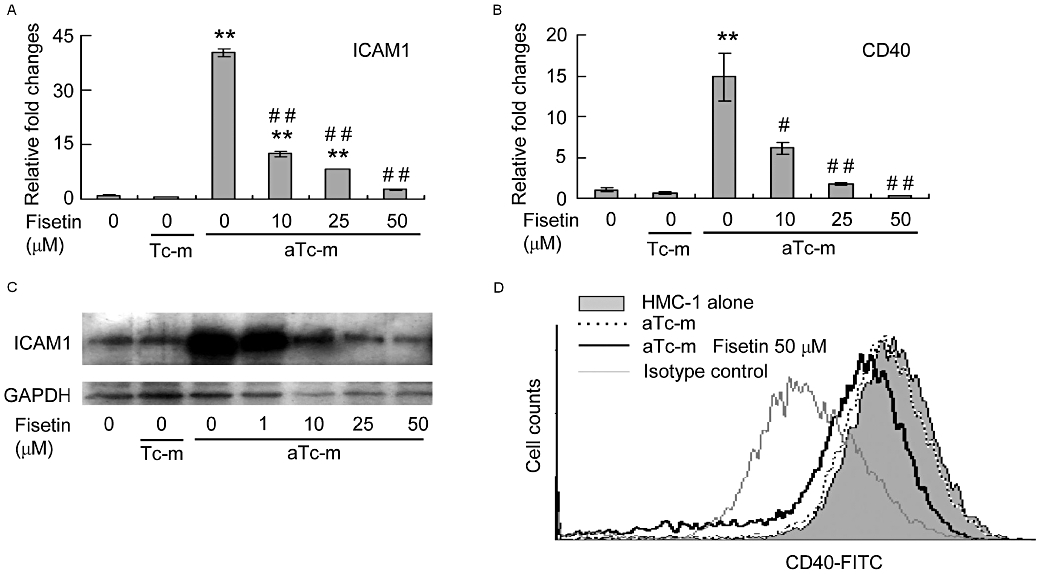
The effect of fisetin on expression of intercellular adhesion molecule-1 (ICAM-1) and CD40 HMC-1 cells stimulated by activated T cell membranes. HMC-1 cells were pre-incubated with or without the indicated concentration of fisetin for 15 min. Resting (Tc-m) or phorbol-12-myristate 13-acetate-activated T cell membranes (aTc-m) were then added and the cells incubated for a further 16 h. (A,B) Gene expression levels were determined by real-time RT-PCR, normalized against glyceraldehyde-3-phosphate dehydrogenase (GAPDH) and plotted relative to those in cells treated with aTc-m alone. Data are expressed as mean ± standard error from three different experiments. **P < 0.01, significant difference from HMC-1 cells alone; and #P < 0.05, ##P < 0.01, significant difference from aTc-m. (C) The level of ICAM-1 protein was determined by Western blot analysis. (D) The level of CD40 protein was determined by a flow cytometer.
Fisetin suppresses activity of the transcriptional factor NF-κB in HMC-1 cells stimulated by activated T cell membranes
Mast cells, and other cells related to inflammation, show expression of a number of pro-inflammatory cytokines, such as IL-1β, IL-8, colony stimulating factor 2 and TNFα, following activation of the ubiquitous nuclear transcription factor NF-κB (Marquardt and Walker, 2000; Azzolina et al., 2003; Peng et al., 2005). ICAM-1 and CD40 are also regulated by activation of the NF-κB signalling pathway, for example, after TNFα and LPS stimulation (Qin et al., 2005; Tsang et al., 2005). As the activated T cell membranes induced expression in HMC-1 cells of TNFα and other genes regulated by NF-κB, we sought to determine NF-κB activity in these cells using a DNA binding activity assay. HCM-1 cells were stimulated with PMA-activated T cell membranes for 4 h, and then the NF-κB-DNA binding activity of nuclear extracts was determined by an elisa of their affinity to immobilized oligonucleotides containing NF-κB p65 consensus binding sites. NF-κB-DNA binding activity was induced in HMC-1 cells stimulated by activated T cell membranes but not in those co-cultured with resting T cell membranes (Figure 7A). Fisetin (50 µM) significantly suppressed NF-κB-DNA binding activity of stimulated HMC-1 cells (Figure 7A). NF-κB is activated by degradation of the inhibitor IκBα, which forms an inactive complex with NF-κB in the cytoplasm. Phosphorylation and subsequent polyubiquitination of IκBα result in its degradation by proteosomes (Alkalay et al., 1995). We therefore analysed phosphorylation of IκBα in stimulated HMC-1 cells and found that it was induced after 5 min incubation in HMC-1 cells co-cultured with PMA-stimulated but not resting T cell membranes (Figure 7B). Fisetin (50 µM) inhibited IκBα phosphorylation in addition to NF-κB-DNA binding activity in HMC-1 (Figure 7B). We suggest that fisetin can inhibit NF-κB activity induced in HMC-1 cells by activated T cell membranes by inhibiting phosphorylation of IκBα.
Figure 7.
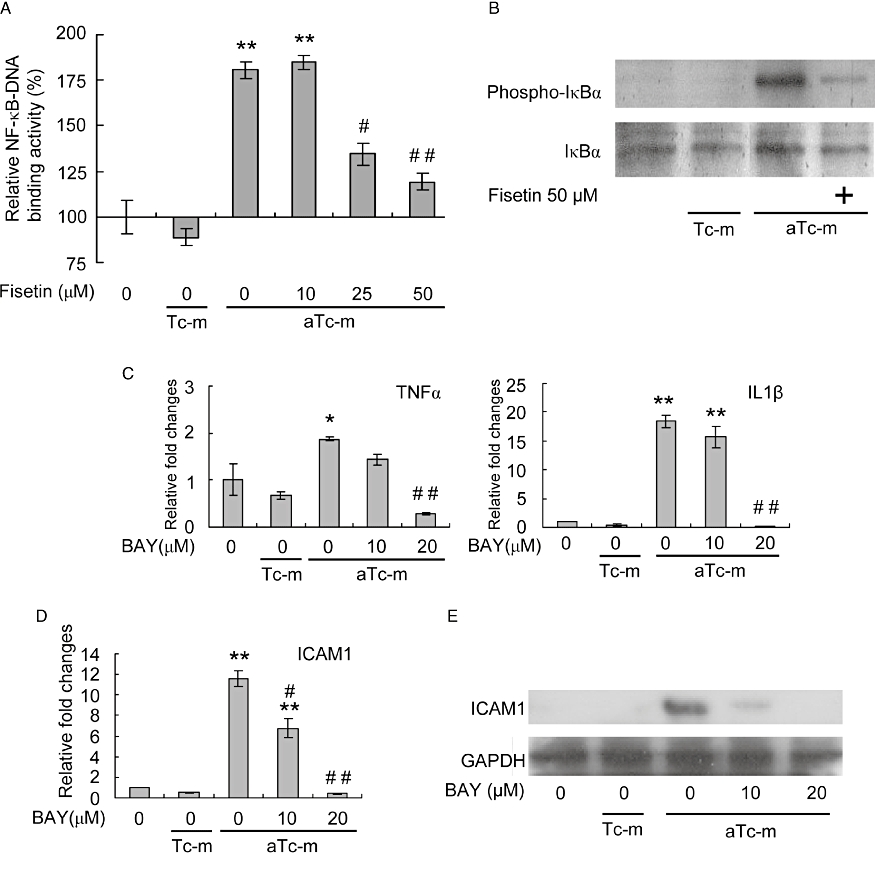
The effect of fisetin on NF-κB-DNA binding activity and IκBα phosphorylation in HMC-1 cells stimulated by activated T cell membranes. (A) HMC-1 cells were pre-incubated with or without the indicated concentration of fisetin for 15 min. Resting (Tc-m) or PMA-activated T cell membranes (aTc-m) were then added and the cells incubated for a further 4 h. DNA binding activity in the nuclear extract was determined using a TransAM NF-κB kit (Active Motif). Relative NF-κB binding activity is expressed as a percentage of that of HMC-1 cells alone. The data are expressed as mean ± standard error from three different experiments. **P < 0.01, significant difference from HMC-1 cells alone; and #P < 0.05, ##P < 0.01, significant difference from aTc-m. (B) HMC-1 cells were pre-incubated with or without 50 µm fisetin for 15 min. Resting (Tc-m) or PMA-activated T cell membranes (aTc-m) were then added and the cells incubated for further 5 min. The levels of IκBα and of phosphorylated IκBα were determined by Western blot analysis. (C,D) HMC-1 cells were pre-incubated with or without the indicated concentration of BAY11-7082 (BAY) for 2 h. Resting (Tc-m) or PMA-activated T cell membranes (aTc-m) were then added and the cells incubated for 16 h. Gene expression levels were determined by real-time RT-PCR, normalized against GAPDH and plotted relative to those in cells treated with aTc-m alone. The data are expressed as means ± standard error from three different experiments. *P < 0.05, **P < 0.01, significant difference from HMC-1 cells alone; and #P < 0.05, ##P < 0.01, significant difference from aTc-m. (E) HMC-1 cells were pre-incubated with or without the indicated concentration of BAY11-7082 (BAY) for 2 h. Resting (Tc-m) or PMA-activated T cell membranes (aTc-m) were then added and the cells incubated for 16 h. The level of ICAM-1 protein was determined by Western blot analysis. GAPDH, glyceraldehyde-3-phosphate dehydrogenase; HMC-1, human mast cell line-1; ICAM-1, intercellular adhesion molecule-1; IL, interleukin; PMA, phorbol-12-myristate 13-acetate; TNFα, tumour necrosis factor-α.
We then examined the effect of the IκBα phosphorylation inhibitor BAY11-7082 on the ability of PMA-activated T cell membranes to induce mRNA and protein expression in HMC-1 cells. BAY11-7082 significantly reduced the levels of TNFα and IL-1β mRNAs, and of mRNA and protein expression of ICAM-1 in HMC-1 cells stimulated by PMA-activated T cell membranes (Figure 7C–E). Our results suggest that the NF-κB pathway plays an important role in gene expression in mast cells stimulated by PMA-activated T cell membranes.
Fisetin suppresses phosphorylation of MAPKs in HMC-1 cells stimulated by activated T cell membranes
In addition to NF-κB, MAPKs regulate expression of pro-inflammatory genes (Wong et al., 2006). It was previously reported that activation of HMC-1 cells by PMA-activated T cells or T cell membranes is associated with phosphorylation of ERK1/2 and p38 MAPK (Brill et al., 2004). We examined the suppressive effect of fisetin on ERK and p38 phosphorylation in HMC-1 cells stimulated by PMA-activated T cell membranes. In these cells, phosphorylation of ERK1/2 and p38 was observed after 20 min stimulation with activated T cell membranes (Figure 8). Fisetin strongly suppressed phosphorylation of ERK1/2 and p38 in HMC-1 cells (Figure 8).
Figure 8.
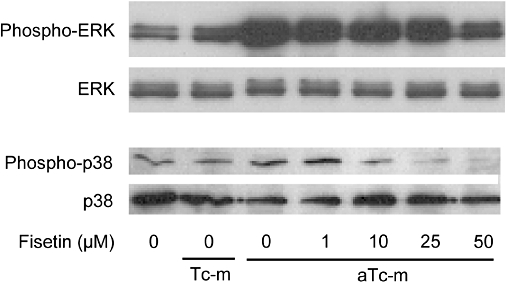
The effect of fisetin on phosphorylation of MAP kinases (MAPKs) [extracellular signal-regulated kinase (ERK)1/2 and p38] in HMC-1 cells. HMC-1 cells were pre-incubated with or without the indicated concentration of fisetin for 15 min. Resting (Tc-m) or phorbol-12-myristate 13-acetate-activated T cell membranes (aTc-m) were added and the cells incubated for a further 20 min. The levels of MAPKs and phosphorylated MAPKs were determined by Western blot analysis.
Discussion
Previous studies identified a novel activation pathway in mast cells that is induced by a direct interaction of the cells with CD3- or PMA-activated T cells or T cell membranes but not by resting T cells (Krishnaswamy et al., 1997; Inamura et al., 1998; Baram et al., 2001). In this study, we showed that fisetin not only suppressed IgE-mediated mast cell degranulation but also mast cell activation mediated by activated T cell membranes. HMC-1 cells that had been stimulated by activated T cell membranes showed induced up-regulation of 34 of the 205 genes present on the DNA microarray, including those for cell surface membrane proteins, cytokines/chemokines, interferon (IFN)-inducible and other proteins, such as granzyme B and BCL2A1 (Figure 4). Fisetin suppressed expression of all genes up-regulated by activated T cell membranes.
Cytokines/chemokines play several roles in inflammatory responses such as leukocyte proliferation and activation, extravasation and chemotaxis (Galli et al., 2008). For example, CC chemokine ligand 2 (CCL2) and CCL3 induce both immediate and delayed skin hypersensitivity reactions in human atopic and non-atopic subjects (Gaga et al., 2008).
Granzymes are serine proteases expressed in cytotoxic lymphocytes such as natural killer cells and cytotoxic T lymphocytes. Cytotoxic lymphocytes achieve apoptosis of target cells via perforin, and granzyme A and/or granzyme B. Perforin- and granzyme A-independent granzyme B has been reported to induce cleavage in the extracellular matrix (of vitronectin, fibronectin and laminin), cell detachment and anoikis in smooth muscle cells and endothelial cells, and to cause remodelling in inflammatory tissues (Choy et al., 2004; Buzza et al., 2005). Granzyme B has been found in vivo in bronchoalveolar lavage fluids from asthmatic patients (Tschopp et al., 2006). Thus, granzyme B has been shown to be a novel allergic inflammation mediator and clearly involved in inflammatory diseases. Although IgE-stimulated skin-derived mast cells express granzyme A- and perforin-independent granzyme B, other types of mast cells, such as HMC-1 cells, LAD 2 cells and human cord blood-derived mast cells, can only be induced to express low levels of granzyme B except with non-physiological stimuli such as A23187/PMA or Compound 48/80 (Pardo et al., 2007; Strik et al., 2007). This study is the first to show that HMC-1 cells can be induced to express granzyme B by the physiological stimuli provided by activated T cell membranes. Perforin is expressed in A23187/PMA-stimulated HMC-1 cells but not in other mast cell lines or primary cultures of mast cells (Strik et al., 2007). Our analysis showed that HMC-1 cells stimulated by activated T cell membranes did not express granzyme A but expressed and produced perforin. We showed that fisetin suppressed the secretion of granzyme B and perforin, induced from HMC-1 cells that have been activated by T cell membranes.
The cell surface membrane proteins ICAM-1 and CD40 play a critical role in adhesion and co-stimulation during cell-to-cell interactions, such as between T cells and antigen-presenting cells (Borrow et al., 1996; Bennett et al., 1998; Ridge et al., 1998; Schoenberger et al., 1998). In this study, we showed that the stimulation of HMC-1 cells by activated T cell membranes induced expression of the ICAM-1 gene and production of its protein, and also expression of the CD40 gene. HMC-1 cells constitutively express CD40, and stimulation by activated T cell membranes did not increase the amount of the CD40 protein on the cells. However, fisetin reduced the amounts of CD40 and ICAM-1 proteins on HMC-1 cells. Deficiency of ICAM-1, which binds to LFA-1, macrophage antigen-1 (Mac-1) and fibrinogen, eliminates immediate-type hypersensitivity and inhibits the subsequent late-phase reaction (Shimada et al., 2003). Deficiency of CD40L results in defective humoral immune responses (Renshaw et al., 1994). Thus, ICAM-1 and CD40 are potential therapeutic targets for regulating human allergic reactions. HMC-1 cells were reported to be stimulated by activated T cells through interaction of ICAM-1/LFA-1 and other co-stimulatory factors, such as the TNF receptor superfamily [lymphotoxin-β receptor and CD137 (4-1BB)] and the TNF superfamily (CD30 ligand, OX40 ligand), with their ligands and receptors (Inamura et al., 1998; Molin et al., 2001; Stopfer et al., 2004; Nishimoto et al., 2005; Fischer et al., 2006; Bachelet and Levi-Schaffer, 2007). Our results suggest that activated T cell membranes induce the expression of ICAM-1 and enhance the adhesion and activation of HMC-1 cells. Mast cell activation induced by activated T cells is inhibited by the introduction of neutralizing anti-ICAM-1 and anti-CD11/18 antibodies (Inamura et al., 1998). Fisetin probably suppressed the activation of mast cells by inhibiting the expression of ICAM-1, CD40 and other cell surface receptors.
Both ICAM-1 and CD40 expression are regulated by the transcriptional factor NF-κB signalling pathway (Qin et al., 2005; Tsang et al., 2005). Activated T cell membranes induced an increase in expression of pro-inflammation genes regulated by NF-κB, such as TNFα, IL-1α, IL-1β and IL-8 (Figure 4). Although the stimulation of mast cells by activated T cells is associated with phosphorylation of MAPKs (ERK1/2 and p38), induced activation of NF-κB by activated T cells has not been reported in HMC-1 cells (Brill et al., 2004). This study provides the first evidence that the activation of HMC-1 cells in response to activated T cell membranes is induced by phosphorylation of IκBα and the subsequent activation of the transcriptional factor NF-κB. In a similar fashion to fisetin, the IκBα phosphorylation inhibitor BAY11-7082 suppressed the mRNA expression of TNFα and IL-1β, and mRNA and protein expression of ICAM-1 in HMC-1 cells stimulated by PMA-activated T cell membranes. Our results suggest that fisetin, by inhibiting NF-κB transcriptional activity, suppressed the gene expression in HMC-1 cells induced by PMA-activated T cell membranes.
Wong et al. (2006) showed that SCF induced ERK activity while TNFα induced both p38 MAPK and NF-κB activity in HMC-1 cells. The ERK inhibitor PD98059 suppressed the SCF-induced expressions of IL-8, CCL1, CCL2 and CCL4, and the p38 MAPK inhibitor SB203580 suppressed the TNFα-induced expressions of IL-8, CCL1 and CCL2 (Wong et al., 2006). Our results showed that the activated T cell membranes induced activation of the MAPKs ERK and p38 in HMC-1 cells. Activation of the MAPKs ERK and p38 were probably associated with the induction of the expressions of IL-8, CCL1, CCL2, CCL4 and some other genes in HMC-1 cells stimulated by activated T cell membranes. It is likely that fisetin suppressed the activated T cell membranes-induced gene expression in HMC-1 cells by inhibiting the NF-κB transcriptional activity and also the ERK and p38 MAPK activities.
Our DNA microarray analysis indicated that expression of IFN-inducible genes was increased in HMC-1 cells stimulated by activated T cell membranes. IFN-inducible gene expression is mainly up-regulated via the Janus kinase/signal transducer and activator of transcription (JAK/STAT) pathway by IFN. Recently, however, GBP1 and IRF1 were reported to be inducible via the NF-κB pathway by IL-1α or IL-1β in endothelial cells and by CD40L in carcinomas (Naschberger et al., 2004; Moschonas et al., 2008). As activated T cell membranes did not induce the phosphorylation of STAT1 (data not shown), IFN-inducible genes may be stimulated via the NF-κB pathway by CD40 or other cell surface receptors.
We have shown here that fisetin strongly suppresses activation of HMC-1 cells by activated T cell membranes. HMC-1 cells stimulated by activated T cell membranes express chemotactic cytokines and chemokines (Bhattacharyya et al., 1998; Salamon et al., 2005; Salamon et al., 2008) (Figure 4); these chemical mediators have been shown to enhance activation of mast cells (Lee et al., 2004; Wong et al., 2006; Palmqvist et al., 2007). Our results suggest that fisetin suppressed activation of HMC-1 cells by activated T cell membranes by inhibiting the activity of NF-κB and MAPKs and thereby suppressing gene expression. Fisetin may also suppress activation of HMC-1 cells by interfering with cell-to-cell interaction through its inhibition of adhesion and spreading of mast cells, and suppression of the expression of cell surface receptors. Thus, fisetin possibly suppresses T cell-mediated inflammatory responses, such as cutaneous delayed-type hypersensitivity and airway inflammation and remodelling in asthma, by inhibition of the mast cell activation induced by activated T cells.
Acknowledgments
This work was financially supported in part by grants from the Ministry of Agriculture, Forestry and Fisheries and by grant-in-aid for Science Research (21500814) from the Ministry of Education, Culture, Sports, Science and Technology. We wish to thank Professor JH Butterfield for supplying HMC-1 cells and for technical advice. We thank Professor A Kirshenbaum for supplying LAD 2 cells and for technical advice. We also thank Yokogawa Electric Co. for the loan of a DNA microarray reader and technical advice.
Glossary
Abbreviations:
- CD40L
CD40 ligand
- ERK
extracellular signal-regulated kinase
- FCS
fetal calf serum
- FITC
fluorescein
- GAPDH
glyceraldehyde-3-phosphate dehydrogenase
- ICAM-1
intercellular adhesion molecule-1
- IL
interleukin
- IMDM
Iscove's modified Dulbecco's medium
- JAK/STAT
Janus kinase/signal transducer and activator of transcription
- LFA-1
lymphocyte function-associated antigen-1
- MAPKs
MAP kinases
- PBS
phosphate-buffered saline
- PMA
phorbol-12-myristate 13-acetate
- RT-PCR
reverse transcription-PCR
- SCF
stem cell factor
- TNFα
tumour necrosis factor-α
Conflict of interest
The authors state no conflict of interest. However, K Nagai and T Fukusima are employed by Mitsubishi Rayon Co., Ltd. Yokogawa Electric Co. gave the loan of a DNA microarray reader.
References
- Alkalay I, Yaron A, Hatzubai A, Orian A, Ciechanover A, Ben-Neriah Y. Stimulation-dependent I kappa B alpha phosphorylation marks the NF-kappa B inhibitor for degradation via the ubiquitin-proteasome pathway. Proc Natl Acad Sci USA. 1995;92:10599–10603. doi: 10.1073/pnas.92.23.10599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arai Y, Watanabe S, Kimira M, Shimoi K, Mochizuki R, Kinae N. Dietary intakes of flavonols, flavones and isoflavones by Japanese women and the inverse correlation between quercetin intake and plasma LDL cholesterol concentration. J Nutr. 2000;130:2243–2250. doi: 10.1093/jn/130.9.2243. [DOI] [PubMed] [Google Scholar]
- Azzolina A, Bongiovanni A, Lampiasi N. Substance P induces TNF-alpha and IL-6 production through NF kappa B in peritoneal mast cells. Biochim Biophys Acta. 2003;1643:75–83. doi: 10.1016/j.bbamcr.2003.09.003. [DOI] [PubMed] [Google Scholar]
- Bachelet I, Levi-Schaffer F. Mast cells as effector cells: a co-stimulating question. Trends Immunol. 2007;28:360–365. doi: 10.1016/j.it.2007.06.007. [DOI] [PubMed] [Google Scholar]
- Baram D, Vaday G, Salamon P, Drucker I, Hershkoviz R, Mekori Y. Human mast cells release metalloproteinase-9 on contact with activated T cells: juxtacrine regulation by TNF-alpha. J Immunol. 2001;167:4008–4016. doi: 10.4049/jimmunol.167.7.4008. [DOI] [PubMed] [Google Scholar]
- Bennett S, Carbone F, Karamalis F, Flavell R, Miller J, Heath W. Help for cytotoxic-T-cell responses is mediated by CD40 signalling. Nature. 1998;393:478–480. doi: 10.1038/30996. [DOI] [PubMed] [Google Scholar]
- Bhattacharyya S, Drucker I, Reshef T, Kirshenbaum A, Metcalfe D, Mekori Y. Activated T lymphocytes induce degranulation and cytokine production by human mast cells following cell-to-cell contact. J Leukoc Biol. 1998;63:337–341. doi: 10.1002/jlb.63.3.337. [DOI] [PubMed] [Google Scholar]
- Biedermann T, Kneilling M, Mailhammer R, Maier K, Sander C, Kollias G, et al. Mast cells control neutrophil recruitment during T cell-mediated delayed-type hypersensitivity reactions through tumor necrosis factor and macrophage inflammatory protein 2. J Exp Med. 2000;192(10):1441–1452. doi: 10.1084/jem.192.10.1441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borrow P, Tishon A, Lee S, Xu J, Grewal I, Oldstone M, et al. CD40L-deficient mice show deficits in antiviral immunity and have an impaired memory CD8+ CTL response. J Exp Med. 1996;183:2129–2142. doi: 10.1084/jem.183.5.2129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brill A, Baram D, Sela U, Salamon P, Mekori Y, Hershkoviz R. Induction of mast cell interactions with blood vessel wall components by direct contact with intact T cells or T cell membranes in vitro. Clin Exp Allergy. 2004;34:1725–1731. doi: 10.1111/j.1365-2222.2004.02093.x. [DOI] [PubMed] [Google Scholar]
- Butterfield J, Weiler D, Dewald G, Gleich G. Establishment of an immature mast cell line from a patient with mast cell leukemia. Leuk Res. 1988;12:345–355. doi: 10.1016/0145-2126(88)90050-1. [DOI] [PubMed] [Google Scholar]
- Buzza M, Zamurs L, Sun J, Bird C, Smith A, Trapani J, et al. Extracellular matrix remodeling by human granzyme B via cleavage of vitronectin, fibronectin, and laminin. J Biol Chem. 2005;280:23549–23558. doi: 10.1074/jbc.M412001200. [DOI] [PubMed] [Google Scholar]
- Choy J, Hung V, Hunter A, Cheung P, Motyka B, Goping I, et al. Granzyme B induces smooth muscle cell apoptosis in the absence of perforin: involvement of extracellular matrix degradation. Arterioscler Thromb Vasc Biol. 2004;24:2245–2250. doi: 10.1161/01.ATV.0000147162.51930.b7. [DOI] [PubMed] [Google Scholar]
- Church M, Levi-Schaffer F. The human mast cell. J Allergy Clin Immunol. 1997;99:155–160. doi: 10.1016/s0091-6749(97)70089-7. [DOI] [PubMed] [Google Scholar]
- Fischer M, Harvima I, Carvalho R, Möller C, Naukkarinen A, Enblad G, et al. Mast cell CD30 ligand is upregulated in cutaneous inflammation and mediates degranulation-independent chemokine secretion. J Clin Invest. 2006;116:2748–2756. doi: 10.1172/JCI24274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman M, Kaliner M. In situ degranulation of human nasal mucosal mast cells: ultrastructural features and cell-cell associations. J Allergy Clin Immunol. 1985;76:70–82. doi: 10.1016/0091-6749(85)90807-3. [DOI] [PubMed] [Google Scholar]
- Gaga M, Ong Y, Benyahia F, Aizen M, Barkans J, Kay A. Skin reactivity and local cell recruitment in human atopic and nonatopic subjects by CCL2/MCP-1 and CCL3/MIP-1alpha. Allergy. 2008;63:703–711. doi: 10.1111/j.1398-9995.2007.01578.x. [DOI] [PubMed] [Google Scholar]
- Galli S, Tsai M, Piliponsky A. The development of allergic inflammation. Nature. 2008;454:445–454. doi: 10.1038/nature07204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilfillan A, Tkaczyk C. Integrated signalling pathways for mast-cell activation. Nat Rev Immunol. 2006;6:218–230. doi: 10.1038/nri1782. [DOI] [PubMed] [Google Scholar]
- Heusel J, Wesselschmidt R, Shresta S, Russell J, Ley T. Cytotoxic lymphocytes require granzyme B for the rapid induction of DNA fragmentation and apoptosis in allogeneic target cells. Cell. 1994;76:977–987. doi: 10.1016/0092-8674(94)90376-x. [DOI] [PubMed] [Google Scholar]
- Hirano T, Higa S, Arimitsu J, Naka T, Shima Y, Ohshima S, et al. Flavonoids such as luteolin, fisetin and apigenin are inhibitors of interleukin-4 and interleukin-13 production by activated human basophils. Int Arch Allergy Immunol. 2004;134:135–140. doi: 10.1159/000078498. [DOI] [PubMed] [Google Scholar]
- Inamura N, Mekori Y, Bhattacharyya S, Bianchine P, Metcalfe D. Induction and enhancement of Fc(epsilon)RI-dependent mast cell degranulation following coculture with activated T cells: dependency on ICAM-1- and leukocyte function-associated antigen (LFA)-1-mediated heterotypic aggregation. J Immunol. 1998;160:4026–4033. [PubMed] [Google Scholar]
- Kawai M, Hirano T, Higa S, Arimitsu J, Maruta M, Kuwahara Y, et al. Flavonoids and related compounds as anti-allergic substances. Allergol Int. 2007;56:113–123. doi: 10.2332/allergolint.R-06-135. [DOI] [PubMed] [Google Scholar]
- Kirshenbaum A, Akin C, Wu Y, Rottem M, Goff J, Beaven M, et al. Characterization of novel stem cell factor responsive human mast cell lines LAD 1 and 2 established from a patient with mast cell sarcoma/leukemia; activation following aggregation of FcεR1 or FcγR1. Leuk Res. 2003;27:677–682. doi: 10.1016/s0145-2126(02)00343-0. [DOI] [PubMed] [Google Scholar]
- Kobori M, Nakayama H, Fukushima K, Ohnishi-Kameyama M, Ono H, Fukushima T, et al. Bitter gourd suppresses lipopolysaccharide-induced inflammatory responses. J Agric Food Chem. 2008;56:4004–4011. doi: 10.1021/jf800052y. [DOI] [PubMed] [Google Scholar]
- Krishnaswamy G, Lakshman T, Miller A, Srikanth S, Hall K, Huang S, et al. Multifunctional cytokine expression by human mast cells: regulation by T cell membrane contact and glucocorticoids. J Interferon Cytokine Res. 1997;17:167–176. doi: 10.1089/jir.1997.17.167. [DOI] [PubMed] [Google Scholar]
- Lee S, Fitzgerald S, Huang S, Li C, Chi D, Milhorn D, et al. Molecular regulation of interleukin-13 and monocyte chemoattractant protein-1 expression in human mast cells by interleukin-1beta. Am J Respir Cell Mol Biol. 2004;31:283–291. doi: 10.1165/rcmb.2004-0089OC. [DOI] [PubMed] [Google Scholar]
- Lew M. Good statistical practice in pharmacology. Problem 2. Br J Pharmacol. 2007;152(3):299–303. doi: 10.1038/sj.bjp.0707372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowin B, Hahne M, Mattmann C, Tschopp J. Cytolytic T-cell cytotoxicity is mediated through perforin and Fas lytic pathways. Nature. 1994;370:650–652. doi: 10.1038/370650a0. [DOI] [PubMed] [Google Scholar]
- Marquardt D, Walker L. Dependence of mast cell IgE-mediated cytokine production on nuclear factor-kappaB activity. J Allergy Clin Immunol. 2000;105:500–505. doi: 10.1067/mai.2000.104942. [DOI] [PubMed] [Google Scholar]
- Mekori Y, Metcalfe D. Mast cells in innate immunity. Immunol Rev. 2000;173:131–140. doi: 10.1034/j.1600-065x.2000.917305.x. [DOI] [PubMed] [Google Scholar]
- Melgarejo E, Medina M, Sánchez-Jiménez F, Botana L, Domínguez M, Escribano L, et al. Epigallocatechin-3-gallate interferes with mast cell adhesiveness, migration and its potential to recruit monocytes. Cell Mol Life Sci. 2007;64:2690–2701. doi: 10.1007/s00018-007-7331-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Middleton EJ, Kandaswami C, Theoharides T. The effects of plant flavonoids on mammalian cells: implications for inflammation, heart disease, and cancer. Pharmacol Rev. 2000;52:673–751. [PubMed] [Google Scholar]
- Molin D, Fischer M, Xiang Z, Larsson U, Harvima I, Venge P, et al. Mast cells express functional CD30 ligand and are the predominant CD30L-positive cells in Hodgkin's disease. Br J Haematol. 2001;114:616–623. doi: 10.1046/j.1365-2141.2001.02977.x. [DOI] [PubMed] [Google Scholar]
- Morimoto Y, Yasuhara T, Sugimoto A, Inoue A, Hide I, Akiyama M, et al. Anti-allergic substances contained in the pollen of Cryptomeria japonica possess diverse effects on the degranulation of RBL-2H3 cells. J Pharmacol Sci. 2003;92(3):291–295. doi: 10.1254/jphs.92.291. [DOI] [PubMed] [Google Scholar]
- Moschonas A, Kouraki M, Knox P, Thymiakou E, Kardassis D, Eliopoulos A. CD40 induces antigen transporter and immunoproteasome gene expression in carcinomas via the coordinated action of NF-kappaB and of NF-kappaB-mediated de novo synthesis of IRF-1. Mol Cell Biol. 2008;28:6208–6222. doi: 10.1128/MCB.00611-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakae S, Suto H, Kakurai M, Sedgwick J, Tsai M, Galli S. Mast cells enhance T cell activation: importance of mast cell-derived TNF. Proc Natl Acad Sci USA. 2005;102(18):6467–6472. doi: 10.1073/pnas.0501912102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakae S, Ho LH, Yu M, Monteforte R, Iikura M, Suto H, et al. Mast cell-derived TNF contributes to airway hyperreactivity, inflammation, and TH2 cytokine production in an asthma model in mice. J Allergy Clin Immunol. 2007;120(1):48–55. doi: 10.1016/j.jaci.2007.02.046. [DOI] [PubMed] [Google Scholar]
- Naschberger E, Werner T, Vicente A, Guenzi E, Töpolt K, Leubert R, et al. Nuclear factor-kappaB motif and interferon-alpha-stimulated response element co-operate in the activation of guanylate-binding protein-1 expression by inflammatory cytokines in endothelial cells. Biochem J. 2004;379:409–420. doi: 10.1042/BJ20031873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishimoto H, Lee S, Hong H, Potter K, Maeda-Yamamoto M, Kinoshita T, et al. Costimulation of mast cells by 4-1BB, a member of the tumor necrosis factor receptor superfamily, with the high-affinity IgE receptor. Blood. 2005;106:4241–4248. doi: 10.1182/blood-2005-04-1358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okayama Y, Ra C, Saito H. Role of mast cells in airway remodeling. Curr Opin Immunol. 2007;19:687–693. doi: 10.1016/j.coi.2007.07.018. [DOI] [PubMed] [Google Scholar]
- Palmqvist C, Wardlaw A, Bradding P. Chemokines and their receptors as potential targets for the treatment of asthma. Br J Pharmacol. 2007;151:725–736. doi: 10.1038/sj.bjp.0707263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pardo J, Wallich R, Ebnet K, Iden S, Zentgraf H, Martin P, et al. Granzyme B is expressed in mouse mast cells in vivo and in vitro and causes delayed cell death independent of perforin. Cell Death Differ. 2007;14:1768–1779. doi: 10.1038/sj.cdd.4402183. [DOI] [PubMed] [Google Scholar]
- Park H, Lee S, Oh J, Lee M, Yoon K, Park B, et al. Anti-inflammatory activity of fisetin in human mast cells (HMC-1) Pharmacol Res. 2007;55:31–37. doi: 10.1016/j.phrs.2006.10.002. [DOI] [PubMed] [Google Scholar]
- Park H, Lee S, Son H, Park S, Kim M, Choi E, et al. Flavonoids inhibit histamine release and expression of proinflammatory cytokines in mast cells. Arch Pharm Res. 2008;31(10):1303–1311. doi: 10.1007/s12272-001-2110-5. [DOI] [PubMed] [Google Scholar]
- Peng Y, Power M, Li B, Lin T. Inhibition of IKK down-regulates antigen + IgE-induced TNF production by mast cells: a role for the IKK-IkappaB-NF-kappaB pathway in IgE-dependent mast cell activation. J Leukoc Biol. 2005;77:975–983. doi: 10.1189/jlb.0204115. [DOI] [PubMed] [Google Scholar]
- Qin H, Wilson C, Lee S, Zhao X, Benveniste E. LPS induces CD40 gene expression through the activation of NF-kappaB and STAT-1alpha in macrophages and microglia. Blood. 2005;106:3114–3122. doi: 10.1182/blood-2005-02-0759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renshaw B, Fanslow W, Armitage R, Campbell K, Liggitt D, Wright B, et al. Humoral immune responses in CD40 ligand-deficient mice. J Exp Med. 1994;180:1889–1900. doi: 10.1084/jem.180.5.1889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ridge J, Di Rosa F, Matzinger P. A conditioned dendritic cell can be a temporal bridge between a CD4+ T-helper and a T-killer cell. Nature. 1998;393:474–478. doi: 10.1038/30989. [DOI] [PubMed] [Google Scholar]
- Salamon P, Shoham N, Gavrieli R, Wolach B, Mekori Y. Human mast cells release Interleukin-8 and induce neutrophil chemotaxis on contact with activated T cells. Allergy. 2005;60:1316–1319. doi: 10.1111/j.1398-9995.2005.00886.x. [DOI] [PubMed] [Google Scholar]
- Salamon P, Shoham N, Puxeddu I, Paitan Y, Levi-Schaffer F, Mekori Y. Human mast cells release oncostatin M on contact with activated T cells: possible biologic relevance. J Allergy Clin Immunol. 2008;121:448–455. e445. doi: 10.1016/j.jaci.2007.08.054. [DOI] [PubMed] [Google Scholar]
- Schoenberger S, Toes R, van der Voort E, Offringa R, Melief C. T-cell help for cytotoxic T lymphocytes is mediated by CD40-CD40L interactions. Nature. 1998;393:480–483. doi: 10.1038/31002. [DOI] [PubMed] [Google Scholar]
- Secor V, Secor W, Gutekunst C, Brown M. Mast cells are essential for early onset and severe disease in a murine model of multiple sclerosis. J Exp Med. 2000;191(5):813–822. doi: 10.1084/jem.191.5.813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimada Y, Hasegawa M, Kaburagi Y, Hamaguchi Y, Komura K, Saito E, et al. L-selectin or ICAM-1 deficiency reduces an immediate-type hypersensitivity response by preventing mast cell recruitment in repeated elicitation of contact hypersensitivity. J Immunol. 2003;170:4325–4334. doi: 10.4049/jimmunol.170.8.4325. [DOI] [PubMed] [Google Scholar]
- Smith T, Weis J. Mucosal T cells and mast cells share common adhesion receptors. Immunol Today. 1996;17:60–63. doi: 10.1016/0167-5699(96)80580-9. [DOI] [PubMed] [Google Scholar]
- Stopfer P, Männel D, Hehlgans T. Lymphotoxin-beta receptor activation by activated T cells induces cytokine release from mouse bone marrow-derived mast cells. J Immunol. 2004;172:7459–7465. doi: 10.4049/jimmunol.172.12.7459. [DOI] [PubMed] [Google Scholar]
- Strik M, de Koning P, Kleijmeer M, Bladergroen B, Wolbink A, Griffith J, et al. Human mast cells produce and release the cytotoxic lymphocyte associated protease granzyme B upon activation. Mol Immunol. 2007;44:3462–3472. doi: 10.1016/j.molimm.2007.03.024. [DOI] [PubMed] [Google Scholar]
- Tsang C, Wong C, Ip W, Lam C. Synergistic effect of SCF and TNF-alpha on the up-regulation of cell-surface expression of ICAM-1 on human leukemic mast cell line (HMC)-1 cells. J Leukoc Biol. 2005;78:239–247. doi: 10.1189/jlb.0704400. [DOI] [PubMed] [Google Scholar]
- Tschopp C, Spiegl N, Didichenko S, Lutmann W, Julius P, Virchow J. Granzyme B, a novel mediator of allergic inflammation: its induction and release in blood basophils and human asthma. Blood. 2006;108:2290–2299. doi: 10.1182/blood-2006-03-010348. [DOI] [PubMed] [Google Scholar]
- Williams C, Grayer R. Anthocyanins and other flavonoids. Nat Prod Rep. 2004;21:539–573. doi: 10.1039/b311404j. [DOI] [PubMed] [Google Scholar]
- Wong C, Tsang C, Ip W, Lam C. Molecular mechanisms for the release of chemokines from human leukemic mast cell line (HMC)-1 cells activated by SCF and TNF-alpha: roles of ERK, p38 MAPK, and NF-kappaB. Allergy. 2006;61:289–297. doi: 10.1111/j.1398-9995.2006.00972.x. [DOI] [PubMed] [Google Scholar]