

Abstract
Background and purpose:
Reduction of intracellular calcium ([Ca2+]i) in smooth muscle cells (SMCs) is an important mechanism by which nitric oxide (NO) dilates blood vessels. We investigated whether modes of Ca2+ mobilization during SMC contraction influenced NO efficacy.
Experimental approach:
Isometric contractions by depolarization (high potassium, K+) or α-adrenoceptor stimulation (phenylephrine), and relaxations by acetylcholine chloride (ACh), diethylamine NONOate (DEANO) and glyceryl trinitrate (GTN) and SMC [Ca2+]i (Fura-2) were measured in aortic segments from C57Bl6 mice.
Key results:
Phenylephrine-constricted segments were more sensitive to endothelium-derived (ACh) or exogenous (DEANO, GTN) NO than segments contracted by high K+ solutions. The greater sensitivity of phenylephrine-stimulated segments was independent of the amount of pre-contraction, the source of NO or the resting potential of SMCs. It coincided with a significant decrease of [Ca2+]i, which was suppressed by sarcoplasmic reticulum (SR) Ca2+ ATPase (SERCA) inhibition, but not by soluble guanylyl cylase (sGC) inhibition. Relaxation of K+-stimulated segments did not parallel a decline of [Ca2+]i. However, stimulation (BAY K8644) of L-type Ca2+ influx diminished, while inhibition (nifedipine, 1–100 nM) augmented the relaxing capacity of NO.
Conclusions and implications:
In mouse aorta, NO induced relaxation via two pathways. One mechanism involved a non-cGMP-dependent stimulation of SERCA, causing Ca2+ re-uptake into the SR and was prominent when intracellular Ca2+ was mobilized. The other involved sGC-stimulated cGMP formation, causing relaxation without changing [Ca2+]i, presumably by desensitizing the contractile apparatus. This pathway seems related to L-type Ca2+ influx, and L-type Ca2+ channel blockers increase the vasodilator efficacy of NO.
Keywords: vasodilatation, nitric oxide, intracellular calcium, smooth muscle cell, mouse aorta, pre-contraction, C57Bl6, SERCA, nifedipine
Introduction
Constriction and dilation of blood vessels in response to environmental demands are controlled by changes in the cytosolic Ca2+ levels ([Ca2+]i) in the vascular smooth muscle cell (SMC). In response to mechanical factors or vasoactive agonists, vessels constrict by the increase of SMC [Ca2+]i either through Ca2+ influx via Ca2+-permeable ion channels and/or Ca2+ release from sarcoplasmic reticulum (SR) Ca2+ stores. Calmodulin interacts with the released [Ca2+]i which finally leads to phosphorylation of myosin and subsequent muscle contraction. Dilation, on the other hand, is often initiated in the endothelial cells with the stimulation of Ca2+-dependent endothelial nitric oxide synthase (eNOS) and release of nitric oxide (NO). The released NO stimulates SMCs to relax either directly and/or indirectly by removal of the elevated [Ca2+]i. NO can also cause dilation by decreasing the sensitivity of the contractile apparatus for [Ca2+]i (Akata, 2007a,b;). Indirect effects of NO involve the activation of soluble guanylyl cyclase (sGC) to produce cGMP, which in turn affects Ca2+ influx and/or efflux; or leads to decreased sensitivity of the contractile apparatus to intracellular Ca2+ via stimulation of myosin phosphatase and the dephosphorylation of myosin (Carvajal et al., 2000). Direct effects of NO include inhibition of SMC Ca2+ release elicited by inositol trisphosphate (IP3) (Ji et al., 1998), cADP-ribose (Yu et al., 2000) or caffeine (Li et al., 2000); acceleration of Ca2+ sequestration into intracellular stores via stimulation of the sarco-endoplasmic reticulum Ca2+ ATPase (SERCA) (Cohen et al., 1999; Adachi et al., 2004), and activation of K+ channels (Bolotina et al., 1994; Quignard et al., 2000).
However, the question remains whether relaxation is dependent on the mechanisms involved in the preceding contraction. The direct and indirect effects of NO on [Ca2+]i in SMCs at least suggest that the efficacy of NO to cause relaxation depends on the ability of the SMC to reduce the elevated [Ca2+]i. This ability differs according to the type of blood vessel (Asano and Nomura, 1999; 2000; Nomura and Asano, 2000), its (patho)physiological condition (atherosclerosis, diabetes, hypertension, etc.) (Lewis et al., 2005; Van Assche et al., 2007) and the type of SMC pre-activation (Plane et al., 1998; Lehen'kyi et al., 2002). As a consequence, the efficacy of NO to cause relaxation is expected to depend upon the specific mechanisms by which Ca2+ is mobilized during contraction.
In order to investigate whether the mechanisms of Ca2+ mobilization during the contraction determine the relaxing capacity of NO, contractions of mouse aortic segments were initiated by increasing [Ca2+]i by two widely used and different stimuli. On the one hand, elevated external K+ depolarises the SMC and activates Ca2+ influx via L-type Ca2+ channels. Both processes, i.e. membrane depolarization and activation of L-type Ca2+ channels, have been described to be involved in blood pressure regulation (Moosmang et al., 2003) and hypertension (Sonkusare et al., 2006). On the other hand, α1-adrenoceptor stimulation with phenylephrine causes intracellular Ca2+ release from SR Ca2+ stores triggered by IP3 receptors (Karaki et al., 1997). Yet, even those contractions are associated with depolarization and opening of L-type Ca2+ channels (Plane et al., 1998; Quignard et al., 2000; Richards et al., 2001; Akata, 2007a). Following high-K+ or -phenylephrine contractions, the relaxing capacity of NO was investigated by studying relaxation by endogenous NO evoked by administration of acetylcholine chloride (ACh), or by sources of exogenous NO, that is diethylamine NONOate (DEANO) or glyceryl trinitrate (GTN), and by modulating Ca2+ influx during depolarization or Ca2+ release from internal stores during agonist activation.
Methods
Aortic segments
The studies were approved by the Ethical Committee of the University of Antwerp, and the investigations conform to the Guide for the Care and Use of Laboratory Animals, published by the US National Institutes of Health (NIH Publication No. 85-23, revised 1996). C57Bl6 mice (n= 68, food and water ad libitum, 12/12 light–dark cycle) were used at the age of 4–5 months. After anaesthesia (sodium pentobarbital, 75 mg·kg−1, i.p.), the thoracic aorta was carefully removed, stripped of adherent tissue and dissected systematically. Starting 3 mm from the origin of the left subclavian artery (where the azygos vein crosses the aorta) down to the diaphragm, the descending thoracic aorta was cut in segments of 2 mm width. Vessels were immersed in Krebs Ringer solution (KR solution, 37°C, 95% O2/5% CO2, pH 7.4) with (in mM): NaCl, 118; KCl, 4.7; CaCl2, 2.5, KH2PO4, 1.2; MgSO4, 1.2; NaHCO3 25, CaEDTA 0.025; and glucose 11.1. High-K+ solution was prepared as KR solution, but 44.1 mM NaCl was replaced with KCl. In all experiments, indomethacin (10 µM) was present to avoid any vasomotor interference caused by prostanoids (Okon et al., 2002).
Isometric tension measurements
Aortic segments were mounted between two parallel tungsten wire hooks in 10 mL organ baths. Tension was measured isometrically with a Statham UC2 force transducer (Gould, Cleveland, OH, USA) connected to a data acquisition system (Powerlab 8/30, ADInstruments, Spechbach, Germany), and contractile force was reported in mN. Segments were gradually stretched until a stable loading tension of 16 mN was attained (optimal pre-load to attain maximal force development by phenylephrine or high K+). Endogenous NO release was induced by the cumulative administration of ACh (10−9 to 10−5 M). Relaxation experiments with exogenous NO (from DEANO or GTN, both 10−9 to 10−5 M) or with the membrane-permeable analogue of cGMP (8-Br-cGMP, 10−9 to 10−5 M) were performed in segments in which NO formation was inhibited with a combination of 300 µM NΩ-nitro-l-arginine methyl ester (l-NAME) and 300 µM NΩ-nitro-l-arginine (l-NNA).
Combined assay of isometric tension and SMC Ca2+
In order to avoid the interference of endothelial Ca2+ signals with the SMC cytosolic Ca2+ assay, the endothelium was mechanically removed by rubbing the interior of the segment with braided silk thread. The lack of a raised intracellular Ca2+ and concomitant relaxation of pre-activated segments, when stimulated with 1 µM ACh, was used as a test for successful removal of endothelial cells. Endothelium-denuded segments were mounted in a wire (40 µm) myograph (Danish Myotechnology A/S, Aarhus, Denmark) above an inverted microscope (Axiovert 200, Carl Zeiss, Zaventem, Belgium) immersed in Krebs–Ringer solution (37°C) and continuously aerated with 95% O2/5% CO2 (pH 7.4). The segment was loaded in situ with 10 µM Fura-2 AM in Krebs–Ringer solution with 0.02% pluronic for 120 min at room temperature. Then, the temperature was increased to 37°C, and the segment was set to its normalized diameter (Mulvany and Halpern, 1976). In short, segments were stretched gradually until the calculated internal diameter reached a value equal to the internal circumference the vessel would have in vivo, when fully relaxed and under a transmural pressure of 100 mm Hg. Before starting an experiment, the horizontal and vertical apertures of the microscope photometer were set on the surface of the strip (covering about 10% of the surface area, 40× objective). Excitation wavelengths (340 and 380 nm for Fura-2) were delivered at a frequency of 1 Hz with a DeltaRAM Multiwavelength Illuminator (Photon Technology International (PTI), Birmingham, NJ, USA). Emission (dichroic mirror, emission filter PO# OROM9712127, WO#A018983, Omega Optical, Inc., Brattleboro, VT, USA) was measured with a microscope photometer (D-104, PTI). The single emission (510 nm) ratio at dual excitation (340 and 380 nm) was used as a relative measure of free [Ca2+]i (relative units) and was analysed with Felix software (PTI). At the end of the experiment, 2 mM MnCl2 was added to the segment to determine background emission values, which were subtracted from the respective emission values during the experiment. Absolute levels of [Ca2+]i were not reported due to the uncertainty of the conventional calibration method in intact tissues (Cohen et al., 1999). Contractile force was measured simultaneously and reported in mN·mm−1.
Data analysis
All results are expressed as mean ± SEM; n represents the number of mice, and s the number of segments. Dose–response curves were fitted with sigmoidal dose–response equations with variable slope, which revealed maximal relaxation responses (Emax) and the negative logarithm of the concentration resulting in 50% of the maximal contraction or relaxation (pEC50) for each vessel segment. Two-way analysis of variance with Bonferroni's post hoc test and unpaired Student's t-test (GraphPad Prism, version 5, GraphPad Software, San Diego, CA, USA) were used to compare means of the different experimental groups. When different segments of the same aorta underwent the same protocol, the average of the different segments was used for statistical comparison between different animals. A 5% level of significance was selected.
Materials
Sodium pentobarbital (Nembutal) was obtained from Sanofi (Brussels, Belgium); indomethacin from Federa (Brussels, Belgium); phenylephrine hydrochloride, ACh, l-NNA, l-NAME, DEANO, cyclopiazonic acid (CPA), nifedipine from Sigma (Bornem, Belgium); Fura 2-AM from Molecular Probes (Invitrogen, Merelbeke, Belgium); GTN from Merck VWR (Leuven, Belgium); verapamil hydrochloride, (±) BAY K8644, 8-Br-cGMP sodium salt from TOCRIS (Bristol, UK); and 1H-[1,2,4]oxadiazolo[4,3-a]quinoxalin-1-one (ODQ) from ASCENT (Weston-Super-Mare, UK).
Results
The relaxing capacity of NO in segments contracted by phenylephrine or high K+
In order to study the relaxing capacity of endogenous NO, the aortic segments were maximally contracted with 50 mM K+ or 3 µM phenylephrine followed by the addition of 10 µM ACh (Figure 1). Relaxations to ACh were complete in segments contracted by 3 µM phenylephrine, but were significantly attenuated in those contracted with 50 mM K+. Moreover, the sensitivity of aortic segments to ACh was significantly lower when the contraction was elicited by high K+ (Figure 1C). When contractions were reduced by lowering the concentration of K+ or phenylephrine, the relaxing capacity of NO was still significantly lower in segments contracted with K+ compared with those contracted with phenylephrine, although both elicited identical force (Figure 2). Similar results were obtained with exogenous NO (10 µM DEANO or 10 µM GTN) in the presence of NOS inhibitors (Figure 1, Table 1); in that condition, application of 10 µM ACh did not evoke relaxation (Figure 1).
Figure 1.
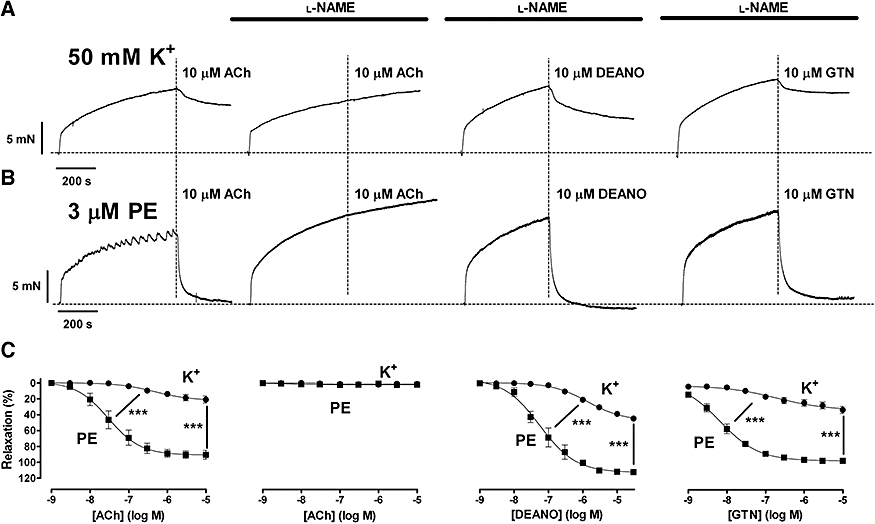
Representative examples of contractions elicited by 50 mM K+ (A) and 3 µM phenylephrine (PE) (B) followed by relaxations with 10 µM acetylcholine chloride (ACh) in the absence of l-NAME/l-NNA, and with 10 µM ACh, 10 µM diethylamine NONOate (DEANO) or 10 µM glyceryl trinitrate (GTN) in the presence of 300 µM l-NAME/l-NNA. (C) Concentration–response relationships for ACh (n= 5) in the absence, and ACh (n= 3), DEANO (n= 7) and GTN (n= 7) in the presence of l-NAME/l-NNA of contractions by 50 mM K+ and 3 µM phenylephrine. Results show mean ± SEM and were obtained in endothelium-intact segments. ***P < 0.001 K+ versus phenylephrine (one-way analysis of variance).
Figure 2.
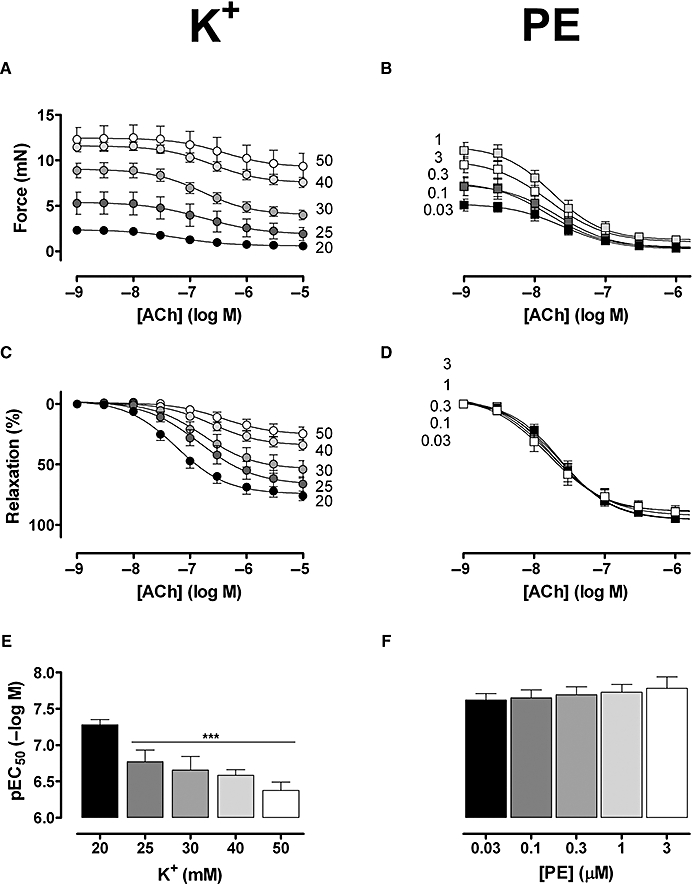
Absolute (A,B) and relative (C,D) force of segments stimulated with 20, 25, 30, 40 and 50 mM K+ (A,C), or 0.03, 0.1, 0.3, 0.3, 3 µM phenylephrine (PE) (B,D) followed by addition of acetylcholine chloride (ACh) (1 × 10−9 to 1 × 10−5 M). (E,F) pEC50 of ACh for contractions induced by increasing [K+] (E) or [phenylephrine] (F). Results show mean ± SEM, n= 6 endothelium-intact segments; ***P < 0.001 phenylephrine versus K+.
Table 1.
Tension induced by 0.3 or 3 µM phenylephrine (PE), and by 25 or 50 mM K+, and respective Emax and pEC50 of relaxations by DEANO
| Contraction constrictor | Tension (mN) | Relaxation Emax (%) | pEC50 (−logM) | |
|---|---|---|---|---|
| PE (µM) | 0.3 | 9.8 ± 0.5 | 103 ± 6 | 7.31 ± 0.15 |
| K+ (mM) | 25 | 10.0 ± 1.0 | 91 ± 6 | 6.41 ± 0.10b |
| PE (µM) | 3.0 | 12.7 ± 1.4a | 104 ± 2 | 7.14 ± 0.17 |
| K+ (mM) | 50 | 17.4 ± 1.3a,b | 54 ± 3a,b | 6.01 ± 0.08a,b |
Results show mean ± SEM, n= 5–7 endothelium-intact segments.
P < 0.001 high versus low concentration of constrictor.
P < 0.001 K+ versus phenylephrine.
DEANO, diethylamine NONOate.
Results indicated that the different relaxing capacity of NO for high K+- or phenylephrine-induced contractions was not dependent on the source of NO, but was dependent on the agonist causing contraction. Hence, all following experiments were performed in segments in which endogenous NO release was inhibited with l-NAME/l-NNA, using the NO donor DEANO.
The vasodilator efficacy of NO was dependent on the amount of pre-contraction for K+, but not for phenylephrine.
Relaxing capacity of NO at depolarized membrane potentials
In order to test whether relaxations induced by NO were affected by the membrane potential of SMCs, segments were contracted by 3 µM phenylephrine in the presence of 50 mM K+, but the depolarization-dependent Ca2+ influx and concomitant contractions were prevented by inhibition of L-type Ca2+ channels with 1 µM nifedipine. As shown in Figure 3B, nifedipine completely prevented force development by 50 mM K+, and inhibited the slow phase of force development by phenylephrine by 70%, from 12.5 ± 0.8 mN to 3.80 ± 1.1 mN (n= 4). However, maximal relaxation and pEC50 of DEANO were not affected by 50 mM K+ plus nifedipine (Figure 3C). These results indicated that the resting membrane potential of the SMC did not determine the relaxing capacity of NO.
Figure 3.
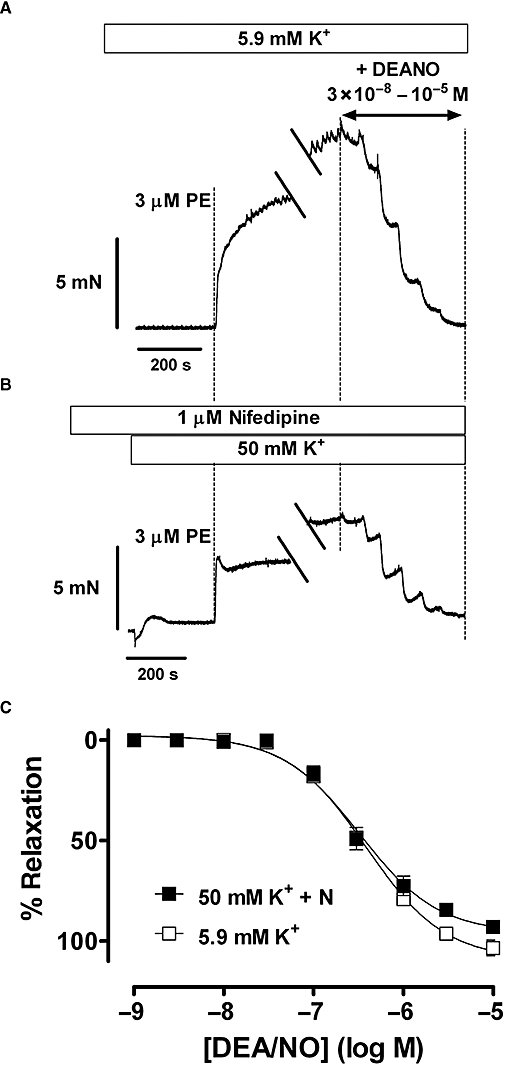
(A) Representative examples of contractions elicited by 3 µM phenylephrine (PE) at normal resting potential (5.9 mM K+), and (B) following depolarization of the membrane potential with 50 mM K+. Contraction by 50 mM K+ was inhibited by pre-incubation of the segment with 1 µM nifedipine (N). When contractions reached a stable level (breaks in tracings), this was followed by the addition of increasing concentrations of diethylamine NONOate (DEANO). (C) Concentration–response relationships for DEANO of contractions by 3 µM phenylephrine at normal resting potential (5.9 mM K+) and depolarized potentials (50 mM K++ N). Relaxation by DEANO amounted to 107 ± 4% with pEC50 of 6.45 ± 0.02 at normal resting potential, whereas at depolarized potentials the maximal relaxation was 98 ± 2% with pEC50 of 6.51 ± 0.10 (n= 4, endothelium-intact segments).
Relaxing capacity of NO with activation of L-type Ca2+ influx
To increase Ca2+ influx during contraction by 3 µM phenylephrine or moderate depolarization with 20 mM K+, segments were incubated with 30 nM BAY K8644, an activator of L-type Ca2+ channels. BAY K8644 slightly raised basal tension and significantly increased the contraction by 20 mM K+, without affecting the phenylephrine contraction (Figure 4A,B). Incubation with BAY K8644 shifted the dose–response curves of DEANO significantly to the right in both situations (Figure 4C,D), and this shift could be reversed by nifedipine (data not shown).
Figure 4.
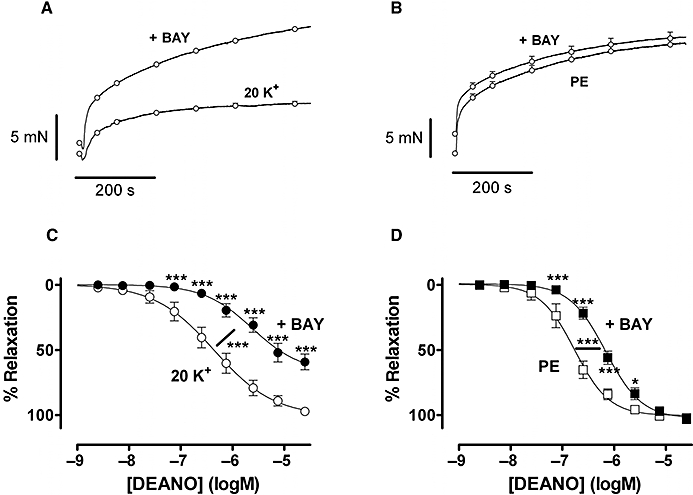
Mean levels of contraction evoked by 20 mM K+ (A) (n= 8) and 3 µM phenylephrine (PE) (B) (n= 7) in the absence and presence of 30 nM BAY K8644 (+Bay). Relaxation by diethylamine NONOate (DEANO) in segments constricted by 20 mM K+ (C) and 3 µM phenylephrine (D) in the absence and presence of 30 nM BAY K8644. All segments were endothelium intact. *P < 0.05, ***P < 0.001 BAY K8644 versus control, repeated measurements analysis of variance.
Relaxing capacity of NO with inhibition of L-type Ca2+ influx
In the next series of experiments, relaxation of contractions by high K+ and phenylephrine was measured following inhibition of Ca2+ influx via L-type Ca2+ channels with nifedipine (Figure 5). Nifedipine (1–30 nM) caused dose-dependent inhibition of the contraction by high K+, but augmented the relaxing capacity of DEANO (Figure 5A,C,E). Similar results were obtained with another L-type Ca2+ channel inhibitor verapamil (10–300 nM, n= 3). Although nifedipine (Figure 5B) and verapamil (data not shown) also attenuated contractions elicited with 3 µM phenylephrine, they did not affect relaxation induced by DEANO (Figure 5B,D,F). Similar results were obtained with another NO donor GTN (n= 3) (data not shown).
Figure 5.
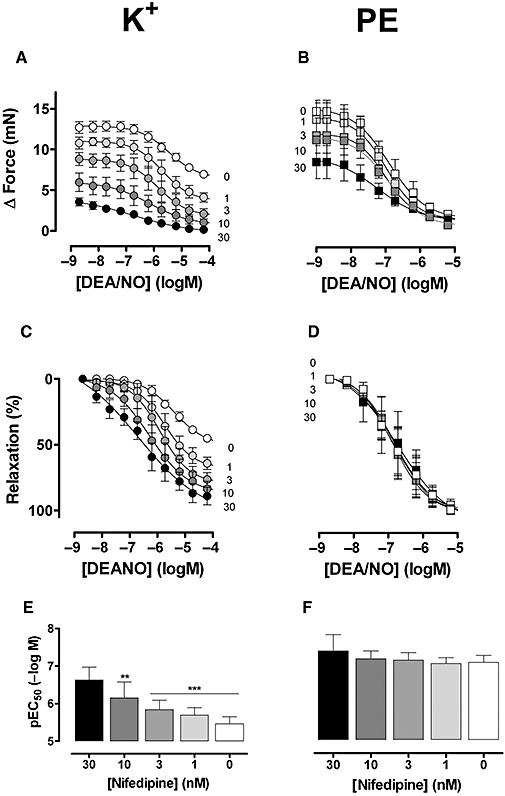
Absolute (A,B) and relative (C,D) force of segments stimulated with 50 mM K+ (A,C) or 3 µM phenylephrine (PE) (B,D) followed by addition of diethylamine NONOate (DEANO) in the absence (C: open symbols) and presence of increasing concentrations of nifedipine (1, 3, 10, 30 and 100 nM). (E) pEC50 of DEANO for contractions induced by 50 mM K+ or 3 µM phenylephrine in the absence (C) and presence of increasing nifedipine concentrations (1, 3, 10, 30 and 100 nM). Results show mean ± SEM, n= 4 endothelium-intact segments. **P < 0.05 and ***P < 0.001 phenylephrine versus K+.
NO and Ca2+ uptake by SERCA
When SERCA was inhibited with 10 µM CPA, force development by 50 mM K+ decreased from 12.9 ± 0.6 mN to 9.5 ± 0.3 mN (−25%, n= 7), whereas force developed by phenylephrine was not affected (from 10.1 ± 1.6 mN to 11.4 ± 1.2 mN; +9%, n= 6). Relaxation of the high K+ contraction by DEANO was not affected by CPA (pEC50 6.30 ± 0.07 and 6.23 ± 0.14, respectively, P > 0.05), but was significantly shifted to the right (pEC50 from 7.01 ± 0.06 to 6.54 ± 0.17, P < 0.05, n= 6) for the phenylephrine contraction without significant change in the amplitude of relaxation (Emax of 110 ± 4 and 104 ± 3%, respectively). These observations suggest that SERCA-mediated re-uptake of Ca2+ into the SR is involved in the NO-mediated relaxation of contractions elicited by phenylephrine, but probably not involved when contractions are induced by high K+.
NO-induced decrease of intracellular Ca2+
Stimulated Ca2+ uptake to the SR by NO was further studied in experiments in which [Ca2+]i was measured in segments contracted with 50 mM K+ or 3 µM phenylephrine, followed by relaxation with 10 µM DEANO in the absence or presence of CPA to inhibit SERCA-mediated Ca2+ uptake. For contractions induced by phenylephrine, relaxation by DEANO was accompanied by a significant (about 70%) decrease of Ca2+ mobilized during contraction. NO-induced relaxation was not accompanied by a Ca2+ decrease when segments were contracted by 50 mM K+ or when segments were contracted by 3 µM phenylephrine in the presence of CPA (Figure 6).
Figure 6.
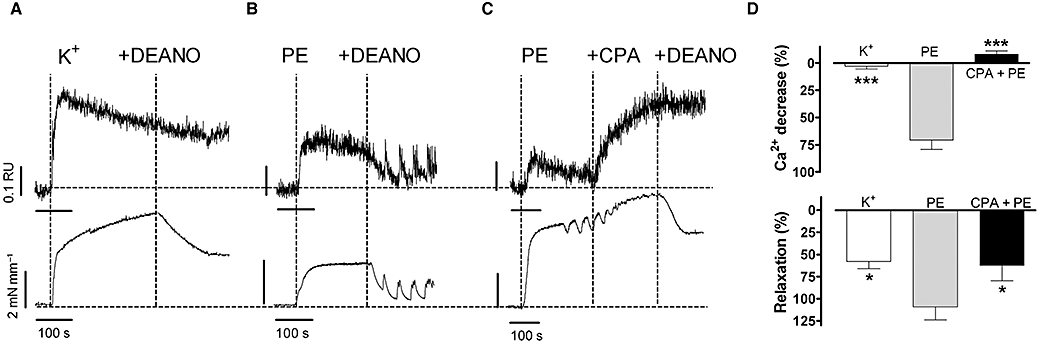
Representative examples of Ca2+ signals (upper tracings) and force signals (bottom tracings) for contractions by (A) 50 mM K+, (B) 3 µM phenylephrine (PE) and (C) 3 µM phenylephrine after SERCA inhibition with 10 µM cyclopiazonic acid (CPA) and following addition of 10 µM diethylamine NONOate (DEANO). (D) Summary of A–C showing % decrease of Ca2+ and % relaxation following addition of 10 µM DEANO (mean ± SEM, n= 4 endothelium-denuded segments). *P < 0.05; ***P < 0.001 versus phenylephrine.
cGMP-mediated relaxations of contractions by phenylephrine and high K+
Relaxation induced by the membrane-permeable analogue of cGMP (8-Br-cGMP) was compared with responses to DEANO. Maximal relaxations by DEANO and 8-Br-cGMP of contractions induced by 50 mM K+ or 3 µM phenylephrine were not significantly different (Table 2). Segments contracted with phenylephrine were significantly more sensitive to DEANO than segments contracted with K+ (ΔpEC50= 1.7 – logM), but the difference in sensitivity was far less pronounced for 8-Br-cGMP (ΔpEC50= 0.29 – logM). These results indicate that the difference in the relaxing capacity of NO for K+ or phenylephrine contractions is mainly situated at the level of direct effects of NO and not at the level of NO-mediated increase of cGMP or the subsequent steps.
Table 2.
Emax (%) and pEC50 for relaxations by DEANO and 8-Br-cGMP of contractions induced by 50 mM K+ and 3 µM phenylephrine (PE)
| DEANO | 8-Br-cGMP | ||
|---|---|---|---|
| Emax (%) | K+ | 54 ± 3 | 47 ± 13 |
| PE | 105 ± 2*** | 102 ± 6*** | |
| pEC50 (–logM) | K+ | 6.04 ± 0.08 | 4.36 ± 0.29 |
| PE | 6.68 ± 0.16*** | 4.65 ± 0.08 |
Results show mean ± SEM, n= 5 endothelium-intact segments.
P < 0.001 phenylephrine versus K+.
DEANO, diethylamine NONOate.
Incubation of aortic segments with 10 µM ODQ, an inhibitor of sGC, completely abolished relaxation to DEANO in K+-constricted segments, and shifted relaxation curves for contractions by phenylephrine to the right (Figure 7). However, for phenylephrine, more than 70% (76 ± 4%) of the maximum DEANO response could still be attained in a cGMP-independent manner.
Figure 7.
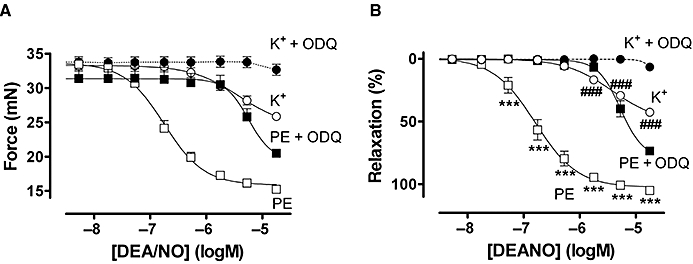
Relaxation expressed as absolute force (A) or % relaxation (B) by diethylamine NONOate (DEANO) of contractions elicited by 50 mM K+ and 3 µM phenylephrine (PE) in the absence and presence of 10 µM ODQ. Results were obtained in endothelium-intact segments and show mean ± SEM of the average of three segments per mouse (n= 4). ***P < 0.001 versus phenylephrine + ODQ, ###P < 0.001 versus K++ ODQ.
As shown in Figure 8, sGC inhibition by ODQ did not affect the decrease of cytosolic Ca2+ or the concomitant relaxation upon addition of DEANO, suggesting that Ca2+ uptake into the SR was directly stimulated by exogenous NO and not dependent on sGC stimulation by NO. Finally, in the presence of ODQ (10 µM) and CPA (10 µM), relaxation of the phenylephrine-induced contraction by NO was also completely absent (data not shown).
Figure 8.
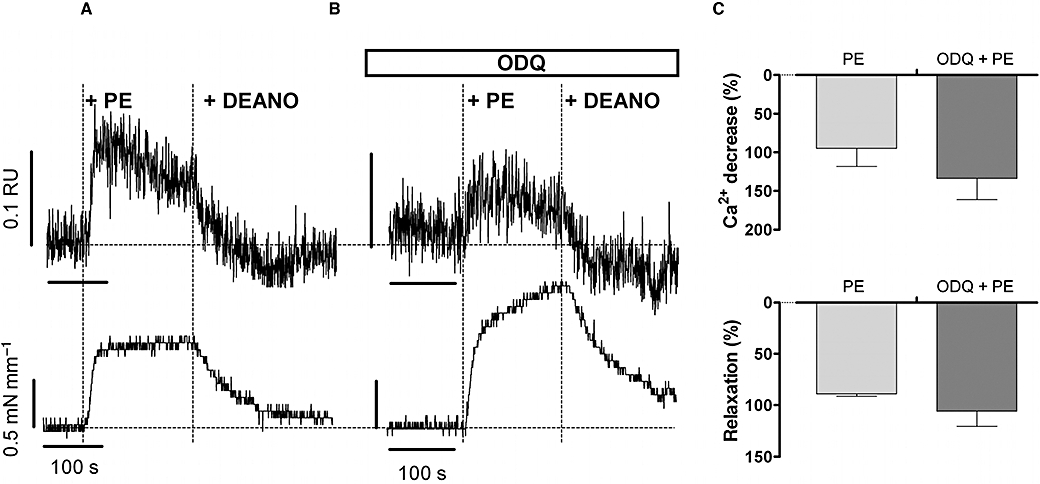
Representative example (n= 4) of Ca2+ signals (upper panels) and force (lower panels) induced by 3 µM phenylephrine (PE) in the absence (A) and presence (B) of 10 µM ODQ followed by application of 10 µM diethylamine NONOate (DEANO). In two segments, ODQ raised baseline Ca2+ and tone, but DEANO evoked a similar decline of phenylephrine-induced Ca2+ and tension. (C) Summary of % decrease of Ca2+ and % relaxation following addition of 10 µM DEANO. Results show mean ± SEM, n= 4 endothelium-denuded segments.
Discussion
The agonist causing contraction determines the vasodilator efficacy of NO
In the present study, vasodilatation evoked by endogenous (ACh) or exogenous (DEANO, GTN) NO was severely attenuated when mouse aortic segments were pre-activated with high K+ rather than with phenylephrine. A further difference was that the vasodilator capacity of NO was equal for different concentrations (and thus different levels of contraction) of phenylephrine, whereas for K+ it was strongly dependent on the concentration used to evoke contraction. Moreover, when K+ and phenylephrine (Figures 2 and 4; Table 1) evoked identical contraction levels, the vasodilator capacity of NO remained different, suggesting that the mechanisms of Ca2+ mobilization during contraction determine the vasodilator efficacy of NO.
Because elevation of external K+ causes depolarization of the membrane potential of endothelial cells and SMCs, one could argue that the attenuated relaxation of endogenous NO at high K+ in comparison with phenylephrine is caused by diminished NO release by the depolarized endothelium and/or by lower NO efficiency in depolarized SMCs. However, the difference was also seen using sources of exogenous NO in the presence of NOS inhibitors. Furthermore, SMC depolarization as the factor underlying the low efficacy of NO was also ruled out. This is because the capacity of NO to reverse phenylephrine-induced contractions was identical in SMCs at normal resting membrane potentials and in depolarized SMCs, at least when, in the latter condition, Ca2+ influx via L-type Ca2+ channels was prevented with nifedipine (see also Scarborough and Carrier, 1984). That the relaxing capacity of NO for phenylephrine contractions was not attenuated at elevated external K+ further indicates that direct activation of Ca2+-dependent K+ channels by NO, as observed in rabbit aorta (Bolotina et al., 1994; Quignard et al., 2000), is not the factor underlying the greater efficacy of NO in phenylephrine-constricted segments.
The different ACh-induced relaxation profile of K+- or phenylephrine-constricted segments cannot be due to altered release of NO in both conditions, because exogenous NO also showed a low efficacy in K+-constricted segments. Furthermore, it is important to note that relaxation by ACh in the mouse aorta was absent with NOS inhibition, thus confirming that ACh-induced relaxation is exclusively mediated by NO, without participation of endothelium-derived hyperpolarizing factor (EDHF) or prostacyclin as previously observed by Crauwels et al. (2000; 2003;). Therefore, the low efficacy of ACh in relaxing K+-constricted rings cannot be due to inhibition of an EDHF-induced relaxation as observed in human small arteries (Buus et al., 2006). Also, altered prostanoid release (Okon et al., 2002) at depolarized potentials (Thapaliya et al., 2000) could be excluded as the underlying factor, because all experiments were performed in the presence of 10 µM indomethacin. As a consequence, the greater efficacy of NO to dilate phenylephrine- compared with high K+-constricted aortic segments can only be attributed to different mechanisms of contraction.
Mechanisms of contractions by high K+ or phenylephrine determine the NO-mediated relaxation
NO-evoked relaxation is mediated by cGMP-dependent and by cGMP-independent pathways (Bolotina et al., 1994; Cohen et al., 1999; Nimmegeers et al., 2007). The cGMP-dependent mechanisms involve reduction in [Ca2+]i and/or a decrease in the sensitivity of the contractile system to Ca2+ (Carvajal et al., 2000). In the present study, relaxation of constrictions initiated by L-type Ca2+ channel activation at high K+ had similar amplitudes when induced by NO or by the membrane-permeable analogue of cGMP. These effects were abolished by the sGC inhibitor ODQ, were not influenced by the SERCA inhibitor CPA and were not associated with a decrease of cytosolic Ca2+. These results indicated that NO-induced relaxation of high K+-constricted segments occurred mainly through activation of sGC and subsequent cGMP-mediated effects, which were independent of [Ca2+]i levels. However, when the initial [Ca2+]i was recruited from inside the SMC, as with phenylephrine, the NO-induced relaxation was paralleled by a time-matched decrease of [Ca2+]i, which was about 70% of the [Ca2+]i mobilized during the preceding contraction. The NO-induced [Ca2+]i decrease was cGMP independent, as it was not affected by ODQ and it was accompanied by 70% or more of the relaxation response observed in the absence of ODQ. Both the [Ca2+]i decline and the relaxation were CPA sensitive, suggesting an important role for Ca2+ uptake into the SR by SERCA (Cohen et al., 1999; Adachi et al., 2004). Therefore, these results indicated that NO-induced relaxation of phenylephrine-mediated constrictions occurred via two pathways: about 70% of the maximum response occurred via a cGMP-independent reduction of [Ca2+]i, and 30% of the response occurred via NO-stimulated cGMP formation, without reduction of [Ca2+]i, possibly involving desensitization. Inhibition of both pathways (ODQ plus CPA) abolished NO-induced relaxations completely. The unique ability of NO to directly stimulate [Ca2+]i decrease when segments were contracted with phenylephrine is suggested to be at the basis of the greater efficacy of NO to dilate phenylephrine- compared with high K+-constricted aortic segments.
A decrease of SMC [Ca2+]i by NO has been observed in different vascular beds of different species (Chen and Rivers, 2001; Ohi et al., 2001; Buus et al., 2006), but has never been associated with the specific intracellular Ca2+ pools mobilized during contraction. Although high K+ and the α-adrenoceptor agonists phenylephrine or noradrenaline elicit comparable sustained vasoconstriction, they mobilize intracellular Ca2+ via different mechanisms (Karaki et al., 1997). Vasoconstriction by high K+ could be fully attributed to the depolarization-induced opening of L-type Ca2+ channel, whereas the relationship between intracellular Ca2+ and contraction seemed to be more complicated for phenylephrine. Phenylephrine induces Ca2+ release from internal storage sites, increases [Ca2+]i in the sub-plasmalemmal region (where Ca2+-dependent mechanisms such as ion channels, ion pumps and enzymes are located), opens receptor- or store-operated Ca2+-permeable cation channels (or directly L-type Ca2+ channels) and finally increases [Ca2+]i around the contractile elements (Karaki et al., 1997; Akata, 2007a,b;). As L-type Ca2+ influx is much more important for high K+-induced than for phenylephrine-elicited contractions, depolarization-dependent L-type Ca2+ influx might be the compromising factor in determining the relaxing capacity of NO in constricted mouse aortic segments.
L-type Ca2+ influx compromises the vasodilator efficacy of NO
Voltage-dependent L-type Ca2+ influx is a major Ca2+ influx pathway in vascular SMCs (Karaki et al., 1997; Berkels et al., 2003; Godfraind, 2005) and also in mouse thoracic aortic segments, because in this preparation nifedipine (1 µM) inhibited contractions by high K+ completely and by phenylephrine partly. Application of the L-type Ca2+ channel blockers nifedipine and verapamil not only directly reduced Ca2+ influx and the concomitant contraction, but, at least for contractions by high K+, also caused a dose-dependent leftward shift of the NO-induced relaxation curves. Indeed, the vasodilator capacity of NO declined when the influx of Ca2+ via L-type Ca2+ channels became more prominent as with elevation of external K+ or as with application of an activator of L-type Ca2+ influx (BAY K8644). Vice versa, the vasodilator capacity of NO increased when the influx of Ca2+ was reduced by lowering external K+ or by inhibition of L-type Ca2+ channels with nifedipine or verapamil. As a result, the vasodilator capacity of NO was strongly dependent on the concentration of external K+ used to evoke depolarization and contraction, which might explain the dependence of the vasodilator capacity of NO on the level of pre-contraction by high K+, but not by phenylephrine.
The present study shows that segments contracted by increase of SMC [Ca2+]i via release of Ca2+ from intracellular stores (phenylephrine) relax better and at lower NO concentrations than segments contracted via L-type Ca2+ influx (high K+). Nevertheless, L-type Ca2+ channels were also involved in the phenylephrine-induced contraction, but inhibition of these channels with nifedipine or verapamil did not affect the vasodilator capacity of NO. Relaxation was, however, compromised by stimulating L-type Ca2+ influx in phenylephrine-constricted segments with BAY K8644, again pointing to the compromising effect of L-type Ca2+ influx on the relaxing capacity of NO.
NO-sensitive Ca2+ pools
Because NO and Ca2+ are both non-homogeneously distributed in the SMC (van Breemen et al., 1995; Rembold and Chen, 1998; Asano and Nomura, 2000; Nomura and Asano, 2000), the interaction between NO and Ca2+ pools may vary according to the mechanisms by which Ca2+ was increased during contraction. SMC Ca2+ pools have been described to display different intracellular localization with important consequences for the relationship between intracellular Ca2+ increase and interaction with the contractile filaments (Karaki et al., 1997). Other complicating events include the large variation in the capacity of Ca2+ domains to store Ca2+ (Asano and Nomura, 2000; Nomura and Asano, 2000); the specific intracellular localization of SERCA, IP3 and ryanodine receptors (Golovina and Blaustein, 1997; Janiak et al., 2001; Wamhoff et al., 2002); the different sensitivity of the stores to inhibition by CPA (Rembold and Chen, 1998; Blaustein et al., 2002); and finally, the specific sensitivity of the Ca2+ pools to NO (Ji et al., 1998; Li et al., 2000; Yu et al., 2000). Therefore, to predict the vasodilator capacity of NO in a specific vascular bed, it is essential to investigate the mechanisms of Ca2+ mobilization during the preceding contraction. For example, in rat tail arteries (Lehen'kyi et al., 2002; 2005; Soloviev et al., 2004), NO caused a greater decrease in [Ca2+]i when segments were contracted with high K+ compared with phenylephrine, whereas the present study described the reverse situation in mouse aorta. Species and vascular bed differences may account for such differences, but according to the present study, it could be directly related to the different mechanisms by which high K+ or phenylephrine mobilize contractile SMC [Ca2+]i in rat tail artery or in mouse aortic SMCs. It has at least been shown that bovine tail artery has about 60% less SR than does rat aorta (Ashida et al., 1988).
Conclusions
According to the present study in mouse aorta, the different relaxing capacity of NO for SMC contractions initiated by depolarization (high K+) or by α1 adrenoceptor stimulation (PE) is not dependent on the source of NO (endogenous or exogenous), but depends mainly on the pathways of [Ca2+]i mobilization during contraction. In mouse aorta, NO induces relaxation via at least two pathways. One pathway involves sGC stimulation by NO with the subsequent elevation of cGMP inducing relaxation without detectable changes of [Ca2+]i. This pathway seems to be related to L-type Ca2+ influx, and the relaxation is most probably due to desensitization of the contractile apparatus to Ca2+ (Carvajal et al., 2000). The other pathway involves the decrease of SMC [Ca2+]i via a non-cGMP-dependent stimulation of SERCA. The concomitant re-uptake of Ca2+ into the SR seems most prominent when the Ca2+ mobilized during contraction originated from the ER. The latter mechanism seems to determine the vasodilator capacity of NO in the mouse aorta. As a consequence, interventions which reduce or increase L-type Ca2+ influx during contraction will influence the relaxing capacity of NO. Whether these observations are relevant in vivo require further investigation. However, our observations may form an alternative explanation for the improved relaxation of atherosclerotic aortic segments in mice treated with the dihydropyridine Ca2+ channel blocker nifedipine (Berkels et al., 2003; Kyselovic et al., 2005), that was initially attributed to enhanced bioavailability of endothelium-derived NO by the authors. Finally, it would be interesting to test whether L-type Ca2+ channel blockers also sensitize resistance arteries to the vasodilator effect of NO.
Acknowledgments
This work was supported by grants from the Fonds voor Wetenschappelijk Onderzoek (FWO, Vlaamse Gemeenschap, project G.0174.06).
Glossary
Abbreviations:
- [Ca2+]i
cytosolic calcium concentration
- CPA
cyclopiazonic acid
- DEANO
diethylamine NONOate
- eNOS
endothelial nitric oxide synthase
- GTN
glyceryltrinitrate
- IP3
inositol trisphosphate
- l-NAME
NΩ-nitro-l-arginine methyl ester
- l-NNA
NΩ-nitro-l-arginine
- ODQ
1H-[1,2,4]oxadiazolo[4,3-a]quinoxalin-1-one
- SERCA
sarco-endoplasmic reticulum calcium ATPase
- SMC
smooth muscle cell
- SR
sarcoplasmic reticulum
Conflicts of interest
None.
References
- Adachi T, Weisbrod RM, Pimentel DR, Ying J, Sharov VS, Schoneich C, et al. S-glutathiolation by peroxynitrite activates SERCA during arterial relaxation by nitric oxide. Nat Med. 2004;10:1200–1207. doi: 10.1038/nm1119. [DOI] [PubMed] [Google Scholar]
- Akata T. Cellular and molecular mechanisms regulating vascular tone. Part 1: basic mechanisms controlling cytosolic Ca2+ concentration and the Ca2+-dependent regulation of vascular tone. J Anesth. 2007a;21:220–231. doi: 10.1007/s00540-006-0487-5. [DOI] [PubMed] [Google Scholar]
- Akata T. Cellular and molecular mechanisms regulating vascular tone. Part 2: regulatory mechanisms modulating Ca2+ mobilization and/or myofilament Ca2+ sensitivity in vascular smooth muscle cells. J Anesth. 2007b;21:232–242. doi: 10.1007/s00540-006-0488-4. [DOI] [PubMed] [Google Scholar]
- Asano M, Nomura Y. Ca2+ buffering action of sarcoplasmic reticulum on Bay k 8644-induced Ca2+ influx in rat femoral arterial smooth muscle. Eur J Pharmacol. 1999;366:61–71. doi: 10.1016/s0014-2999(98)00858-9. [DOI] [PubMed] [Google Scholar]
- Asano M, Nomura Y. Ca(2+) movement from leaky sarcoplasmic reticulum during contraction of rat arterial smooth muscles. Eur J Pharmacol. 2000;404:327–339. doi: 10.1016/s0014-2999(00)00618-x. [DOI] [PubMed] [Google Scholar]
- Ashida T, Schaeffer J, Goldman WF, Wade JB, Blaustein MP. Role of sarcoplasmic reticulum in arterial contraction: comparison of ryanodines's effect in a conduit and a muscular artery. Circ Res. 1988;62:854–863. doi: 10.1161/01.res.62.4.854. [DOI] [PubMed] [Google Scholar]
- Berkels R, Taubert D, Rosenkranz A, Rosen R. Vascular protective effects of dihydropyridine calcium antagonists. Involvement of endothelial nitric oxide. Pharmacology. 2003;69:171–176. doi: 10.1159/000073659. [DOI] [PubMed] [Google Scholar]
- Blaustein MP, Golovina VA, Song H, Choate J, Lencesova L, Robinson SW, et al. Organization of Ca2+ stores in vascular smooth muscle: functional implications. Novartis Found Symp. 2002;246:125–137. [PubMed] [Google Scholar]
- Bolotina VM, Najibi S, Palacino JJ, Pagano PJ, Cohen RA. Nitric oxide directly activates calcium-dependent potassium channels in vascular smooth muscle. Nature. 1994;368:850–853. doi: 10.1038/368850a0. [DOI] [PubMed] [Google Scholar]
- van Breemen C, Chen Q, Laher I. Superficial buffer barrier function of smooth muscle sarcoplasmic reticulum. Trends Pharmacol Sci. 1995;16:98–105. doi: 10.1016/s0165-6147(00)88990-7. [DOI] [PubMed] [Google Scholar]
- Buus NH, Simonsen U, Pilegaard HK, Mulvany MJ. Intracellular smooth muscle [Ca2+] in acetylcholine and nitric oxide-mediated relaxation of human small arteries. Eur J Pharmacol. 2006;535:243–247. doi: 10.1016/j.ejphar.2006.01.061. [DOI] [PubMed] [Google Scholar]
- Carvajal JA, Germain AM, Huidobro-Toro JP, Weiner CP. Molecular mechanism of cGMP-mediated smooth muscle relaxation. J Cell Physiol. 2000;184:409–420. doi: 10.1002/1097-4652(200009)184:3<409::AID-JCP16>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- Chen Y, Rivers RJ. Measurement of membrane potential and intracellular Ca(2+) of arteriolar endothelium and smooth muscle in vivo. Microvasc Res. 2001;62:55–62. doi: 10.1006/mvre.2001.2315. [DOI] [PubMed] [Google Scholar]
- Cohen RA, Weisbrod RM, Gericke M, Yaghoubi M, Bierl C, Bolotina VM. Mechanism of nitric oxide-induced vasodilatation: refilling of intracellular stores by sarcoplasmic reticulum Ca2+ ATPase and inhibition of store-operated Ca2+ influx. Circ Res. 1999;84:210–219. doi: 10.1161/01.res.84.2.210. [DOI] [PubMed] [Google Scholar]
- Crauwels HM, Van Hove CE, Herman AG, Bult H. Heterogeneity in relaxation mechanisms in the carotid and the femoral artery of the mouse. Eur J Pharmacol. 2000;404:341–351. doi: 10.1016/s0014-2999(00)00619-1. [DOI] [PubMed] [Google Scholar]
- Crauwels HM, Van Hove CE, Holvoet P, Herman AG, Bult H. Plaque-associated endothelial dysfunction in apolipoprotein E-deficient mice on a regular diet. Effect of human apolipoprotein AI. Cardiovasc Res. 2003;59:189–199. doi: 10.1016/s0008-6363(03)00353-5. [DOI] [PubMed] [Google Scholar]
- Godfraind T. Antioxidant effects and the therapeutic mode of action of calcium channel blockers in hypertension and atherosclerosis. Philos Trans R Soc Lond B Biol Sci. 2005;360:2259–2272. doi: 10.1098/rstb.2005.1774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golovina VA, Blaustein MP. Spatially and functionally distinct Ca2+ stores in sarcoplasmic and endoplasmic reticulum. Science. 1997;275:1643–1648. doi: 10.1126/science.275.5306.1643. [DOI] [PubMed] [Google Scholar]
- Janiak R, Wilson SM, Montague S, Hume JR. Heterogeneity of calcium stores and elementary release events in canine pulmonary arterial smooth muscle cells. Am J Physiol Cell Physiol. 2001;280:C22–C33. doi: 10.1152/ajpcell.2001.280.1.C22. [DOI] [PubMed] [Google Scholar]
- Ji J, Benishin CG, Pang PK. Nitric oxide selectively inhibits intracellular Ca++ release elicited by inositol trisphosphate but not caffeine in rat vascular smooth muscle. J Pharmacol Exp Ther. 1998;285:16–21. [PubMed] [Google Scholar]
- Karaki H, Ozaki H, Hori M, Mitsui-Saito M, Amano K, Harada K, et al. Calcium movements, distribution, and functions in smooth muscle. Pharmacol Rev. 1997;49:157–230. [PubMed] [Google Scholar]
- Kyselovic J, Martinka P, Batova Z, Gazova A, Godfraind T. Calcium channel blocker inhibits Western-type diet-evoked atherosclerosis development in ApoE-deficient mice. J Pharmacol Exp Ther. 2005;315:320–328. doi: 10.1124/jpet.105.089847. [DOI] [PubMed] [Google Scholar]
- Lehen'kyi VV, Zelensky SN, Stefanov AV, Soloviev AI. Effects of nitric oxide donors on vascular smooth muscles depend on a type of vascular smooth-muscle preactivation. Cardiovasc Toxicol. 2002;2:151–160. doi: 10.1385/ct:2:2:151. [DOI] [PubMed] [Google Scholar]
- Lehen'kyi VV, Zelensky SN, Stefanov AV. Ca2+-sensitivity and cGMP-independent effects of NO in vascular smooth muscle. Nitric Oxide. 2005;12:105–113. doi: 10.1016/j.niox.2005.01.002. [DOI] [PubMed] [Google Scholar]
- Lewis SJ, Bhopatkar MY, Walton TM, Bates JN. Role of voltage-sensitive calcium-channels in nitric oxide-mediated vasodilation in spontaneously hypertensive rats. Eur J Pharmacol. 2005;528:144–149. doi: 10.1016/j.ejphar.2005.10.056. [DOI] [PubMed] [Google Scholar]
- Li N, Zou AP, Ge ZD, Campbell WB, Li PL. Effect of nitric oxide on calcium-induced calcium release in coronary arterial smooth muscle. Gen Pharmacol. 2000;35:37–45. doi: 10.1016/s0306-3623(01)00089-1. [DOI] [PubMed] [Google Scholar]
- Moosmang S, Schulla V, Welling A, Feil R, Feil S, Wegener JW, et al. Dominant role of smooth muscle L-type calcium channel Cav1.2 for blood pressure regulation. EMBO J. 2003;22:6027–6034. doi: 10.1093/emboj/cdg583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulvany MJ, Halpern W. Mechanical properties of vascular smooth muscle cells in situ. Nature. 1976;260:617–619. doi: 10.1038/260617a0. [DOI] [PubMed] [Google Scholar]
- Nimmegeers S, Sips P, Buys E, Brouckaert P, Van de Voorde J. Functional role of the soluble guanylyl cyclase alpha(1) subunit in vascular smooth muscle relaxation. Cardiovasc Res. 2007;76:149–159. doi: 10.1016/j.cardiores.2007.06.002. [DOI] [PubMed] [Google Scholar]
- Nomura Y, Asano M. Ca(2+) uptake function of sarcoplasmic reticulum during contraction of rat arterial smooth muscles. Eur J Pharmacol. 2000;404:315–326. doi: 10.1016/s0014-2999(00)00617-8. [DOI] [PubMed] [Google Scholar]
- Ohi Y, Takai N, Muraki K, Watanabe M, Imaizumi Y. Ca2+-images of smooth muscle cells and endothelial cells in one confocal plane in femoral artery segments of the rat. Jpn J Pharmacol. 2001;86:106–113. doi: 10.1254/jjp.86.106. [DOI] [PubMed] [Google Scholar]
- Okon EB, Golbabaie A, van BC. In the presence of l-NAME SERCA blockade induces endothelium-dependent contraction of mouse aorta through activation of smooth muscle prostaglandin H2/thromboxane A2 receptors. Br J Pharmacol. 2002;137:545–553. doi: 10.1038/sj.bjp.0704884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plane F, Wiley KE, Jeremy JY, Cohen RA, Garland CJ. Evidence that different mechanisms underlie smooth muscle relaxation to nitric oxide and nitric oxide donors in the rabbit isolated carotid artery. Br J Pharmacol. 1998;123:1351–1358. doi: 10.1038/sj.bjp.0701746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quignard J, Feletou M, Corriu C, Chataigneau T, Edwards G, Weston AH, et al. 3-Morpholinosydnonimine (SIN-1) and K(+) channels in smooth muscle cells of the rabbit and guinea pig carotid arteries. Eur J Pharmacol. 2000;399:9–16. doi: 10.1016/s0014-2999(00)00372-1. [DOI] [PubMed] [Google Scholar]
- Rembold CM, Chen XL. The buffer barrier hypothesis, [Ca2+]i homogeneity, and sarcoplasmic reticulum function in swine carotid artery. J Physiol. 1998;513(2):477–492. doi: 10.1111/j.1469-7793.1998.477bb.x. Pt. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards GR, Weston AH, Burnham MP, Feletou M, Vanhoutte PM, Edwards G. Suppression of K(+)-induced hyperpolarization by phenylephrine in rat mesenteric artery: relevance to studies of endothelium-derived hyperpolarizing factor. Br J Pharmacol. 2001;134:1–5. doi: 10.1038/sj.bjp.0704256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scarborough NL, Carrier GO. Nifedipine and alpha adrenoceptors in rat aorta. I. Role of extracellular calcium in alpha-1 and alpha-2 adrenoceptor-mediated contraction. J Pharmacol Exp Ther. 1984;231:597–602. [PubMed] [Google Scholar]
- Soloviev A, Lehen'kyi V, Zelensky S, Hellstrand P. Nitric oxide relaxes rat tail artery smooth muscle by cyclic GMP-independent decrease in calcium sensitivity of myofilaments. Cell Calcium. 2004;36:165–173. doi: 10.1016/j.ceca.2004.02.002. [DOI] [PubMed] [Google Scholar]
- Sonkusare S, Palade PT, Marsh JD, Telemaque S, Pesic A, Rusch NJ. Vascular calcium channels and high blood pressure: pathophysiology and therapeutic implications. Vascul Pharmacol. 2006;44:131–142. doi: 10.1016/j.vph.2005.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thapaliya S, Matsuyama H, Takewaki T. Bradykinin causes endothelium-independent hyperpolarisation and neuromodulation by prostanoid synthesis in hamster mesenteric artery. Eur J Pharmacol. 2000;408:313–321. doi: 10.1016/s0014-2999(00)00776-7. [DOI] [PubMed] [Google Scholar]
- Van Assche T, Fransen P, Guns PJ, Herman AG, Bult H. Altered Ca2+ handling of smooth muscle cells in aorta of apolipoprotein E-deficient mice before development of atherosclerotic lesions. Cell Calcium. 2007;41:295–302. doi: 10.1016/j.ceca.2006.06.010. [DOI] [PubMed] [Google Scholar]
- Wamhoff BR, Bowles DK, Dietz NJ, Hu Q, Sturek M. Exercise training attenuates coronary smooth muscle phenotypic modulation and nuclear Ca2+ signaling. Am J Physiol Heart Circ Physiol. 2002;283:H2397–H2410. doi: 10.1152/ajpheart.00371.2001. [DOI] [PubMed] [Google Scholar]
- Yu JZ, Zhang DX, Zou AP, Campbell WB, Li PL. Nitric oxide inhibits Ca(2+) mobilization through cADP-ribose signaling in coronary arterial smooth muscle cells. Am J Physiol Heart Circ Physiol. 2000;279:H873–H881. doi: 10.1152/ajpheart.2000.279.3.H873. [DOI] [PubMed] [Google Scholar]