

Abstract
Increasing evidence shows aberrant hypermethylation of genes occurring in and potentially contributing to pathogenesis of myeloid malignancies. Several of these diseases, such as myelodysplastic syndromes (MDSs), are responsive to DNA methyltransferase inhibitors. To determine the extent of promoter hypermethylation in such tumors, we compared the distribution of DNA methylation of 14 000 promoters in MDS and secondary acute myeloid leukemia (AML) patients enrolled in a phase 1 trial of 5-azacytidine and the histone deacetylase inhibitor entinostat against de novo AML patients and normal CD34+ bone marrow cells. The MDS and secondary AML patients displayed more extensive aberrant DNA methylation involving thousands of genes than did the normal CD34+ bone marrow cells or de novo AML blasts. Aberrant methylation in MDS and secondary AML tended to affect particular chromosomal regions, occurred more frequently in Alu-poor genes, and included prominent involvement of genes involved in the WNT and MAPK signaling pathways. DNA methylation was also measured at days 15 and 29 after the first treatment cycle. DNA methylation was reversed at day 15 in a uniform manner throughout the genome, and this effect persisted through day 29, even without continuous administration of the study drugs. This trial was registered at www.clinicaltrials.gov as J0443.
Introduction
Aberrant DNA methylation is believed to be a hallmark of preneoplastic and neoplastic conditions (reviewed in Plass,1 Herman and Baylin,2 and Jones and Baylin3). Hypermethylation of promoters rich in CpGs (such as those containing CpG islands) is often associated with gene silencing, which can be epigenetically inherited across cell divisions. Hypermethylation-induced silencing of genes, including tumor suppressors such as HIC1, genes involved in DNA damage repair such as MLH1, and cell-cycle regulators such as CDKN1B, was described in myelodysplastic syndrome (MDS),4–7 although the full extent and nature of aberrant DNA methylation in MDS has been uncertain.
MDS and secondary acute myeloid leukemia (AML) are particularly resistant to standard chemotherapy, suggesting that these entities are biologically distinct from de novo AML. In particular, MDS can be treated with cytosine analog DNA methyltransferase (DNMT) inhibitors such as 5-azacytidine (5AC) and decitabine,8 resulting in improved hematopoiesis and in some cases elimination or reduction of the malignant clone. Accordingly, 5AC increases overall survival and prolongs the time to AML transformation in high-risk MDS.9–12
Promoter hypermethylation of a set of cancer-related genes has been linked to the progression of MDS.13 By examining 1505 CpG sites in 807 genes, Jiang et al13 demonstrated that high-risk MDS (refractory anemia with excess of blasts/AML) showed a higher-than-average methylation level and a higher number of aberrantly methylated CpG sites than either low-risk MDS or normal controls. A total of 51 CpG sites methylated in at least 50% of the patients were found among high-risk MDS, compared with only 7 sites in low-risk MDS. Supporting the hypothesis that cytosine analog DNA methyltransferase inhibitors exert their clinical activity through epigenetic modifications, increased baseline methylation of the CDKN2B promoter was associated with response to 5AC.14 Furthermore, the demethylating effect of the DNMT inhibitors in MDS was shown on selected genes such as CDKN2B15,16 and CDH1.15
The sensitivity of MDS to DNMT inhibitors suggests that aberrant epigenetic programming may play an important role in MDS pathogenesis, which might distinguish it from de novo AML. However, 5AC and decitabine incorporate into DNA and can trigger DNA damage response pathways or blocking replication, complicating the interpretation of their antitumor activities.17–20 To elucidate the extent and pervasiveness of aberrant epigenetic programming in MDS/secondary AML and the effect of epigenetic therapies, we ascertained the extent of DNA methylation across a large set of human promoters, in a group of patients with MDS and secondary AML and compared it with both a cohort of de novo AML cases and CD34+ bone marrow cells from a group of healthy donors.
Methods
Samples
Bone marrow aspirates from 14 patients with MDS, MDS-associated AML (AML with trilineage dysplasia), chronic myelomonocytic leukemia, or relapsed AML enrolled in Johns Hopkins clinical trial J0443 were obtained at baseline and at days +15 to 16 and +28 to 29 during the first cycle of treatment with 5AC at concentrations of 30, 40, or 50 mg/m2 per day subcutaneously for 10 consecutive days. Entinostat (2, 4, 6, or 8 mg/m2) was administered orally on days 3 and 10 of the 5AC treatment. Cycles were repeated every 28 days. Mononuclear cells were isolated with the use of Ficoll-Hypaque, and the CD34+ and CD34− fractions were separated with the use of CD34 MicroBeads (Miltenyi Biotec Inc). The patients' characteristics are summarized in supplemental Table 1 (available on the Blood website; see the Supplemental Materials link at the top of the online article). De novo normal karyotype AML samples were obtained at diagnosis from 15 patients enrolled into the E1900 trial of the Eastern Cooperative Oncology Group. Mononuclear cells were isolated from heparinized bone marrow or peripheral blood specimens by Ficoll-Hypaque density centrifugation and frozen in 10% DMSO plus 90% FBS for later use without selecting for the CD34+ fraction. Institutional Review Board approval was obtained at Johns Hopkins University School of Medicine, at the Albert Einstein College of Medicine, and at Weill Cornell Medical College, and patient informed consent was obtained in accordance with the Declaration of Helsinki. Normal CD34+ bone marrow progenitors were obtained from by selection with CD34 MicroBeads from the bone marrows of 8 healthy donors: 4 from the Translational Trials Development and Support Laboratory, Cincinnati Children's Hospital, and 4 from Allcells. Matched CD34+ and CD34− fractions were obtained from an additional 4 healthy donors through the Stem Cell and Xenograft Core, University of Pennsylvania School of Medicine.
Genomic DNA isolation
High-molecular-weight genomic DNA was isolated from all samples with the use of a standard phenol-chloroform purification followed by an ethanol precipitation. DNA pellets were resuspended in 10 mM Tris, pH 8.0.
DNA methylation analysis by HELP
The HELP (HpaII tiny fragment enrichment by ligation mediated PCR [polymerase chain reaction]) assay was carried out as previously described.21,22 HpaII- and MspI-generated genomic fragments between 200 and 2000 base pairs (Bp) in length were labeled with either Cy3- or Cy5-labeled random primers and then cohybridized onto a human (HG17) custom-designed oligonucleotide array (50-mers) covering 25 626 HpaII amplifiable fragments (HAFs) annotated to 14 214 gene promoters.22 HAFs are defined as genomic sequences contained between 2 flanking HpaII sites found 200 to 2000 bp apart. Each HAF on the array is represented by a probe set consisting of 14 to 15 individual probes, randomly distributed across the microarray slide. Scanning was performed with a GenePix 4000B scanner (Molecular Devices).23 Quality control and array normalization were performed as described in Thompson et al.24 After normalization, background noise was calculated for each channel by determining a threshold of 2.5 median absolute deviations from the median of the random probes' signal. Each normalized channel was subsequently centered by subtracting its noise threshold from the array's signal. The HpaII/MspI (unmethylated/whole genome reference) ratio was then determined for each probe set on array. All microarray data have been submitted to the Gene Expression Omnibus repository25 (accession no. GSE17328).
Quantitative DNA methylation analysis by MassARRAY EpiTyping
Validation of HELP findings was performed by matrix-assisted laser desorption–time-of-flight mass spectrometry with the use of EpiTyper by MassARRAY (Sequenom) on bisulfite-converted DNA as previously described.26 MassARRAY primers were designed to cover the flanking HpaII sites for a given HAF, as well as for any other HpaII sites found up to 2000 bp upstream of the downstream site and up to 2000 bp downstream of the upstream site, to cover all possible alternative sites of digestion within our range of polymerase chain reaction amplification (see supplemental Table 2 for primer sequences).
Microarray data analysis
Unsupervised analysis of HELP data by hierarchical clustering (Euclidean distance and Ward clustering method), principal component analysis and correspondence analysis were performed with the statistical software R Version 2.7.127 and the BioConductor28 package MADE4.29 Supervised analysis was carried out with the use of a t test with a significance level of P less than .0005. In addition, an absolute difference in methylation greater than 1.5 between the means of the 2 groups being compared was required to increase the likelihood of detection of biologically significant changes in methylation levels. After correction for multiple testing with the Benjamini-Hochberg method, all P values remained significant at less than .05.
Gene ontology analysis
Pathway analysis of gene expression software (Hani Goodarzi, Olivier Elemento, and Saeed Tavazoie, manuscript submitted, 2008) was used to perform gene ontology analysis of differentially methylated genes. HAFs on the HELP microarray were annotated to the nearest gene up to a maximum distance of 5 kb from the transcription start site, in both directions.
Sequence analysis
Sequence retrieval.
Sequences were downloaded from the University of California Santa Cruz Genome Browser, selecting the 2 kb upstream of the reported 5′ end. Only genes that were found in the RefSeq annotations were downloaded. A total of 7207 RefSeq sequences were retrieved for the genes that were differentially methylated between MDS and normal CD34+, and 4619 RefSeq sequences were retrieved for the set of control sequences. Likewise, 649 RefSeq sequences were retrieved for the genes identified in the baseline versus posttreatment (day +15) samples, and 5180 RefSeq sequences were retrieved for an associated set of control sequences.
Repeat element analysis.
RepeatMasker 3.1.630 was used to find all human repeats in upstream sequences for the genes from the MDS versus normal CD34+ signature and the baseline versus day 15 after treatment signature and in both sets of associated control sequences randomly selected from among the genes that did not change in each one of the respective analyses. The number of times each element was found was calculated, and the different proportions between each set were estimated, using a Fisher exact test.
Motif analysis
FIRE31 was used to discover motifs that were able to distinguish between 1206 genes that were differentially methylated between the MDS/secondary AML and normal CD34+ samples, and a group of 4619 control sequences. For each gene, we used as the promoter sequence the 2 kb upstream of the transcriptional start site as defined in the University of California Santa Cruz Genome Browser.32
Genomic distribution of DNA methylation
Density plots, using a Gaussian smoothing kernel, marking the positions of HpaII fragments on each chromosome of the genome were calculated for (1) all HpaII fragments on the NimbleGen HELP array, (2) all HpaII fragments that were differentially methylated between MDS/secondary AML and CD34+ samples, and (3) all HpaII fragments that were differentially methylated between baseline samples and the same patients 15 days after starting to take 5AC. Kolmogorov-Smirnov tests were carried out to find those density plots from calculation (2) and calculation (3) that differed significantly from the underlying distribution of all HpaII fragments (calculation 1).
Results
MDS and secondary AML display marked aberrant promoter DNA hypermethylation
DNA methylation was measured with the HELP assay.21 HELP was performed with the use of a customized NimbleGen high-density oligonucleotide microarray containing 25 626 probe sets, each of which represents a HAF, corresponding to approximately 14 000 unique genes. All microarray profiles thus obtained were submitted to quality control algorithms and a HELP-specific normalization procedure.24 Direct methylation sequencing (MassARRAY Epi Typing) was performed for 17 randomly selected HAF from the array on 9 randomly selected samples. The correlation coefficient between percent methylation by MassARRAY EpiTYPER, and log(HpaII/MspI) values was R = −0.77 (supplemental Figure 1).
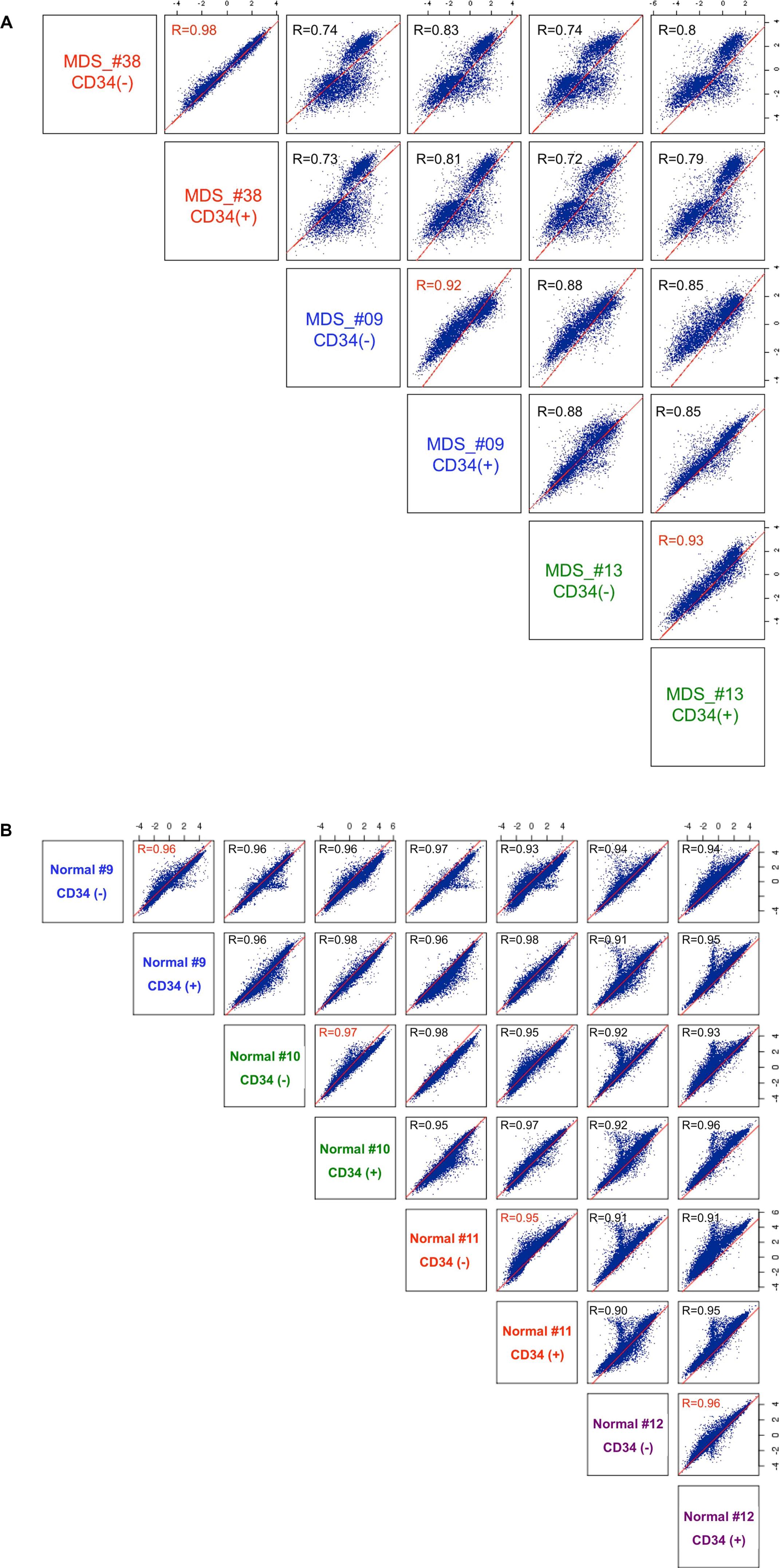
Because purified CD34+ MDS cells were not available for all patients, we first wanted to know whether DNA methylation profiles were similar in CD34+ versus CD34− MDS cells. These cell fractions were obtained from bone marrows of 3 patients from whom we had sufficient high-molecular-weight DNA. DNA methylation profiles were highly similar between the 2 fractions (r ≥ 0.93), and the variability observed between the 2 fractions was less than the interpatient variability (r ≤ 0.83; supplemental Figure 2A). Similarly, 4 matched sample pairs of CD34+ and CD34− fractions were obtained from 4 healthy donors, and their DNA methylation profiles were analyzed by the HELP assay. In all 4 cases a very high correlation was observed between the 2 fractions (r > 0.95; supplemental Figure 2B). The data suggest that both normal and aberrant epigenetic patterns within the marrow are transmitted vertically from progenitors to more differentiated progeny. This allowed subsequent work on MDS/secondary AML samples to be performed in the CD34− fraction, making it possible to study a greater number of cases.
To determine whether there was an aberrant distribution of methylated promoters in patients with MDS and secondary AML, we first compared promoter methylation in 14 such patients at baseline versus bone marrow CD34+ cells from 8 healthy donors. The resulting methylation profiles were first analyzed in an unsupervised fashion with hierarchical clustering (Euclidean distance and Ward clustering method), principal component analysis (PCA), and correspondence analysis (COA). All 3 methods clearly segregated the malignant samples from the normal bone marrow cells (Figure 1A-C). Because methylation was measured as a continuous variable, the degree of the difference of methylation of each gene was quantified to identify those genes in which the greatest and potentially most biologically significant shift in methylation occurred. Along these lines, a supervised analysis comparing MDS and secondary AML specimens to normal CD34+ identified 5845 HpaII fragments differentially methylated between the 2 groups (P < .0005, 2-tailed t test, and methylation difference > 1.5; Figure 1D). Greater than 99% (5838 of 5845) of the HpaII fragments were hypermethylated in the malignant samples compared with normal bone marrow progenitors (Figure 1E). However, significant heterogeneity was detected among the MDS/secondary AML samples, probably reflecting the heterogeneity of this disease.
Figure 1.

MDS displays marked aberrant promoter DNA methylation. (A-C) Unsupervised clustering analysis of 14 MDS samples and 8 CD34+ normal controls with the use of 3 different algorithms: COA, PCA, and hierarchical clustering. (D) A plot of methylation difference between MDS cases and normal CD34+ controls (x-axis) versus statistical significance (y-axis) shows the marked asymmetry of the 2 branches, illustrating the overall tendency to higher methylation levels in the MDS cases. Red points mark probe sets that reached both criteria for differential methylation on our analysis [P < .0005 and absolute fold change in log(HpaII/MspI) > 1.5), whereas blue points mark probe sets that reached statistical significance but did not have an absolute change in log(HpaII/MspI) greater than 1.5. (E) Two-dimensional hierarchical clustering of genes differentially methylated between MDS and normal CD34+ controls, illustrated by a heatmap. Cases are represented in the columns and probe sets in the rows.
De novo AML displays a less extensive aberrant methylation pattern than secondary AML and MDS
To determine the extent of aberrant promoter methylation in de novo AML, genomic DNA from 15 patients with newly diagnosed normal karyotype leukemia was analyzed by HELP and compared with normal bone marrow. Unsupervised clustering analysis as described in the previous section showed that de novo AML also presents with an epigenetic profile distinct from normal bone marrow progenitors (Figure 2A-C). However supervised analysis showed that only 473 fragments (475 genes; supplemental Table 4) were differentially methylated (P < .0005 and methylation difference > 1.5). Of these, 37 fragments (7.8%) were hypomethylated in AMLs with respect to the normal progenitors (Figure 2D-E).
Figure 2.

De novo AML presents with a lesser extent of aberrant promoter DNA methylation and aberrant hypomethylation. (A-C) Unsupervised clustering analysis of 15 de novo AML samples and 8 CD34+ normal controls with COA, PCA, and hierarchical clustering. (D) Plot of DNA methylation difference between AML cases and normal CD34+ controls (x-axis) versus statistical significance (y-axis). As before, red points mark probe sets that reached both criteria for differential methylation on our analysis [P < .0005 and absolute fold change in log(HpaII/MspI) > 1.5], whereas blue points mark probe sets that reached statistical significance but did not have an absolute change in log(HpaII/MspI) greater than 1.5. (E) Two-dimensional hierarchical clustering of genes differentially methylated between de novo AML and normal CD34+ controls identified by a supervised analysis, illustrated using a heatmap. Cases are represented in the columns and probe sets in the rows.
Secondary AML and MDS have distinct epigenetic features that distinguish them from de novo AML
We next compared and contrasted the methylation profiles obtained from patients with MDS and secondary AML at baseline with those of patients with de novo AML. The 3 unsupervised analyses showed that these 2 groups of patients display distinct epigenetic profiles, although the difference was not as clear as when either set of disease specimens was compared with normal CD34+ cells (Figure 3A-C). Many genes showed an increase in methylation in MDS/secondary AML compared with de novo AML specimens (3489 HpaII fragments at P < .0005). However, the difference in magnitude was not as pronounced as when either group was compared with normal CD34+ cells, because 103 of these HpaII fragments (87 unique genes; Table 1) reached a methylation difference cutoff of more than 1.5. (Figure 3D-E). These data indicate that aberrant DNA methylation is more pronounced in MDS and secondary AML than in de novo AML. This was underscored by further PCA analysis, including normal CD34+ cells, in which the projection of the first 3 principal components showed a methylation “gradient,” with normal CD34+ cells on one extreme, de novo AMLs in an intermediate position, and MDS samples in the opposite end (Figure 3F).
Figure 3.

Underlying differences in DNA methylation distinguish MDS from de novo AML. (A-C) Unsupervised clustering analysis of 14 MDS cases and 15 de novo AML samples with 3 different methods: COA, PCA, and hierarchical clustering. (D) Plot of DNA methylation difference between MDS and de novo AML cases (x-axis) versus statistical significance (y-axis). Red points mark probe sets that reached both criteria for differential methylation on our analysis [P < .0005 and absolute fold change in log(HpaII/MspI) > 1.5], whereas blue points mark probe sets that reached statistical significance but did not have a change in log(HpaII/MspI) greater than 1.5. (E) Two-dimensional hierarchical clustering of genes differentially methylated between MDS and de novo identified by supervised analysis, illustrated with a heatmap. Cases are represented in the columns and probe sets in the rows. (F) Three-dimensional representation of PCA analysis, including normal CD34+ cells, de novo AML, and MDS. MDS samples can all be found the furthest away from normal CD34 cells, whereas de novo AML tend to cluster in an intermediate position between the other 2 groups of samples.
Table 1.
Genes differentially methylated between MDS and de novo AML
| Gene symbol | Description |
|---|---|
| ACSL1 | Acyl-CoA synthetase long-chain family member 1 |
| AIM2 | Absent in melanoma 2 |
| AOC3 | Amine oxidase, copper containing 3 precursor |
| ASGR2 | Asialoglycoprotein receptor 2 |
| BRCA2 | Breast cancer 2, early onset |
| CACNA1I | Calcium channel, voltage-dependent, T type, |
| CDR2 | Cerebellar degeneration-related protein 2 |
| EID2 | CREBBP/EP300 inhibitor 2 |
| ELOVL6 | Elongation of very long chain fatty acids-like |
| ESPL1 | Extra spindle poles like 1 |
| ESPNL | Espin-like |
| EXOC7 | Exocyst complex component 7 |
| FHL2 | Four and a half LIM domains 2 |
| FOXP4 | Forkhead box P4 |
| GGNBP2 | Gametogenetin-binding protein 2 (Laryngeal carcinoma- related protein 1) |
| GHRL | Ghrelin precursor |
| GNAZ | Guanine nucleotide binding protein, α z |
| GNG4 | Guanine nucleotide binding protein (G protein) |
| HOXB5 | Homeobox B5 |
| HYAL3 | Hyaluronoglucosaminidase 3 |
| IFNGR2 | Interferon γ receptor 2 precursor |
| ITGAE | Integrin, α E precursor |
| KRTCAP2 | Keratinocyte associated protein 2 |
| LCE3E | Late-cornified envelope 3E |
| LRFN4 | Leucine-rich repeat and fibronectin type III |
| LRP6 | Low-density lipoprotein receptor-related protein |
| MED25 | ARC/mediator transcriptional coactivator |
| MICALL2 | MICAL-like 2 isoform 1 |
| MTMR11 | Myotubularin-related protein 11 (Cisplatin resistance-associated protein) |
| NAT6 | N-acetyltransferase 6 |
| NOTCH4 | Notch4 preproprotein |
| NPEPL1 | Aminopeptidase-like 1 |
| NRTN | Neurturin preproprotein |
| PEX11A | Peroxisomal biogenesis factor 11A |
| PHLDB1 | Pleckstrin homology-like domain, family B |
| PILRA | Paired immunoglobulin-like type 2 receptor α |
| PPARD | Peroxisome proliferative activated receptor-δ isoform |
| PRF1 | Perforin 1 precursor |
| PTPRS | Protein tyrosine phosphatase, receptor type |
| PYDC1 | Pyrin domain containing 1 |
| RAG1AP1 | Recombination activating gene 1 activating |
| RHOT1 | Ras homolog gene family, member T1 |
| RNH1 | Ribonuclease/angiogenin inhibitor |
| SERPINB13 | Serine (or cysteine) proteinase inhibitor, clade |
| SIRT1 | Sirtuin 1 |
| SLC20A2 | Solute carrier family 20, member 2 |
| SLC26A9 | Solute carrier family 26, member 9 isoform a |
| SLC39A13 | Solute carrier family 39 (zinc transporter) |
| SPATS1 | Spermatogenesis-associated serine-rich protein 1 |
| SPRN | Shadow of prion protein |
| ST3GAL3 | Sialyltransferase 6 |
| SULT1A1 | Sulfotransferase family, cytosolic, 1A |
| TGFA | Transforming growth factor, α |
| TIMP3 | Tissue inhibitor of metalloproteinase 3 |
| TMEM33 | Transmembrane protein 33 |
| TMEM41B | Transmembrane protein 41B |
| TMEM52 | Transmembrane protein 52 |
| TRIM46 | Tripartite motif-containing 46 |
| TRPV3 | Transient receptor potential cation channel |
| TSEN2 | tRNA splicing endonuclease 2 homolog |
| ZNF212 | Zinc finger protein 212 |
| ZNF606 | Zic finger protein 606 |
| ZNF74 | Zinc finger protein 74 |
Genes hypermethylated in MDS display specific functional and genomic characteristics
Among the genes aberrantly methylated in MDS and secondary AML compared with normal CD34+ cells (supplemental Table 3), we found many from the WNT signaling pathway, including GSK3β, APC2, SFRP1, SFRP2, SFRP3, FRAT1, nd AXIN2, as well as the genes coding for the WNT proteins 2b, 3, 4, 5, 6, 9, 10, and 16 (supplemental Figure 3). A gene ontology analysis of the 936 most hypermethylated genes (P < .0005 and methylation change > 2) with the use of the pathway analysis of gene expression algorithm (Hani Goodarzi, Olivier Elemento, and Saeed Tavazoie, manuscript submitted, 2008) showed specific enrichment of gene ontology terms related to activation of MAPK activity (GO:0000187) and synaptogenesis (GO:0007416). There was also a specific underrepresentation of genes associated with the GO term GO:0009888 (tissue development). Table 2 shows the genes associated with each of these enriched GO categories, which include many potentially relevant candidate genes for involvement in MDS pathophysiology such as TNF, MADD, DAXX, TGFA, EGFR, and genes from the protocadherin β gene cluster.
Table 2.
Genes associated with the GO categories overrepresented in the signature of differentially methylated genes between MDS and normal CD34+ cells
| Accession nos. | Description | |
|---|---|---|
| GO:0007416 Synaptogenesis | ||
| NRCAM | NM_001037133/NM_005010/NM_001037132 | Neuronal cell adhesion molecule isoforms C, B, and A |
| PCDHB10 | NM_018930 | Protocadherin β 10 precursor |
| PCDHB16 | NM_020957 | Protocadherin β 16 precursor |
| PCDHB9 | NM_019119 | Protocadherin β 9 precursor |
| GO:0043406 Positive regulation of MAP kinase activity | ||
| ADRB2 | NM_000024 | Adrenergic, β-2-, receptor, surface |
| CXCR4 | NM_001008540/NM_003467 | Chemokine (C-X-C motif) receptor 4 isoforms a and b |
| MADD | NM_003682/NM_130471/NM_130472/NM_130474/NM_130470/NM_130473/NM_130475/NM_130476/NM_001135943/NM_001135944 | MAP-kinase activating death domain-containing |
| MAP3K5 | NM_005923 | Mitogen-activated protein kinase kinase kinase |
| PAK1 | NM_002576/NM_001128620 | p21-Activated kinase 1 isoforms 1 and 2 |
| PROK2 | NM_021935/NM_001126128 | Prokineticin 2 isoforms a and b precursor |
| TNF | NM_000594 | Tumor necrosis factor α |
| GO:0000187 Activation of MAPK activity | ||
| ADRB2 | NM_000024 | Adrenergic, β-2-, receptor, surface |
| CXCR4 | NM_001008540/NM_003467 | Chemokine (C-X-C motif) receptor 4 isoforms a and b |
| MADD | NM_003682/NM_130471/NM_130472/NM_130474/NM_130470/NM_130473/NM_130475/NM_130476/NM_001135943/NM_001135944 | MAP-kinase activating death domain-containing |
| MAP3K5 | NM_005923 | Mitogen-activated protein kinase kinase kinase |
| PROK2 | NM_021935/NM_001126128 | Prokineticin 2 isoforms a and b precursor |
| TNF | NM_000594 | Tumor necrosis factor α |
Aberrant methylation was not distributed randomly across chromosomes. Differentially methylated HpaII fragments showed significant regional differences (P < .05, Bonferroni-adjusted) on chromosomes 4 and 14 compared with the genomic distribution of all HpaII fragments from the HELP array. Genomic regions hypermethylated in MDS and secondary AMLs (compared with control CD34+ cells) were less frequently associated with the presence of SINE/Alu elements than a set of control sequences that showed no difference in methylation between the 2 groups (P < .001, Fisher exact test; odds ratio, 0.74; Table 3). SINE/Alu elements are rich in transcription factor binding sites and to be differentially distributed in gene groups associated with different biologic purposes.33 In particular, it was shown that genes involved in development were Alu poor.
Table 3.
Frequency of SINE/Alu occurrence in differentially methylated regions and control sequences
| SINE/Alu absent, n | SINE/Alu present, n | Fisher P | Odds ratio | |
|---|---|---|---|---|
| MDS versus normal CD34+ | 3638 | 3631 | < .001 | 0.74 |
| Control sequences (MDS vs normal CD34+) | 2040 | 2746 | ||
| Baseline versus day 15 | 217 | 438 | < .001 | 1.81 |
| Control sequences (baseline vs day 15) | 2514 | 2800 |
To determine whether these hypermethylated genes shared any common DNA elements, the FIRE motif discovery program was used.31 The HES/hairy binding motif was the most enriched and conserved DNA element among the set of 1206 most hypermethylated genes (P < .001; Figure 4).
Figure 4.

Genes aberrantly methylated in MDS contain common motifs for DNA binding proteins. Motif analysis with the FIRE algorithm showed a significant overrepresentation of several DNA motifs in promoter regions differentially methylated between MDS and normal CD34+ controls. The yellow color in the heatmap reflects overrepresentation of a motif in a given group, and the blue color represents underrepresentation of the motif. For each motif, we also specify (1) location of the motif; (2) statistical significance of the motif expressed as z scores; (3) robustness of the findings, ranging from 0 of 10 to 10 of 10; (4) conservation index; (5) seed that gave rise to the motif; and (6) name of the closest known motif in our motif database.
Treatment with 5AC and entinostat causes sustained and global promoter hypomethylation
We compared baseline DNA methylation profiles of patients treated with a combination of 5AC and entinostat to those obtained at days 15 to 16 and days 28 to 29 of the first cycle of treatment. Unsupervised clustering showed that changes in DNA methylation at day 15 readily segregated the samples based on treatment status, rather than pairing the samples according to patient of origin, indicating that therapy-associated changes in DNA methylation were sufficiently profound to override individual DNA signatures (Figure 5A-C). The supervised analysis showed marked genomewide hypomethylation of the samples by day +15, as reflected by the asymmetry between the 2 arms of the DNA methylation volcano plot (5501 HpaII fragments at P < .0005). Of these, 966 HpaII fragments also reached a methylation difference cutoff of greater than 1.5 (Figure 5D-E). It has been widely reported that bone marrow response in patients treated with 5AC only occurs after several cycles,9,12,34 and this trend also occurred in the current study.35 Therefore, the difference in methylation profile cannot be attributed to normalization of the bone marrow at these early time points. Moreover, of the 9 patients included in this analysis, only 3 showed an eventual hematologic response, again indicating that changes in methylation are not due to replacement of the marrow with normal cells. Unfortunately, the small number of patients available for this analysis was not sufficient to compare and contrast the nature and extent of the demethylation between responders and nonresponders. No significant changes in DNA methylation profile were found for patient no. 6 after treatment, a notable exception. The baseline DNA profile for this patient was much less methylated than the other patients with MDS and secondary AML. It is not clear whether such samples can show further demethylation with these assays. This patient's day +15 sample clustered together with its baseline sample pair, along with all the day +15 samples from the remaining patients.
Figure 5.

Marked promoter DNA hypomethylation is achieved after 15 days of treatment with 5AC. (A-C) Unsupervised clustering analysis (COA, PCA, and hierarchical clustering) of 9 MDS cases for which we obtained pretreatment and posttreatment (day +15) paired samples. (D) Plot of DNA methylation difference between baseline MDS and day +15 after treatment (x-axis) versus statistical significance (y-axis). Red points mark probe sets that reached both criteria for differential methylation on our analysis [P < .0005 and absolute fold change in log(HpaII/MspI) > 1.5], whereas blue points mark probe sets that reached statistical significance but did not have a change in log(HpaII/MspI) greater than 1.5. (E) Two-dimensional hierarchical clustering of genes differentially methylated between pretreatment and day +15 paired samples identified by supervised analysis is shown with a heatmap. Cases are represented in the columns and probe sets in the rows. (F) Density plots representing the relative frequency (y-axis) of promoters with different degrees of methylation (x-axis). Baseline (black) distributions are compared with day +15 of treatment with 5AC (red) and day +29 (blue). For 8 of 9 cases a clear shift toward more hypomethylated values is observed at day 15, and this shift persisted at day 29 for the cases for which we also had samples at that time point.
The treatment protocol included 10 days of 5AC and entinostat followed by an 18-day break. To determine whether DNA hypomethylation was sustained beyond day 15 after treatment, HELP profiles were performed at day 29 after treatment, just before the second cycle of 5AC and entinostat. Hypomethylation persisted at day 29, suggesting that these changes were stable in the treated cells (Figure 5F). Treatment-induced hypomethylation was evenly distributed across all chromosomal regions, indicating that the drugs have a uniform demethylating effect across the genome. Finally, we also analyzed the presence of repeat elements in promoter regions of the differentially methylated genes after treatment. In this case, we found that genomic regions that showed the greatest change between baseline and day +15 of 5AC treatment were more likely to contain SINE/Alu elements than were the regions that did not change after treatment (P < .001, Fisher exact test; odds ratio, 1.81; Table 2).
Discussion
Aberrant DNA methylation of promoters in MDS has previously been reported in specific genes or cohorts of genes.4–7,13,36 Here, with the use of a genomewide approach we found significant and widespread promoter hypermethylation in most MDS and related disorders. Although particular cancer-related genes such as CDKN2A and genes in the WNT signaling pathway were hypermethylated, epigenetic deregulation was not limited to cancer-associated genes but appeared to be a more widespread phenomenon (see supplemental Table 3). The extent of hypermethylation and the heterogeneity in the genes methylated in MDS and secondary AML suggests that general mechanisms that normally protect promoters from becoming methylated may be disrupted in these disease sates, resulting in a broad and partially random invasion of promoters by methylCpGs. The effect is not completely stochastic because certain chromosomal regions were more prone to hypermethylation than others. Although the mechanism through which loss of promoter hypomethylation occurs remains unclear, it seems possible that abnormalities in the function of transcription factors that bind to specific DNA elements contained in the aberrantly hypermethylated promoters (such as HES, and E2F isoforms) might at least partially contribute to the observed aberrant DNA methylation patterns. Aberrant gene hypermethylation in de novo AML was less marked than in MDS and secondary AML (475 promoters in de novo AML vs 5845 in MDS). Furthermore, although there was an overall tendency toward hypermethylation in de novo AMLs, there were more aberrantly hypomethylated loci in AML than in MDS (39 of 473 vs 7 of 5845; P < .001, Fisher exact test). Thus, although epigenetic deregulation is a hallmark of cancer, specific patterns and distributions of epigenetic deregulation may characterize different forms of hematologic malignancies, potentially contributing to the biology of each disease.
Direct comparison between de novo AML and secondary AML/MDS showed that hypermethylation was more pervasive in the latter group. Specifically, 81.6% of genes of the genes differentially methylated between these 2 groups were hypermethylated in the MDS/secondary AML cohort. With the use of principal component analysis, MDS/secondary AML and de novo AML were clearly distinct from normal CD34+ cells with MDS/secondary AML cases grouping at the extreme of a gradient, leaving the de novo AMLs in an intermediate position. It is tempting to speculate that the pervasive hypermethylation found in MDS and secondary AML may be related to the response of these patients to demethylating agents. Prospective clinical studies comparing the methylation state of responders and nonresponders to therapy will be required to confirm this idea. Among the genes uniquely affected in MDS and secondary AML were those encoding certain transcription factors (HOXB5 and FOXP4), genes involved in DNA damage repair (BRCA2), notch signaling genes (NOTCH4), and PPARD. The latter genes have been implicated in attenuating colon carcinogenesis37; hence, loss of their expression because of hypermethylation could be linked to disease aggressiveness.
Despite some limited in vitro and in vivo evidence that DNMT inhibitors can lead to loss of methylation of specific cancer-related genes,15,37,38,39 the global effect of DNMT inhibitors on overall DNA methylation has not been clearly established. With the use of an array-based approach to study more than 14 000 promoters, we found that 5501 HpaII fragments consistently showed a reduction in methylation after treatment with a combination of 5AC and entinostat, and 966 of these promoters were strongly demethylated (Figure 5D). In contrast to previous reports in which the hypomethylating effect of 5AC was found to be transient, recovering to almost baseline levels before each treatment cycle,36 in this study, this effect persisted even 2 weeks after the cessation of therapy. Whether this persistence is due to the assay of a greater number of promoters or due to the effect of entinostat to augment the efficacy of 5AC is not yet certain. However, this finding suggests that a new epigenetic pattern can be established in bone marrow cells, which can be successfully transmitted to daughter cells through several generations.
Finally, the full effect of the hypermethylation found in MDS and AML for epigenetic programming remains to be determined. Future studies of DNA methylation of promoters will need to be correlated with critical histone modifications such as dimethylation of H3K4 (active genes) and trimethylation H3K27 (inactive genes) and levels of expression of genes. Measurement of these genes before and after 5AC-based therapy would determine the importance of specific gene demethylation in disease response.
Supplementary Material
Acknowledgments
This work was supported by an American Society of Hematology Fellow Scholar Award (M.E.F.), grant R01 CA104348 (A.M.), grant R01 HD044078 (J.M.G.), grant R21 CA110507 (S.D.G. and J.D.L.), grant K24 CA111717 (S.D.G.), grant U01CA70095 (S.D.G., M.S.T., and A.M.), grant U24 CA114737 (E.P.), National Cancer Institute grant CA21115 (E.P.), the Chemotherapy Foundation (A.M.), the Samuel Waxman Cancer Research Foundation (A.M. and J.D.L.), the G&P Foundation (A.M.), the Burroughs Welcome Clinical Scientist Award (J.D.L.), and the Leukemia & Lymphoma Society Scholar Award (A.M.).
Footnotes
An Inside Blood analysis of this article appears at the front of this issue.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Authorship
Contribution: M.E.F. conceived, designed, and performed the research, analyzed the data, and wrote the manuscript; L.S. analyzed the data and wrote the manuscript; Y.L. and A.J. performed the research; T.E.F., E.P., and M.S.T. contributed research material and contributed to data interpretation; H.F. contributed research material; J.M.G. conceived and designed the research and contributed analytical tools; H.C. conceived and designed the research, contributed research material, and contributed to data interpretation; J.D.L. conceived and designed the research, contributed to data interpretation, and wrote the manuscript; S.D.G. conceived and designed the research, contributed research material, analyzed the data, and wrote the manuscript; and A.M. conceived and designed the research, analyzed the data, and wrote the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Ari Melnick, Weill Cornell Medical College, 525 E 68th St, WMC Box 113 (C-620), New York, NY 10065; e-mail: amm2014@med.cornell.edu.
References
- 1.Plass C. Cancer epigenomics. Hum Mol Genet. 2002;11(20):2479–2488. doi: 10.1093/hmg/11.20.2479. [DOI] [PubMed] [Google Scholar]
- 2.Herman JG, Baylin SB. Gene silencing in cancer in association with promoter hypermethylation. N Engl J Med. 2003;349(21):2042–2054. doi: 10.1056/NEJMra023075. [DOI] [PubMed] [Google Scholar]
- 3.Jones PA, Baylin SB. The fundamental role of epigenetic events in cancer. Nat Rev Genet. 2002;3(6):415–428. doi: 10.1038/nrg816. [DOI] [PubMed] [Google Scholar]
- 4.Tessema M, Langer F, Dingemann J, Ganser A, Kreipe H, Lehmann U. Aberrant methylation and impaired expression of the p15(INK4b) cell cycle regulatory gene in chronic myelomonocytic leukemia (CMML). Leukemia. 2003;17(5):910–918. doi: 10.1038/sj.leu.2402891. [DOI] [PubMed] [Google Scholar]
- 5.Brakensiek K, Langer F, Schlegelberger B, Kreipe H, Lehmann U. Hypermethylation of the suppressor of cytokine signalling-1 (SOCS-1) in myelodysplastic syndrome. Br J Haematol. 2005;130(2):209–217. doi: 10.1111/j.1365-2141.2005.05590.x. [DOI] [PubMed] [Google Scholar]
- 6.Christiansen DH, Andersen MK, Pedersen-Bjergaard J. Methylation of p15INK4B is common, is associated with deletion of genes on chromosome arm 7q and predicts a poor prognosis in therapy-related myelodysplasia and acute myeloid leukemia. Leukemia. 2003;17(9):1813–1819. doi: 10.1038/sj.leu.2403054. [DOI] [PubMed] [Google Scholar]
- 7.Aggerholm A, Holm MS, Guldberg P, Olesen LH, Hokland P. Promoter hypermethylation of p15INK4B, HIC1, CDH1, and ER is frequent in myelodysplastic syndrome and predicts poor prognosis in early-stage patients. Eur J Haematol. 2006;76(1):23–32. doi: 10.1111/j.1600-0609.2005.00559.x. [DOI] [PubMed] [Google Scholar]
- 8.Griffiths E, Gore S. DNA Methyltransferase and histone deacetylase inhibitors in the treatment of myelodysplastic syndromes. Semin Hematol. 2008;45(1):23–30. doi: 10.1053/j.seminhematol.2007.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Silverman LR, Demakos EP, Peterson BL, et al. Randomized controlled trial of azacitidine in patients with the myelodysplastic syndrome: a study of the Cancer and Leukemia Group B. J Clin Oncol. 2002;20(10):2429–2440. doi: 10.1200/JCO.2002.04.117. [DOI] [PubMed] [Google Scholar]
- 10.Silverman LR, McKenzie DR, Peterson BL, et al. Further analysis of trials with azacitidine in patients with myelodysplastic syndrome: studies 8421, 8921, and 9221 by the Cancer and Leukemia Group B. J Clin Oncol. 2006;24(24):3895–3903. doi: 10.1200/JCO.2005.05.4346. [DOI] [PubMed] [Google Scholar]
- 11.Fenaux P. Inhibitors of DNA methylation: beyond myelodysplastic syndromes. Nat Clin Pract Oncol. 2005;2(suppl 1):S36–S44. doi: 10.1038/ncponc0351. [DOI] [PubMed] [Google Scholar]
- 12.Fenaux P, Mufti GJ, Hellstrom-Lindberg E, et al. Efficacy of azacitidine compared with that of conventional care regimens in the treatment of higher-risk myelodysplastic syndromes: a randomised, open-label, phase III study. Lancet Oncol. 2009;10(3):223–232. doi: 10.1016/S1470-2045(09)70003-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jiang Y, Dunbar A, Gondek LP, et al. Aberrant DNA methylation is a dominant mechanism in MDS progression to AML. Blood. 2009;113(6):1315–1325. doi: 10.1182/blood-2008-06-163246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Raj K, John A, Ho A, et al. CDKN2B methylation status and isolated chromosome 7 abnormalities predict responses to treatment with 5-azacytidine. Leukemia. 2007;21(9):1937–1944. doi: 10.1038/sj.leu.2404796. [DOI] [PubMed] [Google Scholar]
- 15.Gore SD, Baylin S, Sugar E, et al. Combined DNA methyltransferase and histone deacetylase inhibition in the treatment of myeloid neoplasms. Cancer Res. 2006;66(12):6361–6369. doi: 10.1158/0008-5472.CAN-06-0080. [DOI] [PubMed] [Google Scholar]
- 16.Kantarjian H, Oki Y, Garcia-Manero G, et al. Results of a randomized study of 3 schedules of low-dose decitabine in higher-risk myelodysplastic syndrome and chronic myelomonocytic leukemia. Blood. 2007;109(1):52–57. doi: 10.1182/blood-2006-05-021162. [DOI] [PubMed] [Google Scholar]
- 17.Karpf AR, Moore BC, Ririe TO, Jones DA. Activation of the p53 DNA damage response pathway after inhibition of DNA methyltransferase by 5-Aza-2′-deoxycytidine. Mol Pharmacol. 2001;59(4):751–757. [PubMed] [Google Scholar]
- 18.Zhu W-G, Hileman T, Ke Y, et al. 5-Aza-2′-deoxycytidine activates the p53/p21Waf1/Cip1 pathway to inhibit cell proliferation. J Biol Chem. 2004;279(15):15161–15166. doi: 10.1074/jbc.M311703200. [DOI] [PubMed] [Google Scholar]
- 19.Palii SS, Van Emburgh BO, Sankpal UT, Brown KD, Robertson KD. DNA methylation inhibitor 5-Aza-2′-deoxycytidine induces reversible genome-wide DNA damage that is distinctly influenced by DNA methyltransferases 1 and 3B. Mol Cell Biol. 2008;28(2):752–771. doi: 10.1128/MCB.01799-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kuo HK, Griffith JD, Kreuzer KN. 5-Azacytidine induced methyltransferase-DNA adducts block DNA replication in vivo. Cancer Res. 2007;67(17):8248–8254. doi: 10.1158/0008-5472.CAN-07-1038. [DOI] [PubMed] [Google Scholar]
- 21.Khulan B, Thompson R, Ye K, et al. Comparative isoschizomer profiling of cytosine methylation: the HELP assay. Genome Res. 2006;16(8):1046–1055. doi: 10.1101/gr.5273806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Figueroa ME, Reimers M, Thompson RF, et al. An integrative genomic and epigenomic approach for the study of transcriptional regulation. PLoS One. 2008;3(3):e1882. doi: 10.1371/journal.pone.0001882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Selzer RR, Richmond TA, Pofahl NJ, et al. Analysis of chromosome breakpoints in neuroblastoma at sub-kilobase resolution using fine-tiling oligonucleotide array CGH. Genes Chromosomes Cancer. 2005;44(3):305–319. doi: 10.1002/gcc.20243. [DOI] [PubMed] [Google Scholar]
- 24.Thompson RF, Reimers M, Khulan B, et al. An analytical pipeline for genomic representations used for cytosine methylation studies. Bioinformatics. 2008;24(9):1161–1167. doi: 10.1093/bioinformatics/btn096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Edgar R, Domrachev M, Lash AE. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002;30(1):207–210. doi: 10.1093/nar/30.1.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ehrich M, Nelson MR, Stanssens P, et al. Quantitative high-throughput analysis of DNA methylation patterns by base-specific cleavage and mass spectrometry. Proc Natl Acad Sci U S A. 2005;102(44):15785–15790. doi: 10.1073/pnas.0507816102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Version 2.8. Vienna: R Foundation for Statistical Computing; 2008. R: A Language and Environment for Statistical Computing [computer program]. [Google Scholar]
- 28.Gentleman R, Carey V, Bates D, et al. Bioconductor: open software development for computational biology and bioinformatics. Genome Biol. 2004;5(10):R80. doi: 10.1186/gb-2004-5-10-r80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Culhane AC, Thioulouse J, Perriere G, Higgins DG. MADE4: an R package for multivariate analysis of gene expression data. Bioinformatics. 2005;21(11):2789–2790. doi: 10.1093/bioinformatics/bti394. [DOI] [PubMed] [Google Scholar]
- 30.Smit A, Hubley R, Green P. RepeatMasker Open-3.0. 1996-2004. [Accessed July 2009]. http://www.repeatmasker.org.
- 31.Elemento O, Slonim N, Tavazoie S. A universal framework for regulatory element discovery across all genomes and data types. Mol Cell. 2007;28(2):337–350. doi: 10.1016/j.molcel.2007.09.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Karolchik D, Kuhn RM, Baertsch R, et al. The UCSC Genome Browser Database: 2008 update. Nucleic Acids Res. 2008;36:D773–D779. doi: 10.1093/nar/gkm966. Database issue. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Polak P, Domany E. Alu elements contain many binding sites for transcription factors and may play a role in regulation of developmental processes. BMC Genomics. 2006;7:133. doi: 10.1186/1471-2164-7-133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Silverman LR, Verma A, Odchimar-Reissig R, et al. A phase I trial of the epigenetic modulators Vorinostat, in combination with azacitidine (azaC) in patients with the myelodysplastic syndrome (MDS) and acute myeloid leukemia (AML): a study of the New York Cancer Consortium [abstract]. Blood. 2008;112(11) Abstract 3656. [Google Scholar]
- 35.Fandy TE, Herman JG, Kerns K, et al. Early epigenetic changes and DNA damage do not predict clinical response in an overlapping schedule of 5-azacytidine and entinostat in patients with myeloid malignancies. Blood. 2009;114(13):2764–2773. doi: 10.1182/blood-2009-02-203547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Stresemann C, Bokelmann I, Mahlknecht U, Lyko F. Azacytidine causes complex DNA methylation responses in myeloid leukemia. Mol Cancer Ther. 2008;7(9):2998–3005. doi: 10.1158/1535-7163.MCT-08-0411. [DOI] [PubMed] [Google Scholar]
- 37.Harman FS, Nicol CJ, Marin HE, Ward JM, Gonzalez FJ, Peters JM. Peroxisome proliferator-activated receptor-delta attenuates colon carcinogenesis. Nat Med. 2004;10(5):481–483. doi: 10.1038/nm1026. 2004. [DOI] [PubMed] [Google Scholar]
- 38.Cameron EE, Bachman KE, Myohanen S, Herman JG, Baylin SB. Synergy of demethylation and histone deacetylase inhibition in the reexpression of genes silenced in cancer. Nat Genet. 1999;21(1):103–107. doi: 10.1038/5047. [DOI] [PubMed] [Google Scholar]
- 39.Mund C, Hackanson B, Stresemann C, Lubbert M, Lyko F. Characterization of DNA demethylation effects induced by 5-Aza-2′-Deoxycytidine in patients with myelodysplastic syndrome. Cancer Res. 2005;65(16):7086–7090. doi: 10.1158/0008-5472.CAN-05-0695. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}