

Abstract
Pioneering work with the Bcr-Abl inhibitor, imatinib, demonstrated a requirement for constant Bcr-Abl inhibition to achieve maximal therapeutic benefit in treating chronic myeloid leukemia (CML), establishing a paradigm that has guided further drug development for this disease. Surprisingly, the second-generation Bcr-Abl inhibitor, dasatinib, was reported to be clinically effective with once-daily dosing, despite a short (3- to 5-hour) plasma half-life. Consistent with this observation, dasatinib treatment of progenitor cells from chronic-phase CML patients for 4 hours, followed by washout, or continuously for 72 hours both resulted in an induction of apoptosis and a reduction in the number of clonogenic cells. Such acute treatments with clinically achievable dasatinib concentrations also irreversibly committed Bcr-Abl+ CML cell lines to apoptotic cell death. Potent transient Bcr-Abl inhibition using the alternative inhibitor, nilotinib, also resulted in cell death. These findings demonstrate that in vitro assays designed to model in vivo pharmacokinetics can predict clinical efficacy. Furthermore, they challenge the widely held notion that continuous target inhibition is required for optimal efficacy of kinase inhibitors.
Introduction
Chronic myeloid leukemia (CML) is characterized by a 9;22 chromosomal translocation, which generates the oncogenic Bcr-Abl protein.1 Because constitutive Bcr-Abl kinase activity is essential for its transforming capacity, Bcr-Abl is an ideal target for directed molecular therapy.2,3 Several Bcr-Abl inhibitors, first imatinib,4 and more recently dasatinib5 and nilotinib,6 were developed and are now used to treat CML.
Preclinical data have suggested that exposure of CML cell lines to 1 μM imatinib (achieving > 90% inhibition of Bcr-Abl) for at least 16 hours is required to irreversibly commit them to apoptotic death7 (and B.J.D., unpublished data, June 1999). The requirement for prolonged imatinib exposure was confirmed in mouse studies where once-daily dosing led to inhibition of tumor growth, but not to eradication of disease.4,7,8 Because the half-life of imatinib in mice is 4 hours, 3-times-daily dosing was required for continuous inhibition of Bcr-Abl kinase activity over a 24-hour time course.4,7,8
Subsequent clinical studies showed significant responses with trough doses more than 1 μM,9 leading to the consensus that continuous Bcr-Abl inhibition is required for maximal therapeutic benefit. Accumulated data have since confirmed a consistent association between higher trough imatinib plasma concentration and better response rates.10,11 Similarly, the extent of Bcr-Abl inhibition, as monitored by in vivo phosphorylation of the adaptor protein CrkL, was predictive of response.12 The Bcr-Abl–dependent phosphorylation of CrkL13 serves as an established biomarker of Bcr-Abl activity that can be observed as a phosphorylation-dependent gel shift,14–16 or using a CrkL pY207-directed antibody.17
Compared with imatinib, both dasatinib and nilotinib have enhanced potency against Bcr-Abl and both exhibit activity against most imatinib-resistant Bcr-Abl mutants.18 However, several observations suggest that the prevailing dogma of continuous kinase inhibition for the treatment of CML may not apply to dasatinib. Whereas imatinib and nilotinib are relatively stable in plasma (half-life > 15 hours),9,19 clinical pharmacokinetic studies revealed that dasatinib has a plasma half-life of 3 to 5 hours.20 Further, dasatinib clearance was accompanied by rephosphorylation of CrkL.21–23 This clinical observation mirrors preclinical murine studies, in which the plasma half-life of dasatinib was less than 1 hour and CrkL phosphorylation reached pretreatment levels by 24 hours.24 Despite its short plasma half-life, responses to dasatinib in both preclinical CML xenograft models and phase 1 clinical evaluation were noted even with once-daily dosing.24,25 A follow-up study in patients with chronic-phase CML found equivalent efficacy of once- versus twice-daily dosing, although longer follow-up is required to ascertain whether the responses are equally durable.26 These data suggest that, for dasatinib, continuous Bcr-Abl inhibition is not required to eliminate CML cells. We therefore designed experiments to analyze whether transient exposure of CML cells to dasatinib at clinically relevant concentrations followed by washout would irrevocably commit cells to undergo apoptosis.
Methods
Bcr-Abl–positive human leukemic cell lines, LAMA-84, KYO-1, and K562, as well as the Bcr-Abl–negative leukemic cell line HEL, were maintained in RPMI 1640 medium containing 4 mM l-glutamine, 100 units/mL penicillin, 100 μg/mL streptomycin, and 10% fetal bovine serum.
For washout experiments, cell lines were cultured for 0.5 to 8 hours, as indicated, in media either untreated or containing dasatinib (10, 25, 50, or 100 nM) or nilotinib (100, 500, 1000, or 5000 nM), followed by 3 washes with warm media over 30 minutes. Inhibitor stock solutions were prepared as previously described.18 After washout, cells were cultured in inhibitor-free media until analyzed for apoptosis at 72 hours by staining with annexin V-phycoerythrin and 7-amino-actinomycin D.18 Apoptosis values were measured in triplicate. Viable cell counts were also obtained at 72 hours using the Viacount Assay (Guava Technologies). For comparison, cells exposed to each inhibitor concentration for 72 hours without washout were also analyzed.
Samples were obtained from peripheral blood of 3 patients with newly diagnosed (untreated) chronic-phase CML and one patient with myeloid blast-crisis CML with only T315I mutant BCR-ABL detectable, after informed consent was obtain in accordance with the Declaration of Helsinki as approved by the Institutional Review Boards at Oregon Health & Science University and the University of Leipzig. Mononuclear cells were isolated by density gradient centrifugation, and progenitor cells were subsequently obtained by immunomagnetic cell separation using magnetic-activated cell sorting CD34-directed microbeads (Miltenyi Biotech). For washout experiments, progenitor cells were cultured as for cell lines. In addition, at 72 hours, clonogenic granulocyte/macrophage progenitor cells were enumerated by plating in cytokine-supplemented methylcellulose culture (H4534; StemCell Technologies) in the absence of inhibitor. Colony number was quantified after 14 to 16 days.
Bcr-Abl activity was monitored in 3 ways. For LAMA-84, KYO-1, and K562 cell lines, aliquots were obtained just before washout and at 0.5, 2, and 4 hours after washout for immunoblotting. Identically loaded (1.5 × 104 cells/lane) 4% to 15% Criterion gels (Bio-Rad) were run under conditions where phosphorylated and unphosphorylated CrkL comigrate. Membranes were probed in parallel with antibodies against total CrkL (C-20; Santa Cruz Biotechnology) and phospho-CrkL(pY207) (Cell Signaling Technology). For patient samples, Bcr-Abl activity was assessed at 24 hours by staining with phospho-tyrosine directed antibody (4G10-FITC; Upstate Biotechnology) and quantified by flow cytometry. When cell numbers permitted, the sample was immunoblotted in parallel. Specifically, 1.5 × 106 cells per lane were run on a 15% 15-cm gel to allow for separation of phosphorylated and unphosphorylated CrkL bands and immunoblotted with antibodies against total CrkL (C-20; Santa Cruz Biotechnology). Phosphorylated (upper band) and unphosphorylated (lower band) CrkL were quantified using ImageQuant 5.2 software (Molecular Dynamics), and percentage phosphorylation was reported.
All graphs were generated and statistical analysis was performed using Prism software (GraphPad). P values were calculated using 2-way analysis of variance with a Bonferroni posttest comparison with untreated.
Results and discussion
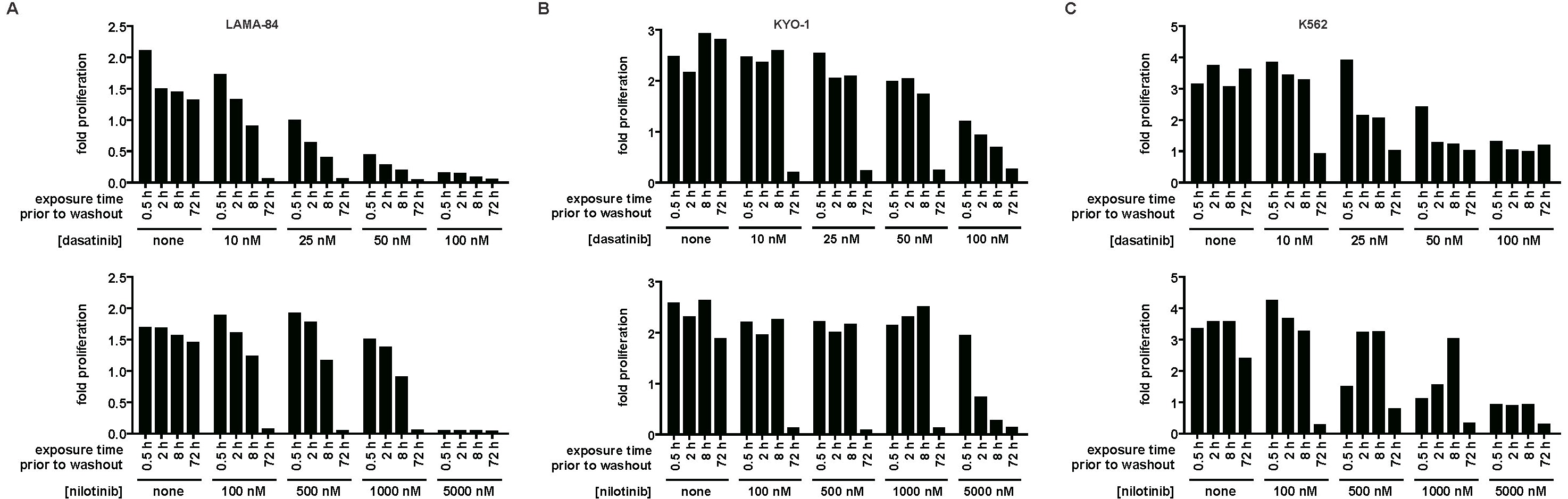
The minimal dasatinib treatment time for reduction of CML cell viability was determined in 3 cell lines. LAMA-84 (Figure 1A), KYO-1 (Figure 1B), and K562 cells (Figure 1C) were treated for increasing durations with a range of dasatinib concentrations followed by thorough washout and culture in the absence of dasatinib. Treatment of CML cells with dasatinib resulted in a concentration- and time-dependent increase in cells undergoing apoptosis (Figure 1A-C). A similar time- and concentration-dependent decrease in proliferation was also observed (supplemental Figure 1, available on the Blood website; see the Supplemental Materials link at the top of the online article). The striking ability for dasatinib to induce apoptosis after transient exposure was a Bcr-Abl–dependent phenomenon, as apoptosis was not observed in an identically treated Bcr-Abl–negative leukemic cell line (supplemental Figure 2).
Figure 1.

Response of Bcr-Abl–positive CML cells to treatment duration with various concentrations of dasatinib. LAMA-84 (A), KYO-1 (B), and K562 (C) cells were treated with 0, 10, 25, 50, or 100 nM dasatinib for the duration indicated, followed by washout, and monitored at 72 hours for annexin V staining. Averages ± SEM are reported for multiple independent experiments (LAMA-84, n = 3; KYO-1, n = 2; K562, n = 2). (D-F) Bcr-Abl activity was monitored with an antibody to phosphorylated CrkL (pY207) after treatment of LAMA-84 (D), KYO-1 (E), and K562 (F) cells with 0, 10, 25, 50, or 100 nM dasatinib for the duration indicated and for 4 hours after inhibitor washout. Total CrkL was examined in parallel as a total protein control. Experiments were performed at least twice for each cell line, and a representative dataset is shown.
The proportion of apoptotic LAMA-84 cells began to significantly rise above the untreated control with dasatinib exposures as brief as 0.5 hour at the lowest (10 nM) dose (n = 3, P < .001). Remarkably, near-maximal apoptosis levels were observed with exposure to 100 nM dasatinib (within the maximum plasma concentration range20), independent of exposure time (Figure 1A).
These experiments were designed to closely mimic relevant clinical parameters (such as clinically achievable dasatinib concentrations), including the observed inhibition and reactivation of Bcr-Abl with once-daily dasatinib, as monitored by CrkL phosphorylation.21 Phosphorylated CrkL was assayed by phosphorylation-dependent gel shift21; however, to increase throughput, we used the pY207 phospho-specific CrkL antibody and a total CrkL loading control. The dose-response to dasatinib observed using each of these methods was comparable (supplemental Figure 3). Importantly, CrkL phosphorylation was restored to levels at or near pretreatment levels by 4 hours after washout and is thus an appropriate mimic of once-daily dasatinib treatment (Figure 1D-F).
The efficacy of transient dasatinib treatment is in sharp contrast to previous work in which constant exposure to imatinib was required to commit cells to apoptosis.7 Thus, we aimed to determine whether the discrepancy could be explained by the increased potency of dasatinib relative to imatinib against Bcr-Abl or whether it was the result of some other property of dasatinib. Achieving equal potency to dasatinib (325 times more potent) would require imatinib administration at up to 32.5 μM (16 042 ng/mL), well above (∼ 4 to 6-fold) peak plasma concentrations at clinical doses.9 Nilotinib is a structurally related imatinib derivative with increased potency. Therefore, we examined the effect of nilotinib treatments on cellular apoptosis in parallel with dasatinib.
Consistent with requirement for potent Bcr-Abl inhibition for efficacious transient inhibitor treatment, 5000 nM nilotinib induced apoptosis with transient exposure (Figure 2A-C). However, the response to nilotinib treatment differed slightly from dasatinib. Although nilotinib is approximately 1-log less potent than dasatinib against Bcr-Abl,5,6,18 a 2-log higher dose of nilotinib was required before apoptosis above background after a 0.5-hour exposure was observed (10 nM dasatinib, n = 3, P < .001; and 1000 nM nilotinib, n = 3, P < .001). As with LAMA-84 cells, K562 and KYO-1 cells did not respond to 100 nM nilotinib, although intermediate nilotinib concentrations induced an inconsistent response (Figure 2B-C). With no difference in the duration and degree of Bcr-Abl inhibition, as monitored by CrkL phosphorylation (Figures 1D-F, 2D-F), the underlying cause for the minor differences in response between dasatinib and nilotinib at short exposure times remains an open question, with differential kinase domain conformational binding mode6,27–29 and degree of target specificity6,8,29,30 being notable differences between the 2 compounds. Despite these differences, our data demonstrate that potent transient inhibition of Bcr-Abl using either of 2 inhibitors with unrelated structural scaffolds induces apoptosis in CML cells. Clinically, this is most relevant to dasatinib administration, as the nilotinib concentrations that were efficacious in our assay approach or exceed maximum plasma levels, and the long plasma half-life of nilotinib precludes the observation of transient nilotinib treatment in the clinic.19
Figure 2.

Response of Bcr-Abl–positive CML cells to treatment duration with various concentrations of nilotinib. LAMA-84 (A), KYO-1 (B), and K562 (C) cells were treated with 0, 100, 500, 1000, or 5000 nM nilotinib for the duration indicated, followed by washout, and monitored at 72 hours for annexin V staining. Averages ± SEM are reported for multiple independent experiments (LAMA-84, n = 3; KYO-1, n = 2; K562, n = 2). (D-F) Bcr-Abl activity was monitored with an antibody to phosphorylated CrkL (pY207) after treatment of LAMA-84 (D), KYO-1 (E), and K562 (F) cells with 0, 100, 500, 1000, or 5000 nM nilotinib for the duration indicated and for 4 hours after inhibitor washout. Total CrkL was examined in parallel as a total protein control. Experiments were performed at least twice for each cell line, and a representative dataset is shown.
Our data demonstrate that the in vitro CML model can be predictive of a clinical phenomenon. However, the 3 lines used are all derived from blast-crisis CML. Thus, the studies were extended to assess the effect of transient dasatinib exposure on primary progenitor cells isolated from patients with chronic-phase CML. Myeloid progenitors from chronic-phase CML can be cultured in vitro and have reduced proliferation on dasatinib treatment.31 These cells also respond to Bcr-Abl inhibition by dasatinib with inhibition-induced apoptosis when cultured in the absence of exogenous growth factors.32 An apoptotic response to 4-hour dasatinib treatment was observed in 3 independently tested CML samples (CML 1-3; Figure 3A). The degree of response was similar to K562 cells cultured under the same conditions. In addition, primary CML samples treated transiently with 100 nM dasatinib had a reduced absolute number of viable cells after 72 hours, compared with untreated (Figure 3B) and a reduced number of cells with granulocyte/macrophage clonogenic potential (Figure 3D). Transiently treated CML samples reactivated Bcr-Abl to a similar extent as K562 cells (Figure 3C), as monitored by mean phosphotyrosine level per cell. For one patient (CML 3), for which sufficient cells were available, CrkL phosphorylation could be assessed by immunoblot and was also found to be restored (Figure 3E). In contrast, cells from a CML patient positive for Bcr-AblT315I (CML R) did not respond to 100 nM dasatinib treatment, either continuously or with washout (Figure 3A-D), indicating that the effect of acute dasatinib treatment on primary CML cells requires Bcr-Abl inhibition.
Figure 3.

Response of primary CML progenitor cells to treatment duration with dasatinib. Progenitor cells isolated from 3 chronic-phase CML patients (CML 1-3) and a blast-crisis CML patient with 100% Bcr-AblT315I (CML R) were cultured in 100 nM dasatinib with (+) or without (−) washout at 4 hours, as indicated, or in the absence of dasatinib as an untreated control. K562 cells were treated identically to primary cells for comparison (gray bars, mean ± SEM from 2 independent experiments). (A) Percentage annexin V+ cells at 72 hours. Drug-specific apoptosis was calculated by subtracting the percentage annexin+ cells in the untreated arm. (B) Proliferation over 72 hours calculated as the fold expansion of viable cells during the 72-hour culture period, expressed as a percentage of untreated control. For K562, mean ± SEM of 2 independent experiments is reported. (C) Bcr-Abl activity at 24 hours, as measured by total phosphotyrosine level per cell and expressed as a percentage of the level in untreated cells. MFI indicates mean fluorescence intensity. (D) Proliferation of granulocyte/macrophage colony-forming cells. The number of cells at 72 hours that could lead to colony formation were enumerated and expressed as a percentage of untreated control. Colony-forming cell number was assayed in triplicate and mean ± SEM reported. (E) Bcr-Abl activity at 24 hours in CML 3 as assayed by CrkL phosphorylation. Western blot of total CrkL (bottom panel) showing phosphorylated (top band) and unphosphorylated (bottom band) forms. Top and bottom bands were quantified and the percentage phosphorylated plotted. (B-E) Dotted line represents the value for the untreated control.
In contrast to the paradigm established with imatinib, we demonstrate that continuous inactivation of Bcr-Abl with clinically relevant concentrations of dasatinib is not required to induce cell death and that acute treatment with dasatinib is effective against Bcr-Abl–positive cell lines and primary cells. In LAMA-84 cells, both dasatinib and nilotinib induce a dose-dependent increase in apoptosis with exposure time. This is consistent with a study that found increasing sensitivity to imatinib with increasing exposure time, albeit on a longer time frame than we observed with dasatinib.33 In this scenario, transient inhibition of an oncogenic kinase above a specific threshold would commit cells to apoptosis. Sharma et al have proposed a unifying mechanism termed “oncogenic shock” for tumor cell death as a result of a transient intracellular signaling imbalance on oncogene inhibition.34 Alternatively, extremely low amounts of residual Bcr-Abl inhibition below a threshold detectable by phospho-CrkL could still support a sustained signaling event leading to cell death.
Our work supports the potential for pulse treatment with some oncogene inhibitors, and in particular with extremely potent Bcr-Abl inhibitors in treating CML. Pulse treatments have been highly efficacious in other contexts, specifically to maximize concentration-dependent bacterial killing and minimize time-dependent toxicity exhibited by aminoglycosides.35 Similarly, clinical application of dasatinib pulse-dosing may reduce associated toxicities, which include adverse hematologic and gastrointestinal effects, fluid retention, pleural effusion, and headache.25 In a study of chronic-phase patients treated with 70 mg twice daily, the majority of patients required dose reduction (66%) or interruption (83%) resulting from toxicity.36 In a comparison of 100 mg once daily, 50 mg twice daily, 140 mg once daily, and 70 mg twice daily (the recommended dose), each dosing schedule was found to have similar time to, and duration of, cytogenic response with a minimum follow-up of 6 months; however, 100 mg once daily had reduced adverse events, including a significant reduction in instances of pleural effusion (P = .024).26
It remains a formal possibility that a long-acting metabolite of dasatinib is present in vivo and could be responsible for the clinical efficacy of once-daily dasatinib; however, we demonstrate that such a metabolite need not be invoked, as short exposures to dasatinib itself are sufficient for in vitro efficacy. Although the evidence to date favors once-daily dasatinib dosing, we did observe that the degree of apoptosis after transient exposure increased with increasing dasatinib concentration. Clones bearing Bcr-Abl kinase domain mutations that confer decreased sensitivity to dasatinib may not respond as well to transient exposure. Whether the frequency of mutant clone emergence will be increased in the once-daily treatment arm as the trial continues is yet undetermined.
Beyond supporting a revised paradigm for Bcr-Abl inhibition in treating CML, our results emphasize that, even among inhibitors directed against a common target, differences in target potency and toxicity profile necessitate determination of optimal dosing strategies for each inhibitor. For dasatinib, sole reliance on drug pharmacodynamics was not an effective comprehensive strategy for establishing an efficacious dosing schedule. Compared across relevant cell lines, in vitro efficacy of pulse-dosing from preclinical studies may be used to inform clinical trials.
While our manuscript was in review, an article was published highlighting the efficacy of transient Bcr-Abl inhibition.23 Our work provides further evidence of this effect and extends the findings to culture of primary CML cells.
Supplementary Material
Acknowledgments
This work was supported by grants from the Leukemia & Lymphoma Society and the National Institutes of Health (5R01CA65823 and 5T32HL007781).
Footnotes
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Authorship
Contribution: J.L.S., T.O., L.T.A, T.L., and C.A.E. performed research; J.L.S., T.O., M.W.D., and B.J.D. designed research and analyzed data; J.L.S. and B.J.D. wrote the paper; and all authors reviewed the manuscript.
Conflict-of-interest disclosure: M.W.D. is a consultant for Novartis and Bristol-Myers Squibb. B.J.D. is an unpaid consultant for Bristol-Myers Squibb. Oregon Health & Science University has clinical trial contracts with Novartis and Bristol-Myers Squibb to pay for patient costs, nurse and data manager salaries, and institutional overhead. B.J.D. does not derive salary, nor does his laboratory receive funds from these contracts. The remaining authors declare no competing financial interests.
Correspondence: Brian J. Druker, Oregon Health & Science University, 3181 SW Sam Jackson Park Rd, Portland, OR 97239; e-mail: drukerb@ohsu.edu.
References
- 1.Rowley JD. A new consistent chromosomal abnormality in chronic myelogenous leukaemia identified by quinacrine fluorescence and Giemsa staining [letter]. Nature. 1973;243(5405):290–293. doi: 10.1038/243290a0. [DOI] [PubMed] [Google Scholar]
- 2.Daley GQ, Van Etten RA, Baltimore D. Induction of chronic myelogenous leukemia in mice by the P210bcr/abl gene of the Philadelphia chromosome. Science. 1990;247(4944):824–830. doi: 10.1126/science.2406902. [DOI] [PubMed] [Google Scholar]
- 3.Lugo TG, Pendergast AM, Muller AJ, Witte ON. Tyrosine kinase activity and transformation potency of bcr-abl oncogene products. Science. 1990;247(4946):1079–1082. doi: 10.1126/science.2408149. [DOI] [PubMed] [Google Scholar]
- 4.Buchdunger E, Zimmermann J, Mett H, et al. Inhibition of the Abl protein-tyrosine kinase in vitro and in vivo by a 2-phenylaminopyrimidine derivative. Cancer Res. 1996;56(1):100–104. [PubMed] [Google Scholar]
- 5.Shah NP, Tran C, Lee FY, Chen P, Norris D, Sawyers CL. Overriding imatinib resistance with a novel ABL kinase inhibitor. Science. 2004;305(5682):399–401. doi: 10.1126/science.1099480. [DOI] [PubMed] [Google Scholar]
- 6.Weisberg E, Manley PW, Breitenstein W, et al. Characterization of AMN107, a selective inhibitor of native and mutant Bcr-Abl. Cancer Cell. 2005;7(2):129–141. doi: 10.1016/j.ccr.2005.01.007. [DOI] [PubMed] [Google Scholar]
- 7.le Coutre P, Mologni L, Cleris L, et al. In vivo eradication of human BCR/ABL-positive leukemia cells with an ABL kinase inhibitor. J Natl Cancer Inst. 1999;91(2):163–168. doi: 10.1093/jnci/91.2.163. [DOI] [PubMed] [Google Scholar]
- 8.Druker BJ, Tamura S, Buchdunger E, et al. Effects of a selective inhibitor of the Abl tyrosine kinase on the growth of Bcr-Abl positive cells. Nat Med. 1996;2(5):561–566. doi: 10.1038/nm0596-561. [DOI] [PubMed] [Google Scholar]
- 9.Peng B, Hayes M, Resta D, et al. Pharmacokinetics and pharmacodynamics of imatinib in a phase I trial with chronic myeloid leukemia patients. J Clin Oncol. 2004;22(5):935–942. doi: 10.1200/JCO.2004.03.050. [DOI] [PubMed] [Google Scholar]
- 10.Larson RA, Druker BJ, Guilhot F, et al. Imatinib pharmacokinetics and its correlation with response and safety in chronic-phase chronic myeloid leukemia: a subanalysis of the IRIS study. Blood. 2008;111(8):4022–4028. doi: 10.1182/blood-2007-10-116475. [DOI] [PubMed] [Google Scholar]
- 11.Picard S, Titier K, Etienne G, et al. Trough imatinib plasma levels are associated with both cytogenetic and molecular responses to standard-dose imatinib in chronic myeloid leukemia. Blood. 2007;109(8):3496–3499. doi: 10.1182/blood-2006-07-036012. [DOI] [PubMed] [Google Scholar]
- 12.White D, Saunders V, Grigg A, et al. Measurement of in vivo BCR-ABL kinase inhibition to monitor imatinib-induced target blockade and predict response in chronic myeloid leukemia. J Clin Oncol. 2007;25(28):4445–4451. doi: 10.1200/JCO.2006.09.9499. [DOI] [PubMed] [Google Scholar]
- 13.de Jong R, ten Hoeve J, Heisterkamp N, Groffen J. Tyrosine 207 in CRKL is the BCR/ABL phosphorylation site. Oncogene. 1997;14(5):507–513. doi: 10.1038/sj.onc.1200885. [DOI] [PubMed] [Google Scholar]
- 14.Nichols GL, Raines MA, Vera JC, Lacomis L, Tempst P, Golde DW. Identification of CRKL as the constitutively phosphorylated 39-kD tyrosine phosphoprotein in chronic myelogenous leukemia cells. Blood. 1994;84(9):2912–2918. [PubMed] [Google Scholar]
- 15.Oda T, Heaney C, Hagopian JR, Okuda K, Griffin JD, Druker BJ. Crkl is the major tyrosine-phosphorylated protein in neutrophils from patients with chronic myelogenous leukemia. J Biol Chem. 1994;269(37):22925–22928. [PubMed] [Google Scholar]
- 16.ten Hoeve J, Arlinghaus RB, Guo JQ, Heisterkamp N, Groffen J. Tyrosine phosphorylation of CRKL in Philadelphia+ leukemia. Blood. 1994;84(6):1731–1736. [PubMed] [Google Scholar]
- 17.Roumiantsev S, Krause DS, Neumann CA, et al. Distinct stem cell myeloproliferative/T lymphoma syndromes induced by ZNF198-FGFR1 and BCR-FGFR1 fusion genes from 8p11 translocations. Cancer Cell. 2004;5(3):287–298. doi: 10.1016/s1535-6108(04)00053-4. [DOI] [PubMed] [Google Scholar]
- 18.O'Hare T, Walters DK, Stoffregen EP, et al. In vitro activity of Bcr-Abl inhibitors AMN107 and BMS-354825 against clinically relevant imatinib-resistant Abl kinase domain mutants. Cancer Res. 2005;65(11):4500–4505. doi: 10.1158/0008-5472.CAN-05-0259. [DOI] [PubMed] [Google Scholar]
- 19.Kantarjian H, Giles F, Wunderle L, et al. Nilotinib in imatinib-resistant CML and Philadelphia chromosome-positive ALL. N Engl J Med. 2006;354(24):2542–2551. doi: 10.1056/NEJMoa055104. [DOI] [PubMed] [Google Scholar]
- 20.Bullock JM, Noory C, Men A, Ramchandani R, Kenna L. Clinical pharmacology review: Spycel, dasatinib [Application no. 21-986]. U.S. Food and Drug Administration, Center for Drug Evaluation and Research. 2006 [Google Scholar]
- 21.Bixby DL, Talpaz M. Efficacy of various doses and schedules of second-generation tyrosine kinase inhibitors. Clin Lymphoma Myeloma. 2008;8:S95–S106. doi: 10.3816/CLM.2008.s.005. [DOI] [PubMed] [Google Scholar]
- 22.Talpaz M, Kantarjian HM, Paquette R, et al. A phase 1 study of BMS-354825 in patients with imatinib-resistant and intolerant chronic phase chronic myeloid leukemia (CML): results from CA180002 [abstract]. J Clin Oncol. 2005;23(suppl 16):6564S. Abstract 6519. [Google Scholar]
- 23.Shah NP, Kasap C, Weier C, et al. Transient potent BCR-ABL inhibition is sufficient to commit chronic myeloid leukemia cells irreversibly to apoptosis. Cancer Cell. 2008;14(6):485–493. doi: 10.1016/j.ccr.2008.11.001. [DOI] [PubMed] [Google Scholar]
- 24.Luo FR, Yang Z, Camuso A, et al. Dasatinib (BMS-354825) pharmacokinetics and pharmacodynamic biomarkers in animal models predict optimal clinical exposure. Clin Cancer Res. 2006;12(23):7180–7186. doi: 10.1158/1078-0432.CCR-06-1112. [DOI] [PubMed] [Google Scholar]
- 25.Talpaz M, Shah NP, Kantarjian H, et al. Dasatinib in imatinib-resistant Philadelphia chromosome-positive leukemias. N Engl J Med. 2006;354(24):2531–2541. doi: 10.1056/NEJMoa055229. [DOI] [PubMed] [Google Scholar]
- 26.Shah NP, Kantarjian HM, Kim DW, et al. Intermittent target inhibition with dasatinib 100 mg once daily preserves efficacy and improves tolerability in imatinib-resistant and -intolerant chronic-phase chronic myeloid leukemia. J Clin Oncol. 2008;26(19):3204–3212. doi: 10.1200/JCO.2007.14.9260. [DOI] [PubMed] [Google Scholar]
- 27.Schindler T, Bornmann W, Pellicena P, Miller WT, Clarkson B, Kuriyan J. Structural mechanism for STI-571 inhibition of abelson tyrosine kinase. Science. 2000;289(5486):1938–1942. doi: 10.1126/science.289.5486.1938. [DOI] [PubMed] [Google Scholar]
- 28.Tokarski JS, Newitt JA, Chang CY, et al. The structure of Dasatinib (BMS-354825) bound to activated ABL kinase domain elucidates its inhibitory activity against imatinib-resistant ABL mutants. Cancer Res. 2006;66(11):5790–5797. doi: 10.1158/0008-5472.CAN-05-4187. [DOI] [PubMed] [Google Scholar]
- 29.Lombardo LJ, Lee FY, Chen P, et al. Discovery of N-(2-chloro-6-methyl- phenyl)-2-(6-(4-(2-hydroxyethyl)-piperazin-1-yl)-2-methylpyrimidin-4-ylamino)thiazole-5-carboxamide (BMS-354825), a dual Src/Abl kinase inhibitor with potent antitumor activity in preclinical assays. J Med Chem. 2004;47(27):6658–6661. doi: 10.1021/jm049486a. [DOI] [PubMed] [Google Scholar]
- 30.Rix U, Hantschel O, Durnberger G, et al. Chemical proteomic profiles of the BCR-ABL inhibitors imatinib, nilotinib and dasatinib reveal novel kinase and non-kinase targets. Blood. 2007;110(12):4055–4063. doi: 10.1182/blood-2007-07-102061. [DOI] [PubMed] [Google Scholar]
- 31.Copland M, Hamilton A, Elrick LJ, et al. Dasatinib (BMS-354825) targets an earlier progenitor population than imatinib in primary CML but does not eliminate the quiescent fraction. Blood. 2006;107(11):4532–4539. doi: 10.1182/blood-2005-07-2947. [DOI] [PubMed] [Google Scholar]
- 32.Nguyen TK, Rahmani M, Harada H, Dent P, Grant S. MEK1/2 inhibitors sensitize Bcr/Abl+ human leukemia cells to the dual Abl/Src inhibitor BMS-354/825. Blood. 2007;109(9):4006–4015. doi: 10.1182/blood-2006-09-045039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mow BM, Chandra J, Svingen PA, et al. Effects of the Bcr/abl kinase inhibitors STI571 and adaphostin (NSC 680410) on chronic myelogenous leukemia cells in vitro. Blood. 2002;99(2):664–671. doi: 10.1182/blood.v99.2.664. [DOI] [PubMed] [Google Scholar]
- 34.Sharma SV, Gajowniczek P, Way IP, et al. A common signaling cascade may underlie “addiction” to the Src, BCR-ABL, and EGF receptor oncogenes. Cancer Cell. 2006;10(5):425–435. doi: 10.1016/j.ccr.2006.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nicolau DP, Freeman CD, Belliveau PP, Nightingale CH, Ross JW, Quintiliani R. Experience with a once-daily aminoglycoside program administered to 2184 adult patients. Antimicrob Agents Chemother. 1995;39(3):650–655. doi: 10.1128/AAC.39.3.650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kantarjian H, Pasquini R, Hamerschlak N, et al. Dasatinib or high-dose imatinib for chronic-phase chronic myeloid leukemia after failure of first-line imatinib: a randomized phase 2 trial. Blood. 2007;109(12):5143–5150. doi: 10.1182/blood-2006-11-056028. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}