

SUMMARY
Objective
To determine haplotype background of common mutations in the genes encoding surfactant proteins B and C (SFTPB and SFTPC) and to assess recombination in SFTPC.
Study design
Using comprehensive resequencing of SFTPC and SFTPB, we assessed linkage disequilibrium (LD) (D’), and computationally inferred haplotypes. We computed average recombination rates and Bayes factors (BFs) within SFTPC in a population cohort and near SFTPC (±50kb) in HapMap cohorts. We then biochemically confirmed haplotypes in families with sporadic SFTPC mutations (n=11) and in individuals with the common SFTPB mutation (121ins2, n=34).
Results
We detected strong evidence (weak LD and BFs > 1400) for an intragenic recombination hot spot in both genes. The 121ins2 SFTPB mutation occurred predominantly (89%) on 2 common haplotypes. In contrast, no consistent haplotypes were associated with mutated SFTPC alleles. Sporadic SFTPC mutations arose on the paternal allele in 4 of 5 families; the remaining child had evidence for somatic recombination on the mutated allele.
Conclusions
In contrast to SFTPB, disease alleles at SFTPC do not share a common haplotype background. Most sporadic mutations in SFTPC occurred on the paternal allele, but somatic recombination may be an important mechanism of mutation in SFTPC.
Keywords: interstitial lung disease, children, genetics, haplotype, linkage disequilibrium
INTRODUCTION
Pulmonary surfactant is a phospholipid-protein complex synthesized by pulmonary alveolar type II cells that lines alveoli at the air-liquid interface, maintains alveolar patency at end expiration, and is required for successful fetal-neonatal pulmonary transition. Mutations in genes encoding two hydrophobic surfactant associated proteins, surfactant proteins-B and -C (SP-B and SP-C), result in surfactant dysfunction and lung disease in children 1. The 9.7kb SP-B gene (SFTPB) encodes a 381 amino acid proprotein that undergoes sequential proteolytic cleavages and glycosylation to yield a 79 amino acid mature peptide. The 3.5 kb SP-C gene (SFTPC) encodes an alternatively spliced 191 or 197 amino acid proprotein (proSP-C) which undergoes sequential proteolytic cleavages and palmitoylation to yield a 35 amino acid mature peptide 2. Rare, recessive loss of function mutations in SFTPB are completely penetrant and result in lethal respiratory distress syndrome in the newborn period 3,4 while dominantly expressed mutations in SFTPC are variably penetrant and may be silent or result in acute or chronic lung disease in individuals ranging from the newborn to adult 5-7.
The most common mutation in SFTPB (>60% of mutated alleles) results from a frameshift due to net insertion of 2 basepairs at codon 121 (121ins2) 3,8. The frequency of the 121ins2 mutation in the general population is 0.1%, and its frequency is enriched >100 fold in cohorts of term infants with severe neonatal respiratory distress 4,9. In a European population, the 121ins2 mutation occurred on 2 common SFTPB haplotypes, suggesting a founder effect and an older mutation despite evidence for high intragenic recombination and low linkage disequilibrium (LD) 10,11. No sporadic mutations in SFTPB in affected families studied have been identified to date.
In contrast, sporadic SFTPC mutations with variable phenotypes occur in approximately 55% of families studied to date. The most common SFTPC mutation results from a nonsynonymous T>C transition at g.1295, resulting in a threonine substitution for isoleucine at codon 73 (I73T) and accounts for approximately 25% of mutations in SFTPC 12. A previous study found no common haplotype background for I73T, suggesting that SFTPC may also have a high degree of recombination 13. To determine whether haplotype background or recombination contributes to sporadic mutation in SFTPC, we assessed linkage disequilibrium (LD) (D’) in families with sporadic SFTPC mutations and in individuals with the common SFTPB mutation (121ins2). We also computationally inferred and biochemically confirmed haplotypes and computed average recombination rates and Bayes factors (BFs) within SFTPC in a population-based cohort and near SFTPC (±50kb) in HapMap cohorts. We report that in contrast to SFTPB, we observed a male germline mutation predominance in SFTPC as well as an example of somatic recombination associated with high sporadic mutation frequency.
MATERIALS AND METHODS
Patient Populations and SFTPC and SFTPB Resequencing
We bidirectionally resequenced SFTPC from lymphocyte-derived DNA obtained from two cohorts of individuals: 1) a “population cohort” of 278 infants ≥ 34 weeks gestation [117 African-American individuals (AA) and 161 European-American individuals (EA)], and 2) a “family collection” comprised of 11 children of European descent with lung disease due to mutations in SFTPC and their families (34 individuals from 11 families, 3 of whom have been included in previous publications13-15). We bidirectionally resequenced SFTPB in individuals carrying the 121ins2 mutation including newborns with SP-B deficiency (n=11 homozygous for 121ins2, and 5 compound heterozygous for 121ins2 and another loss of function mutation) and in 14 heterozygous individuals of predominantly of European descent identified through population based screening of DNA samples obtained from the newborn screening programs of Missouri, Norway and South Africa, as previously described 10,16. In all, 41 alleles from 30 individuals were analyzed. All SFTPB amplification primers are available at http://genome.wustl.edu/activity/med_seq/primers. Amplification primers for SFTPC can be located in the on-line supplementary material.
Linkage Disequilibrium (LD); SFTPC and SFTPB Haplotypes
We used sequence data from the population cohort to estimate pairwise LD (as defined by D’) between all SNPs in SFTPC. Haplotypes were inferred computationally in the population cohort using PHASE and all loci with minor allele frequency (MAF) >5% (n= 22 and 30 loci in AA and EA, respectively) 17 18. Haplotypes in the family collection were inferred for all single nucleotide polymorphisms (SNPs) (including the mutations) using both PHASE and pedigree information. We also inferred haplotypes using common polymorphisms within 50 kb of SFTPC from the HapMap Yoruba (YRI) and European descent (CEU) populations (HapMap Project data release #21 http://www.hapmap.org/ , a catalog of common genetic variants that provides data about haplotypes in populations). To estimate background recombination rate and determine hot spot location, we used PHASE to compute average recombination rates and Bayes factors (BFs), where a BF>10 is evidence of a recombination hot spot 10,19.
To infer haplotypes associated with 121ins2 mutation in SFTPB, we ran PHASE with loci with MAF >5% (n=11) and the 121ins2 mutation on SFTPB sequence data from the 34 individuals with at least one 121ins2 allele, combined with sequence data from 875 European American individuals obtained through the State of Missouri Newborn Screening Program 10.
To screen for evidence of gene conversion or recombination events associated with de novo mutations, we directly ascertained the haplotypes in both the affected child and both parents by cloning long range PCR (LR-PCR) products spanning the mutated SNP from members of the family collection with sporadic SFTPC mutations. After amplification and gel purification (Montage gel extraction kit, Millipore, Bedford, MA) of a single 6.9 kb SFTPC fragment, we ligated the fragment into pCR-XL-TOPO vector (Invitrogen, Carlsbad, CA) according to the manufacturer’s instructions. Multiple clones were picked from each individual and DNA was extracted using the High Pure Plasmid Isolation Kit (Roche Diagnostics, Indianapolis, IN). We sequenced multiple clones from each individual across all heterozygous sites to directly resolve haplotypes across SFTPC in that individual. Computationally inferred 121ins2 bearing haplotypes at SFTPB were also confirmed by amplification and cloning of a single 8.5 kb SFTPB LR-PCR.
Demethylation Transitions in SFTPC and SFTPB
To evaluate the role of demethylation transitions as a mechanism for mutations, we compared the incidence of demethylation transitions from the ancestral allele at CG sites (...CR... where R=A/G base change or ...YG... where Y=C/T base change) in SFTPB and SFTPC as compared to SFTPD in the Seattle SNPs database (http://pga.gs.washington.edu/ ). We determined information about the ancestral allele at each SNP from dbSNP or from a BLAST search of sequence around the SNP against the chimp genome when not available in dbSNP (http://130.14.29.110/BLAST/ ).
Study Approval and Statistical Analysis
The Washington University Human Research Protection Office approved this study. Anonymized blood samples were obtained under a protocol to evaluate the epidemiology of surfactant-associated mutations in diverse ethnic groups. We assembled, reviewed, and edited all sequence data, and we formatted all confirmed polymorphic sites for further statistical and haplotype analysis as previously described 10. We analyzed all data using Statistical Analysis System (SAS, v. 9.3.1) (SAS, Inc., Cary NC).
RESULTS
SFTPC LD, Haplotypes, and Recombination
In a population cohort of 278 infants ≥ 34 weeks gestation [117 African-American individuals (AA) and 161 European-American individuals (EA)], we identified 90 SNPs in SFTPC, including 5 SNPs in coding regions (2 synonymous and 3 non-synonymous), 34 SNPs in intronic regions, and 51 SNPs in the promoter region. We detected no known deleterious variants in this cohort. In a family cohort of 11 children of European descent with lung disease due to mutations in SFTPC and their families (34 individuals from 11 families), we found 8 potentially deleterious variants in children with interstitial lung disease (Table I): these variants were non-synonymous and were predicted to be damaging by 2 homology-based software tools (SIFT or Polyphen; http://blocks.fhcrc.org/sift/SIFT.html; http://genetics.bwh.harvard.edu/pph/), or involved splice sites (donor/acceptor) that could alter exon splicing. Our failure to find these variants in 556 alleles in the population cohort and their locations in evolutionarily conserved regions suggest they may functionally account for the interstitial lung disease in these children.
Table I.
SFTPB and SFTPC Mutations Examined
| Gene | SNP position |
DNA | Function | Observed Alleles |
Independent Alleles |
Sporadic or Inherited |
Allele | Phenotype |
|---|---|---|---|---|---|---|---|---|
| SFTPB | g.1549 | C>GAA | 121ins2 frameshift |
41 | 41 | Inherited | Unknown | |
| SFTPC | g.921 | A>C | H64P | 1 | 1 | Sporadic | Paternal | Onset 1mo; died 14 yrs |
| SFTPC | g.1295 | T>C | I73T | 7 | 4 | Sporadic | Paternal | Onset 4mo; lung transplant at 12 yrs 14 |
| Inherited | From father; (asymptomatic) |
Intermittent symptoms between 7wks and 20 mo; asymptomatic at 3 yrs 15 |
||||||
| Unknown | Unknown | Onset 2mo; lung transplant at 5 yrs |
||||||
| Unknown | Unknown | Onset 14 mo; O2 for 8 mo; asymptomatic at 6 yrs |
||||||
| SFTPC | g.1348 | ACTGCCACC/- | Del ex 91- 93 |
1 | 1 | Sporadic | Somatic recombination |
Onset 3 mo; lung transplant 14 mo 13 |
| SFTPC | g.1634 | G>A | c.350-1G>A | 1 | 1 | Sporadic | Paternal | Onset 4mo; O2 at night; 12 yrs |
| SFTPC | g.1639 | T>C | L110P | 1 | 1 | Sporadic | Paternal | Onset 1 mo; noninvasive ventilation at 2 yrs |
| SFTPC | g.2187 | C>G | L181V | 2 | 1 | Inherited | From mother; (asymptomatic) |
onset 6 wks; died at 18 mo |
| SFTPC | g.2211 | T>G | C189G | 1 | 1 | Unknown | Unknown | onset 1 week; O2 until 2 years; asymptomatic at 3 years |
| SFTPC | g.2211 | T>C | C189R | 2 | 1 | Inherited | From father (symptomatic) |
onset birth; Follow-up unknown |
Despite its small genomic size, we found weak LD within and near SFTPC in both African-American and European-American populations, which suggests that SFTPC includes a recombination hot spot. Computationally inferred haplotype diversity using the common SNPs in the population cohort revealed 104 and 136 haplotypes in the AA and EA populations, respectively, among 117 and 195 individuals from each population, which suggests significant amounts of intragenic recombination. PHASE inferred a very high background recombination rate in the resequencing population, so there was little evidence of a hotspot considering this 6 kb region (low BF of 5.3 and 0.24 respectively). However, when we expanded the genomic context by using HapMap data to examine recombination across 100 kb surrounding SFTPC we obtained BF of 1446 and 1416 in the CEU and YRI populations, respectively (Figure I). Thus, a strong recombination hotspot spans most, if not all, of the SFTPC transcript. We found no common haplotype associated with either the mutated or nonmutated alleles in either the children with known SFTPC mutations or their family members (Table II), suggesting absence of a founder effect.
Figure. Detection of recombination hotspot in SFTPC.
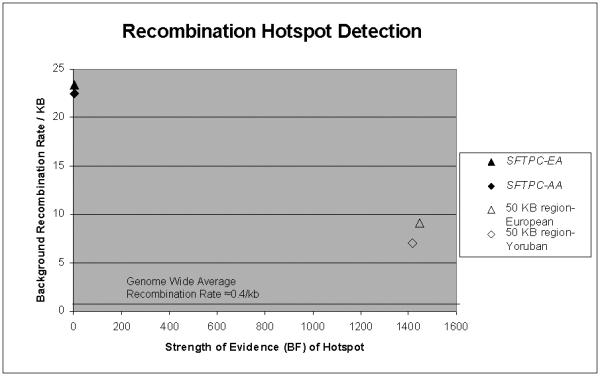
With examining recombination only within the 6 kb of SFTPC using the “population cohort” (filled symbols), the background recombination rate is markedly elevated (20-25 events/kb), making it difficult to detect a hotspot (low BF, 0.24 and 5.3, European (EA) and African (AA) descent, respectively). In contrast, with examining recombination across 100 kb surrounding and including SFTPC using HapMap data (open symbols), background recombination is still elevated (6-10 events/kb), but permits detection of markedly elevated values for the Bayes factor (1446 and 1416, European [CEU] and Yoruba [YRI], respectively) . For comparison, the genome wide average recombination rate is approximately 0.4 events / kb.
Table II.
PHASE SFTPC Haplotypes in European American Population
|
Haplotype Name |
Mutation | Allele | Frequency |
|---|---|---|---|
| A | No |  |
22/136 (16.2%) |
| B | No | 19/136 (14.0%) |
|
| C | No | 15/136 (11.0%) |
|
| D | g.921 | 1/136 | |
| E | g.1295 | 5/136 | |
| F | g.1295 | 1/136 | |
| G | g.1295 | 1/136 | |
| H | g.1348 | 1/136 | |
| I | g.1634 | 1/136 | |
| J | g.1639 | 1/136 | |
| K | g.2187 | 2/136 | |
| L | g.2211 (C189G) |
1/136 | |
| M | g.2211 (C189R) |
2/136 |
Most common and mutational haplotypes seen from 392 alleles. Haplotypes are constructed using SNP sites and family mutations: g.-3183, -2547, -2373, -2372, -1393, -1187, -107, 104, 498, 579, 752, 921, 980, 1295, 1348, 1634, 1639, 1723, 1893, 1975, 2074, 2187, 2203, 2211, 2426, 2443, 2569, 2619, 2620, and 2732 where g.+1 is transcription start site. The mutations are shaded. Haplotype “E” includes individuals from 2 separate families while Haplotype “I” was inherited from mother to child and Haplotype “M” from father to child each within a single family. “-”: variant is an insertion or deletion at the specified locus.
Evidence for Somatic SFTPC Mutation
Among children with interstitial lung disease due to mutations in SFTPC, we found 5 of 11 to have alleles not inherited from either parent that presumably represent sporadic mutations. Biochemical haplotype determination revealed that the sporadic mutation occurred on the paternal allele in 4 of 5 families. Because the transmitted paternal haplotype could be completely phased, we conclude that none of these mutations appears to have occurred coincident with a meiotic recombination event in the paternal germ line. Neither haplotype found in the fifth child occurred in either parent (Table III), and both haplotypes were confirmed in multiple independent clones (mutated haplotype in two clones and nonmutated in three). However, the observed haplotypes are consistent with a somatic recombination event between the inherited maternal and paternal chromosomes. There were no clinical characteristics that distinguished this child’s presentation from those of the other children (Table I).
Table III.
Haplotypes of family demonstrating somatic recombination/mutation event in the child
| Individual | Haplotype | |
|---|---|---|
| Mother | Non-transmitted |  |
| Transmitted | ||
| Child | Mutated | |
| Non-mutated | ||
| Father | Transmitted | |
| Non-transmitted | ||
Haplotypes were constructed using SNPs g.+498, 980, 1348, 1723, 1893, 1975, 2074, and 2203; where g.+1 is the transcription start site. The mutation, a deletion of codons 91-93, is marked by the asterisk.
SFTPB Haplotypes
We identified five SFTPB haplotypes associated with the 121ins2 mutation, two of which represented 90.2% of all 121ins2 haplotypes in this cohort (37/41). A third haplotype accounted for 4.9% of the haplotypes (2/41) and was seen in 2 unrelated families. The other two haplotypes were each seen once in the same individual. We confirmed all 5 haplotypes biochemically. Using 6 SNPs from a study in a Western European population by Tredano et al. (g.-18, 1013, 1580, 4546/4550, 4559/4563, 4564/4568), we found the same two common 121ins2 bearing haplotypes that they reported plus two unique haplotypes in this study 11 (Table IV).
Table IV.
SFTPB 121ins2 Haplotypes
|
Haplotype Name |
121ins2 Allele | Frequency |
|---|---|---|
| A | GAIC*TCATAT | 21/41(51.2%) |
| B | GCIA*TCATAT | 16/41 (39.0%) |
| C | GAIC*TAATAT | 2/41 (4.9%) |
| D | GAIC*CCATAT | 1/41 (2.4%) |
| E | GCIC*CCATAT | 1/41 (2.4%) |
Five 121ins2 haplotypes seen from 41 alleles. Haplotypes are constructed using SNP sites: -384, -18, 750, 1013, 1549, 1580, 4550, 4563, 4568, 5028, 5795 where +1 is transcription start site. I = insertion
121ins2
Demethylation of SFTPB and SFTPC
Neither SFTPB nor SFTPC showed a significant excess of demethylation transitions from the ancestral allele, when compared to SFTPD.
DISCUSSION
SFTPB and SFTPC have important biologic and genomic similarities. Both genes are small (<10 kb) and encode hydrophobic proteins that are critical for function of the pulmonary surfactant. Comprehensive resequencing of SFTPB and SFTPC revealed excess low frequency variation and evidence of intragenic recombination hot spots in both genes 10,20. These similarities raise the possibility that intragenic recombination hotspots are mechanistically related to increased rare variant frequency.
However, mutation inheritance and expression are different in SFTPB and SFTPC. Mutations in SFTPB are inherited in an autosomal recessive manner and are completely penetrant. Heterozygous carriers are asymptomatic during infancy and adulthood 21. Because SFTPB disease alleles can persist silently in the population, affected individuals are more likely to inherit two disease alleles (one from each parent) than to inherit one disease allele and spontaneously mutate the other allele. Consistent with this finding is the limited number of haplotypes upon which the most common SFTPB mutation, 121ins2, occurs, an observation that suggests a founder effect for this mutation, with all observed 121ins2 alleles sharing a single common ancestor in recent history.
In contrast, SFTPC mutations are dominant with variable penetrance and approximately 50% of affected patients have sporadic mutations. Because a single mutated SFTPC allele may result in a lung disease phenotype, we observed a number of sporadic mutations in the clinically affected population but no mutations in the population cohort. In contrast to the founder effect observed in SFTPB, the most common SFTPC mutation (I73T) likely represents a recurrent mutation, because it is observed on a variety of haplotype backgrounds 13.
We sought to determine whether the mechanism of sporadic mutations in SFTPC is related to methylation or intragenic recombination. We found no evidence that either SFTPC or SFTPB is associated with an elevated rate of demethylation associated transitions at CpG dinucleotides in either SFTPB or SFTPC, a common mechanism of mutations across the genome 22. In examining families with sporadic mutations in SFTPC, we began with the hypothesis that the mutations arose as a consequence of meiotic recombination or gene conversion events in the parental germline. In meiosis, cells must orient chromosomes accurately on the meiotic spindle via physical connection by deliberately causing DNA damage and relying on a subsequent repair process 23. It is through this DNA repair process, during the resolution of Halladay junctions, that the potential for mutation events arises. Because we could resolve the parental alleles in 4 of the 5 children with a sporadic mutation, we conclude that meiotic recombination events within SFTPC were not associated with the mutation. Thus, neither CpG demethylation nor meiotic recombination appears to account for the high de novo mutation rate in SFTPC.
The fact that the SFTPC sporadic mutations were observed on the paternal allele in four of the five families is consistent with male mutation bias, first proposed by Haldane in 1935. A male bias in the origin of new mutations should occur if the number of cell divisions between fertilized egg and gamete is higher in males than in females, as observed in humans 24. This bias has been previously demonstrated in Multiple Endocrine Neoplasia 2B patients in whom Carlson et al. demonstrated that 25 of 25 sporadic cases occurred due to mutations on the paternally derived chromosome 25. Although we also observe a trend toward male bias, our sample of sporadic mutations is too small to reject the null hypothesis of equal mutation rates in both sexes.
Our data for the four paternally derived sporadic alleles are not consistent with a recombination or large gene conversion event in the area of the mutation, because paternal haplotypes of markers flanking the mutation bearing haplotype are intact. Thus, paternal meiotic recombination/gene conversion is not obviously driving the recurrent mutations. Gene conversion frequently occurs on a scale of 500bp-2kb 26, so the possibility remains that these mutations arose during undetected gene conversion events, but the trend toward paternal bias suggests that the mutations arose during mitotic divisions.
The del91-93 mutation in the fifth child appears to be a somatic mutation because the two haplotypes recovered from the child, although complementary, are not consistent with either parent. Rather, the haplotypes carried by the child appear to be the products of recombination between a maternal and a paternal haplotype, which could only occur after fertilization. Somatic (or mitotic) recombination also involves repair of double strand breaks and is a frequent occurrence in the mammalian genome as homologous recombination occurs 10-6 to 10-5 times per cell cycle between repeated DNA sequences. If recombination occurs in roughly 1-5kb segments, approximately 10 spontaneous homologous recombination events occur per cell in each cell cycle 27. It is not clear whether the meiotic recombination hotspot in SFTPC is also a somatic recombination hotspot, but if it is, then somatic mutations in SFTPC may contribute to pediatric interstitial lung disease more than previously recognized and may explain some of the variability in disease expression.
Somatic recombination as a mechanism for mutation in non-cancerous diseases has been reported in neurofibromatosis 1 and 2, McCune-Albright syndrome, Paroxysmal nocturnal hemoglobinuria, and males with incontinentia pigmenti. Disorders with proven somatic and germ-line mosaicism include osteogenesis imperfecta II, Duchenne muscular dystrophy, Hunter syndrome, retinoblastoma, neurofibromatosis 2, congenital contractural arachnodactyly, hemophilia B, and hemophilia A 28. In 15 patients with idiopathic atrial fibrillation, Gollob et al. found four mutations in the connexin 40 gene, three of which were present in cardiac tissue only, consistent with somatic mutation. The fourth mutation was found in both heart tissue and lymphocyte DNA, consistent with a germ-line mutation 29.
We believe that the somatic mutation in our patient occurred at a very early stage of development because the majority of both lung and blood cells must have arisen from the mutated cell. Because the child came to clinical attention with interstitial lung disease, both endodermal-derived lung tissue and mesodermal-derived peripheral blood lymphocytes must carry the mutation. The evidence of somatic mutation in this family raises the possibility that mutations at a later stage of development than the one in this family may be detectable to a different extent in lung tissue and peripheral blood. For example, Beck et al. identified a patient with spontaneous early-onset familial Alzheimer’s Disease due to a somatic mutation in the presenilin-1 gene that was detectable in 8% of peripheral lymphocytes and 14% of the cerebral cortex, but enough to cause phenotypic disease 30. As peripheral blood lymphocytes are the most common DNA source used for resequencing, individuals without detectable mutations by resequencing of lymphocyte-derived DNA may carry somatic mutations that could be identified through the resequencing of tissue-specific DNA. Furthermore, this evidence suggests that mutations previously thought to be sporadic may actually be inherited from a parent who carried the mutated allele in the germline but not in lymphocytes. Thus, in the absence of an identifiable mutation in lymphocyte-derived DNA, sequencing SFPTC directly from lung tissue of patients with both sporadic and familial ILD may provide specific insight into the etiology of disease. Studies are underway to determine the concordance of SFTPC genotype between lung tissue and lymphocyte-derived DNA.
Further studies may help to elucidate whether mitosis associated mutation could contribute to other diseases with high sporadic mutation rates, including Tuberous Sclerosis Complex (autosomal dominant with mutation rate approximately 2/3) 31, Multiple Endocrine Neoplasia Type 2B (autosomal dominant, 50% sporadic mutations) 32, Cornelia de Lange syndrome (autosomal dominant, 99% sporadic mutations) 33, and epidermolytic hyperkeratosis (autosomal dominant, 50% sporadic) 34. Better understanding of the mechanisms of mutation may aid in diagnosis of patients with previously negative screening results and will allow for better prospective guidance and reproductive counseling for affected families.
Supplementary Material
ACKNOWLEDGEMENTS
The authors would like to thank Yumi Kasai, PhD of the Genome Sequencing Center at Washington University for overseeing the resequencing of SFTPC in the population cohort, Kimberly Fitzroy for sequencing the 121ins2 carriers, and Peter van Asperen, MD of the University of Sydney, Australia, James Chmiel, MD of Case Western Reserve University, Cleveland, OH, and Jan Kouwenberg, MD, of HAGA Juliana Children’s Hospital, The Hague, Netherlands, for providing DNA samples and clinical data.
This work was supported by grants from the National Heart, Lung, and Blood Institute (HL 82747 and HL 65174 to F.S.C., HL 65385 to A.H.), the Saigh Foundation (FSC and AH), NIH Training Grant T32-HD043010 (ADM), Discovery Labs Pulmonary Research Grant (ADM), NIH Training Grant T32-HD041925 (JW), and Washington University Genome Sequencing Center Pilot Scale Sequencing Program (AH).
Footnotes
This work was presented, in part, at the Pediatric Academic Societies’ Annual Meeting, Toronto, Canada, May 2007 (E-PAS2007:616292.10).
Conflict of interest statement: Dr. McBee’s support from Discovery Labs, Inc. did not introduce any bias into this work. None of the authors has a conflict of interest.
REFERENCES
- 1.Whitsett J, Weaver TE. Mechanisms of disease: hydrophobic surfactant proteins in lung function and disease. N Engl J Med. 2002;347:2141–2148. doi: 10.1056/NEJMra022387. [DOI] [PubMed] [Google Scholar]
- 2.Beers M, Lomax CA, Russo SJ. Synthetic processing of Surfactant Protein C by alveolar epithelial cells. J Biol Chem. 1998;273(24):15287–15293. doi: 10.1074/jbc.273.24.15287. [DOI] [PubMed] [Google Scholar]
- 3.Nogee L, Garnier G, Dietz HC, Singer L, Murphy AM, deMello DE, Colten HR. A mutation in the surfactant protein B gene responsible for fatal neonatal respiratory disease in multiple kindreds. J Clin Invest. 1994;93:1860–1863. doi: 10.1172/JCI117173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cole F, Hamvas A, Rubinstein P, King E, Trusgnich M, Nogee LM, deMello DE, Colten HR. Population-based estimates of surfactant protein B deficiency. Pediatrics. 2000;105:538–541. doi: 10.1542/peds.105.3.538. [DOI] [PubMed] [Google Scholar]
- 5.Nogee L, Dunbar AE, III, Wert SE, Askin F, Hamvas A, Whitsett JA. A mutation in the surfactant protein C gene associated with familial interstitial lung disease. N Engl J Med. 2001;344:573–579. doi: 10.1056/NEJM200102223440805. [DOI] [PubMed] [Google Scholar]
- 6.Thomas A, Lane K, Phillips J, Prince M, Markin C, Speer M, Schwartz DA, Gaddipati R, Marney A, Johnson J, Roberts R, Haines J, Stahlman M, Loyd JE. Heterozygosity for a surfactant protein C gene mutation associated with usual interstitial pneumonitis and cellular nonspecific interstitial pneumonitis in one kindred. Am J Resp Crit Care Med. 2002;165(13221328) doi: 10.1164/rccm.200112-123OC. [DOI] [PubMed] [Google Scholar]
- 7.Tredano M, Griese M, Brasch F, Schumacher S, de Blic J, Marque S, Houdayer C, Elion J, Couderc R, Bahuau M. Mutation of SFTPC in infantile pulmonary alveolar proteinosis with or without fibrosing lung disease. Am J Med Genet. 2004;126A:18–26. doi: 10.1002/ajmg.a.20670. [DOI] [PubMed] [Google Scholar]
- 8.Nogee L, de Mello DE, Dehner LP, Colten HR. Brief report: deficiency of pulmonary surfactant protein B in congenital alveolar proteinosis. N Engl J Med. 1993;328:406–410. doi: 10.1056/NEJM199302113280606. [DOI] [PubMed] [Google Scholar]
- 9.Shulenin SNL, Annilo T, Wert SE, Whitsett JA, Dean M. ABCA3 Gene Mutations in Newborns with Fatal Surfactant Deficiency. N Engl J Med. 2004;350:1296–1303. doi: 10.1056/NEJMoa032178. [DOI] [PubMed] [Google Scholar]
- 10.Hamvas A, Wegner DJ, Carlson CS, Bergmann KR, Trusgnich MA, Fulton L, Kasai Y, An P, Mardis ER, Wilson RK, Cole FS. Comprehensive genetic variant discovery in the surfactant protein B gene. Pediatr Res. 2007;62:170–175. doi: 10.1203/PDR.0b013e3180a03232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tredano M, Cooper DN, Stuhrmann M, Christodoulou J, Chuzhanova NA, Roudot-Thoraval F, Boelle PY, Elion J, Jeanpierre M, Feingold J, couderc R, Bahuau M. Origin of the prevalent SFTPB indel g.1549C>GAA (121ins2) mutation causing surfactant protein B (SP-B) deficiency. Am J Med Genet A. 2006;140(1):62–69. doi: 10.1002/ajmg.a.31050. [DOI] [PubMed] [Google Scholar]
- 12.Hamvas A. Inherited surfactant protein-B deficiency and surfactant protein-C associated disease: Clinical features and evaluation. Semin Perinatol. 2006;30:316–326. doi: 10.1053/j.semperi.2005.11.002. [DOI] [PubMed] [Google Scholar]
- 13.Cameron H, Somaschini M, Carrera P, Hamvas A, Whitsett JA, Wert SE, Deutsch G, Nogee LM. A Common Mutation in the Surfactant Protein C Gene Associated with Lung Disease. J Pediatr. 2005;146:370–375. doi: 10.1016/j.jpeds.2004.10.028. [DOI] [PubMed] [Google Scholar]
- 14.Hamvas A, Nogee LM, White FV, Schuler P, Hackett BP, Huddleston CB, Mendeloff EN, Hsu FF, Wert SE, Gonzales LW, Beers MF, Ballard PL. Progressive lung disease and surfactant dysfunction with a deletion of surfactant protein C gene. Am J Resp Cell Mol Biol. 2004;30:771–776. doi: 10.1165/rcmb.2003-0323OC. [DOI] [PubMed] [Google Scholar]
- 15.Bullard J, Nogee LM. Heterozygosity for ABCA3 mutations modifies the severity of lung disease associated with a surfactant protein-C gene (SFTPC) mutation. Pediatr Res. 2007;62:176–179. doi: 10.1203/PDR.0b013e3180a72588. [DOI] [PubMed] [Google Scholar]
- 16.Hamvas A, Trusgnich M, Brice H, Baumgartner J, Hong Y, Nogee LM, Cole FS. Population-based screening for rare mutations: high throughput DNA extraction and amplification from Guthrie cards. Pediatr Res. 2001;50:666–668. doi: 10.1203/00006450-200111000-00021. [DOI] [PubMed] [Google Scholar]
- 17.Stephens M, Donnelly P. A comparison of bayesian methods for haplotype reconstruction from population genotype data. Am J Hum Genet. 2003;73:1162–1169. doi: 10.1086/379378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Stephens M, Smith NJ, Donnelly P. A new statistical method for haplotype reconstruction from population data. Am J Hum Genet. 2001;68:978–989. doi: 10.1086/319501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Crawford D, Bhangale T, Li N, Hellenthal G, Rieder MJ, Nickerson DA, Stephens M. Evidence for substantial fine-scale variation in recombination rates across the human genome. Nature Genetics. 2004;36(7):700–706. doi: 10.1038/ng1376. [DOI] [PubMed] [Google Scholar]
- 20.Kruglyak L, Nickerson DA. Variation is the spice of life. Nat Genet. 2001;27:234–236. doi: 10.1038/85776. [DOI] [PubMed] [Google Scholar]
- 21.Yusen R, Cohen AH, Hamvas A. Normal lung function in subjects heterozygous for surfactant protein B deficiency. Am J Resp Crit Care Med. 1999;159:411–414. doi: 10.1164/ajrccm.159.2.9806033. [DOI] [PubMed] [Google Scholar]
- 22.Hess S, Blake JD, Blake RD. Wide variations in neighbor-dependent substitution rates. J Mol Biol. 1994;236(4):1022–1033. doi: 10.1016/0022-2836(94)90009-4. [DOI] [PubMed] [Google Scholar]
- 23.Neale M, Keeney S. Clarifying the mechanics of DNA strand exchange in meiotic recombination. Nature. 2006;442:153–158. doi: 10.1038/nature04885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Berlin S, Brandstrom M, Backstrom N, Axelsson E, Smith N, Ellegren H. Substitution rate heterogeneity and the male mutation bias. J Mol Evol. 2006;62:226–233. doi: 10.1007/s00239-005-0103-6. [DOI] [PubMed] [Google Scholar]
- 25.Carlson K, Bracamontes J, Jackson CE, Clark R, Lacroix A, Wells SA, Goodfellow PJ. Parent-of-origin effects in multiple endocrine neoplasia type 2B. Am J Hum Genet. 1994;55:1076–1082. [PMC free article] [PubMed] [Google Scholar]
- 26.Ardlie K, Liu-Cordero SN, Eberle MA, Daly M, Barrett J, Winchester E, Lander ES, Krugylak L. Lower-than-expected linkage disequilibrium between tightly linked markersin humans suggests a role for gene conversion. Am J Hum Genet. 2001;69(3):582–589. doi: 10.1086/323251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Helleday T. Pathways for mitotic homologous recombination in mammalian cells. Mut Res. 2003;532:103–115. doi: 10.1016/j.mrfmmm.2003.08.013. [DOI] [PubMed] [Google Scholar]
- 28.Erickson R. Somatic gene mutation and human disease other than cancer. Mut Res. 2003;543:125–136. doi: 10.1016/s1383-5742(03)00010-3. [DOI] [PubMed] [Google Scholar]
- 29.Gollob M, Jones DL, Krahn AD, Danis L, Gong XQ, Shao Q, Liu X, Veinot JP, Tang AS, Stewart AF, Tesson F, Klein GJ, Yee R, Skanes AC, Guiraudon GM, Ebihara L, Bai D. Somatic mutations in the connexin 40 gene (GJA5) in atrial fibrillation. N Eng J Med. 2006;345(25):2677–2688. doi: 10.1056/NEJMoa052800. [DOI] [PubMed] [Google Scholar]
- 30.Beck J, Poulter M, Campbell TA, Uphill JB, Adamson G, Gedes JF, Revesz T, Davis MB, Wood NW, Collinge J, Tabrizi SJ. Somatic and germline mosaicism in sporadic early-onset Alzheimer’s disease. Hum Mol Genet. 2004;13(12):1219–1224. doi: 10.1093/hmg/ddh134. [DOI] [PubMed] [Google Scholar]
- 31.Franz D. Diagnosis and management of Tuberous Sclerosis Complex. Semin Pediatr Neurol. 1998;5(4):253–268. doi: 10.1016/s1071-9091(98)80004-1. [DOI] [PubMed] [Google Scholar]
- 32.Lee N, Norton JA. Multiple endocrine neoplasia type 2B--genetic basis and clinical expression. Surgical Oncology. 2000;9:111–118. doi: 10.1016/s0960-7404(00)00038-4. [DOI] [PubMed] [Google Scholar]
- 33.Russell K, Ming JE, Patel K, Jukofsky L, Magnusson M, Krantz ID. Dominant paternal transmission of Cornelia de Lange Syndrome: a new case and review of 25 previously reported familial recurrences. Am J Med Genet. 2001;104:267–276. doi: 10.1002/ajmg.10066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lacz NLSR, Kikiczak G. Epidermolytic hyperkeratosis: a keratin 1 or 10 mutational event. Int J Dermatol. 2005;44:1–6. doi: 10.1111/j.1365-4632.2004.02364.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.