

Abstract
Huntington's disease is a progressive neurodegenerative disorder caused by a polyglutamine expansion near the N-terminus of huntingtin. The mechanisms of polyglutamine neurotoxicity, and cellular responses are not fully understood. We have studied gene expression profiles by cDNA array using an inducible PC12 cell model expressing an N-terminal huntingtin fragment with expanded polyglutamine (Htt-N63-148Q). Mutant huntingtin Htt-N63 induced cell death and increased the mRNA and protein levels of activating transcription factor 3 (ATF3). Mutant Htt-N63 also significantly enhanced ATF3 transcriptional activity by a promoter-based reporter assay. Overexpression of ATF3 protects against mutant Htt-N63 toxicity and knocking down ATF3 expression reduced Htt-N63 toxicity in a stable PC12 cell line. These results indicated that ATF3 plays a critical role in toxicity induced by mutant Htt-N63 and may lead to a useful therapeutic target.
Keywords: Activating transcription factor 3, Mutant huntingtin, Polyglutamine, Huntington's disease, Cell death
1. Introduction
Huntington's disease (HD) is a progressive neurodegenerative disorder characterized by movement disorders and cognitive dysfunction resulting from an abnormal polyglutamine (poly Q) expansion in huntingtin (Htt). HD affects both sexes with the same frequency and is inherited in an autosomal dominant manner. (Borrell-Pages et al., 2006; Gatchel and Zoghbi, 2005; Ross, 2002). The molecular mechanisms underlying expanded poly Q-induced neuronal cell death remain poorly understood. Increasing evidence suggests that neuronal dysfunction may result from altered gene expression in an HD brain, with a potential role for a number of transcription factors (Cui et al., 2006; Ross and Thompson, 2006). The effects on transcription can occur through interaction of the mutant Htt with transcriptional activators and repressors (Cui et al., 2006; Jiang et al., 2003; Ross and Thompson, 2006). In addition, Htt may function as a transcriptional co-repressor by interacting with complexes that contain nuclear co-repressor proteins (Cornett et al., 2006; Gatchel and Zoghbi, 2005; Jiang et al., 2003; Nucifora et al., 2001; Steffan et al., 2000; Sugars and Rubinsztein, 2003). The full transcriptional gene expression profile however, is not completely understood.
Previously, we generated a stable inducible PC12 cell model in which expression of the N-terminal fragment of human Htt with an expanded polyglutamine region (Htt-N63-148Q) caused direct cell toxicity (Igarashi et al., 2003). To study the transcriptional gene expression profile, we performed a cDNA array analysis. We found that mutant Htt-N63 induced a change in gene expression profiles. Interestingly, we found that Htt-N63-148Q significantly up-regulated activating transcription factor 3 (ATF3) expression.
ATF3 is a member of the mammalian activation transcription factor/cAMP responsive element-binding (CREB) protein family of transcription factors and has a potential role in the stress response (Hai and Hartman, 2001). Previous studies indicate that the mRNA level of ATF3 is greatly increased when the cells are exposed to stress signals. In animal experiments, the signals include ischemia, ischemia coupled with reperfusion, wounding, axotomy, toxicity, and seizures; in cultured cells, the signals include serum factors, cytokines, genotoxic agents, cell death-inducing agents, and the adenoviral protein E1A (Allen-Jennings et al., 2001; Cai et al., 2000; Seijffers et al., 2007; Yoshida et al., 2008). The consequences of inducing ATF3 during stress responses are not clear. Here we found that expression of Htt-N63-148Q as a toxic protein increased ATF3 expression at mRNA and protein levels and enhanced ATF3 transcriptional activity. Overexpression of ATF3 protected against Htt-N63-148Q toxicity, and knock down of ATF3 increased Htt-N63-148Q toxicity. These results suggest a protective role for ATF3 in mutant Htt-N63-induced toxicity in a stable PC12 cell model.
2. Results
2.1 Induction of mutant Htt-N63 expression induces a transcriptional factor change in gene expression profiles
Gene expression profiling was carried out with a stable inducible PC12 cell line which previously described in Igarashi (Igarashi et al., 2003), designed to express an N-terminal Htt fragment with an expanded length polyQ region (Htt N63-148Q-myc) fused to a C-terminal myc tag (Cooper et al., 1998). To design microarray experiments in PC12 cells, it was important to establish the time period when measurable cell death occurred following the induction of expanded Htt expression. Therefore, we examined the time course of Htt N63-148Q expression and evaluated cell viability by using LDH assay (Fig. 1 A, B). To induce Htt expression the PC12 cells were differentiated and incubated in medium lacking doxycycline over a 6-day period. As shown in Fig 1A, expression of expanded Htt-N63-148Q-myc was observed on day 1, increased by day 2, and reached a peak on day 4. In contrast, in the non-inducing condition with doxycycline, cells did not express Htt. We further found that expression of Htt-N63-148Q significantly induced cell death after 4 and 6 day induction, consistent with a previous report (Igarashi et al., 2003).
Fig 1.
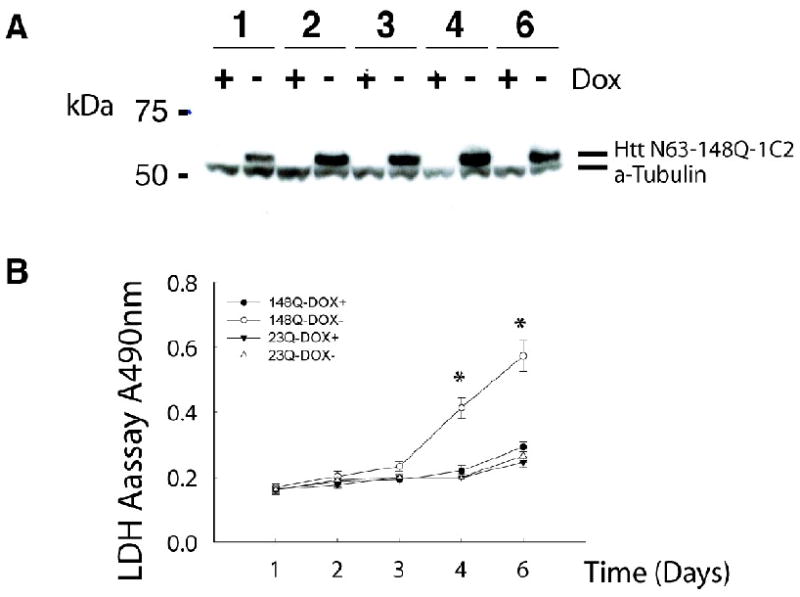
Induction of mutant Htt expression in PC12 cells induces cell death. (A) Expression of mutant Htt-N63 in stable inducible PC12 cells detected by Western blot with use of an expanded polyQ-selective 1C2 antibody. The lower band in all samples represents ˆ- tubulin, shown as a loading control. (B) Htt N63-148Q-myc induced cell death compared with non-inducing conditions meadured by LDH toxicity assays; in contrast, Htt N63-23Q-myc did not. *P<0.001 (by Student's t test), compare to non-inducing conditions.
In order to study Htt-N63-148Q-induced early changes in gene expression of transcription regulators, we conducted cDNA arrays using total RNA from the cells with or without expressing mutant Htt at two time points: day 3 and day 4 post induction. We use the uninduced cells as controls. A total of 2588 genes showed more than 1.2-fold changes in PC12 cells expressing polyQ-expanded Htt on day 4, compared to uninduced “dox+” cells. Among these, 1396 genes were up-regulated and 1190 genes were down-regulated. The mRNA levels of 20 transcription-related genes were increased up to 1.5-fold of that of control cells on day 4 after induction of mutant Htt expression. We found that ATF3 increased significantly (24.5-fold, day 4, Table 1) at the top rank among the other genes. Even after a 3 day induction, ATF3 increased up to 5-fold over that in non-inducing conditions.
Table 1.
Transcriptional factor changes in gene expression cause by expanded polyQ Htt in PC12 cells. The mRNA levels of 20 transcription-related genes were increased up to 1.5-fold of that of control cells on day 4 after induction of mutant Htt expression. Data shows fold change of mutant Htt induction condition compare to non-induction condition.
| GenBank accession no. | Gene Product | Gene symbol | Fold change |
|---|---|---|---|
| NM_012912 | Activating transcription factor 3 | ATF3 | 24.559456 |
| NM_012953 | Fos-like antigen 1 | Fosl1 | 3.69567 |
| NM_021835 | v-jun sarcoma virus 17 oncogene homolog (avian) | v-Jun | 2.642766 |
| NM_012551 | Early growth response 1 | Egr-1 | 2.353988 |
| NM_024134 | DNA-damage inducible transcript 3 | Ddit3 | 2.06334 |
| NM_012954 | Fos-like antigen 2 | Fosl2 | 1.91767 |
| L38247 | Pancreatic and duodenal homeobox gene 1 | 1.85513 | |
| NM_053727 | Nuclear factor, interleukin 3, regulated | Nfil3 | 1.820948 |
| NM_031642 | Core promoter element binding protein | Klf6 | 1.75217 |
| NM_022715 | Major vault protein | Mvp | 1.749057 |
| BE113920 | Signal transducer and activator of transcription 3 | 1.709783 | |
| BF565718 | Core promoter element binding protein | 1.666381 | |
| NM_031599 | Eukaryotic translation initiation factor 2 alpha kinase 3 | Eif2ak3 | 1.655477 |
| AF013965 | Zinc finger protein 265 | 1.644508 | |
| BI289392 | Eukaryotic elongation factor-2 kinase | 1.590502 | |
| NM_053611 | Nuclear protein 1 | Nupr1 | 1.547344 |
| BE329377 | Jun D proto-oncogene | 1.519918 | |
| AA957545 | T-box 3 (ulnar mammary syndrome) | -1.500172 | |
| AI044110 | Thymopoietin | -1.502719 | |
| NM_031633 | Forkhead box M1 | Foxm1 | -1.851786 |
2.2 Mutant Htt increases ATF3 expression and enhances ATF3 transcriptional activity in a stable inducible PC12 cell model
To verify the cDNA array result that mutant Htt increased ATF3 expression, we used RT-qPCR and Western blot analysis. We found that there was an early increase in ATF3 mRNA beginning as soon as day 1, with a peak on day 4 after induction of mutant Htt expression (Fig 2A). ATF3 mRNA in cells expressing mutant Htt-148Q at day 4 was increased 14-fold over that of induction in control cells.
Fig 2.
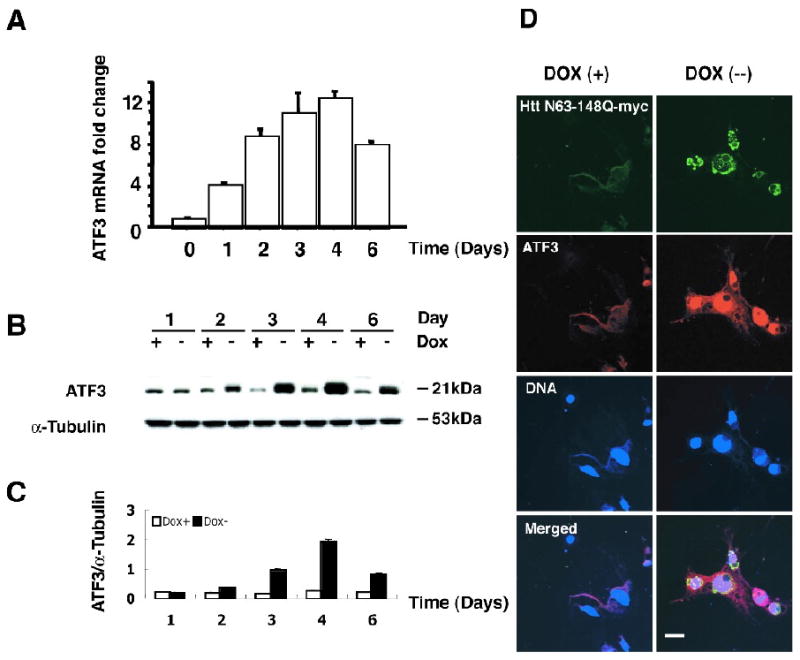
Mutant Htt-N63 increases ATF3 mRNA and protein levels. (A) RT-qPCR analysis was carried out in the presence or absence of mutant Htt-N63 expression. Data are reported fold as a-fold increase in ATF3 mRNA in Htt-expressing cells (Dox-) compared to the un-induced condition (Dox+) and show a peak in ATF3 mRNA 4 days after induction of mutant Htt expression. (B) Western blot analysis of endogenous ATF3 levels following induction of Htt N63-148Q-myc expression shows an increase in ATF3 over time, with a peak on day 4 after induction. Blots were probed with an antibody to ATF3. α-Tubulin is shown as a loading control. (C) Quantification of these data is shown in the lower panel. (D) Confocal images of Htt N63-148Q-myc stable inducible cells showing myc epitope (green), ATF3 (red), and Hoescht (blue) staining in the absence (Dox+, upper panels) or presence (Dox-, lower panels) of mutant Htt expression. Cells were fixed and immunostained 4 days after induction of mutant Htt expression. (Scale bar: 10μm)
To determine the effect of mutant Htt-N63 expression on endogenous protein levels of ATF3, total cell lysates from Htt-N63-148Q cells were collected over a 6-day period and analyzed by Western blotting. Endogenous levels of ATF3 remained at a steady state in the absence of Htt-N63-148Q expression. Following induction of Htt-N63-148Q-myc expression, ATF3 protein noticeably increased by day 2, and reached a peak on day 4 (Fig 2B, C). No significant change in ATF3 protein was observed for cells expressing Htt-N63-23Q-myc (data not shown).
Next, endogenous ATF3 levels were assessed by immunofluorescence microscopy. Confocal images from a representative experiment on day 4 after induction of Htt expression demonstrated faint ATF3 staining in the absence of mutant N63-148Q-myc expression (Dox (+), Fig 2D). In contrast, strong ATF3 staining was observed in response to mutant Htt N63-148Q-myc expression (Dox (-), Fig 2D). Using the transient transfection method, we transfected wild type htt (htt-N63-23Q) or mutant htt (htt-N63-148Q) into PC12 cells. Mutant htt, but not wild type htt, increased endogenous ATF3 expression (Fig. 6). These results are consistent with the data we show in the stable cell line.
Fig 6.
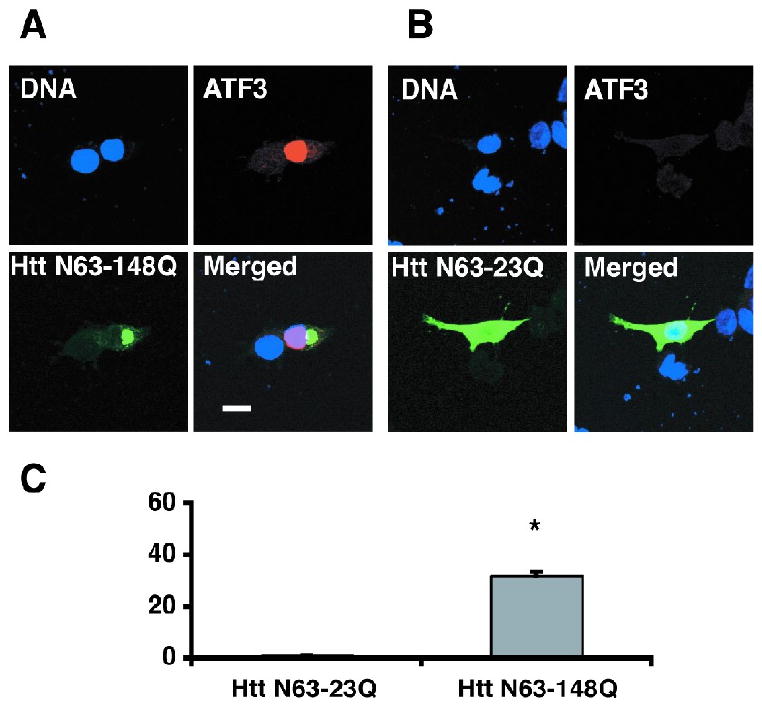
An increase in endogenous ATF3 protein is associated with mutant Htt expression and aggregation in transiently transfected PC12 cells. Cells were fixed and immunostained 48 h after transfection with an antibody to myc epitope for Htt detection (green) and an antibody to ATF3 (red.) Nuclear staining with Hoescht is shown in blue. (A) A cell expressing mutant Htt-N63-148Q aggregates shows increased ATF3 expression. (B) A cell expressing normal Htt-N63-23Q with diffused staining for htt shows no increase in ATF3 protein. Quantification of these data is shown in C. *P<0.001. 100 transfected cells were assessed in each individual experiment. Data are presented as mean +/-SEM of three separate experiments. (Scale bar: 10μm.)
To determine whether mutant Htt N63 expression results in an increase in ATF3 transcriptional activity, we performed a luciferase reporter assay. The reporter construct comprised an ATF3 promoter region, including putative consensus elements for Ap-1, SRF, and ATF/CRE binding (Cai et al 2000; Liang et al., 1996), a TATA sequence, and the luciferase reporter gene (Fig 3A). ATF3 promoter activity was increased in response to Htt-N63-148Q-myc expression (Fig 3B). A negative control lacking the ATF3 promoter region (Delta ATF) showed no increase in luciferase activity with expression of mutant Htt. Transfection of a plasmid containing two point mutations known to block ATF/CRE binding (ATFm) resulted in a decrease in luciferase activity. These observations demonstrate that mutant Htt-induced ATF3 promoter activity required an intact ATF/CRE binding region. In contrast, no change in ATF3 promoter activity was observed in cells expressing Htt N63 with a normal length polyQ region. Taken together, these results establish that expression of mutant, but not wild type, Htt-N63-148Q in this PC12 cell model led to an increase in ATF3 transcription via ATF3/CRE DNA binding.
Fig 3.
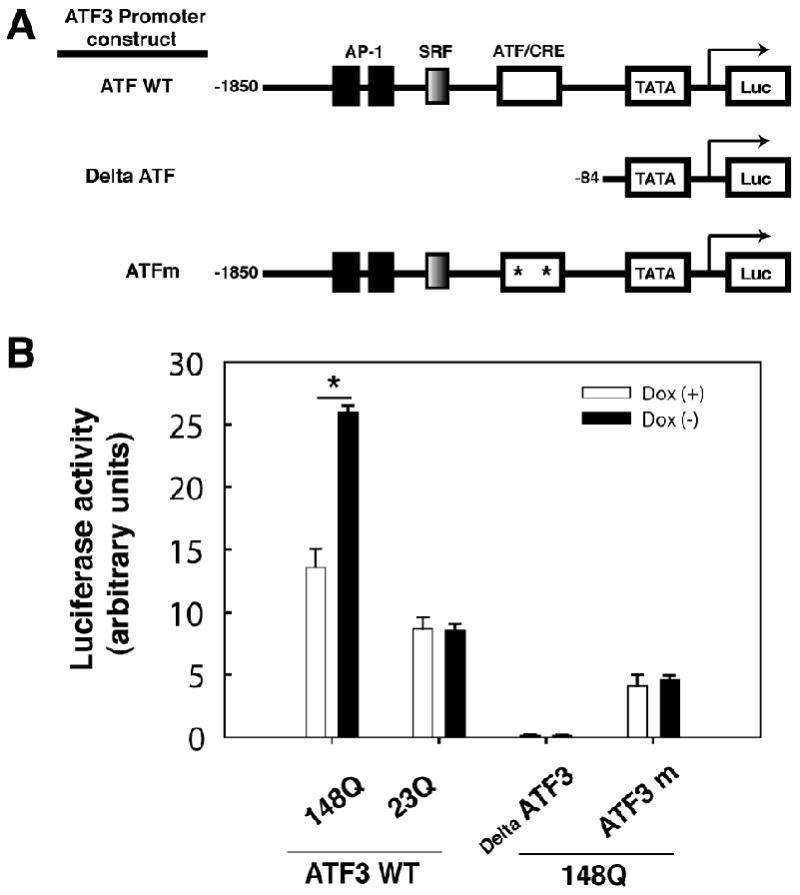
Mutant htt-N63 increases ATF3 transcriptional activity (A) Schematic diagram of ATF3 promoter plasmids with putative consensus elements for AP-1 and SRF, ATF/CRE and TATA binding sites, and the luciferase reporter gene. (B) Luciferase activity measured in Htt N63 PC12 cells transfected with ATF3 reporter plasmids in the presence (Dox+) or absence (Dox-) of doxycycline. Data were normalized against pTK-renilla activity. P < 0.05 by Student's t-test. Statistically significant differences between expressing wild type Htt (Htt-N63-23Q) and mutant Htt (Htt-N63-148Q) in stable PC12 cell line
2.3 Elevated ATF3 levels protect against mutant Htt-induced cell death in the stable inducible PC12 cell model
To study the ATF3 role in mutant Htt N63-induced cell death, we transfected ATF3 into PC12 cells stable expressing Htt- N63-23Q or Htt-N63-148Q (Fig. 4A). Cell viability was assessed using the LDH-based assay. We found that overexpression of ATF3 reduced mutant Htt-induced cell toxicity compared to that of cells untransfected with ATF3 (Fig. 4B). Overexpression of ATF3 did not alter the cells stable expressing Htt-23Q (data not shown). To further investigate a protective function of ATF3, we used specific siRNA to knock down expression of ATF3. siRNA specific for ATF3 significantly reduced the levels of cellular ATF3 protein 4 days after transfection compared to a nonspecific RNA control (Fig 5A). Knockdown of ATF3 in cells expressing Htt- N63-148Q resulted in increased cell death compared to that of cells transfected with control RNA (Fig 5B). These data indicate that knockdown of endogenous ATF3 protein can compromise cell viability in response to mutant Htt: overexpression of ATF3 decreased mutant Htt-induced cell toxicity, suggesting that ATF3 has a protective role in mutant Htt-induced cell death.
Fig 4.
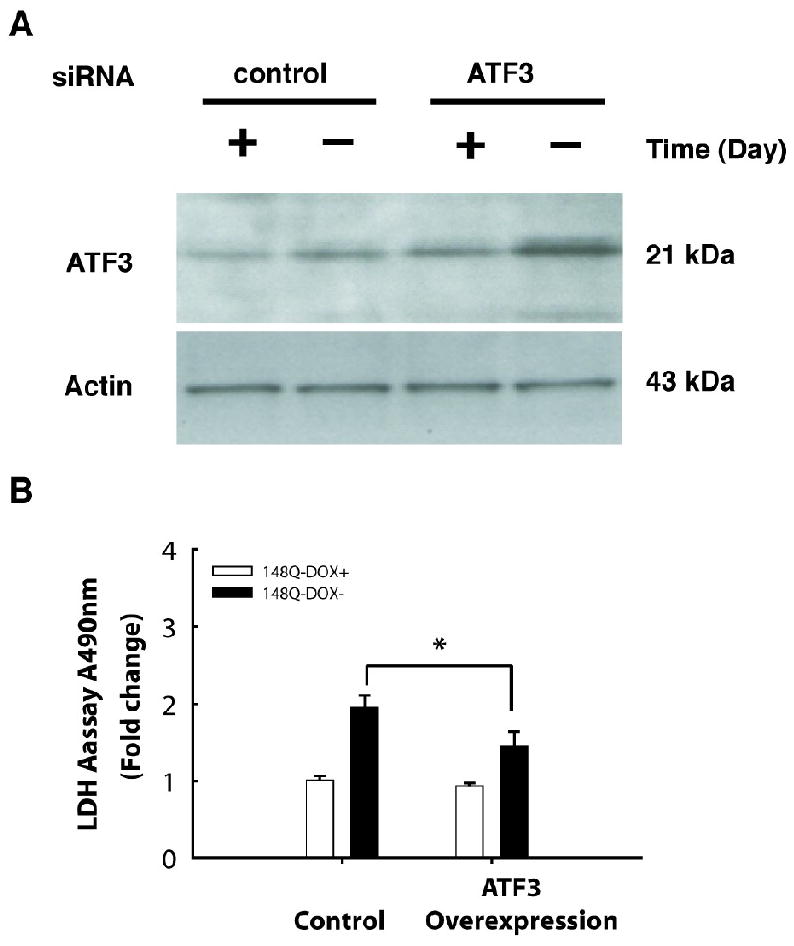
Overexpression of ATF3 reduced mutant Htt-N63 induced toxicity. (A) Htt N63-148Q stable cells were transfected with vector or ATF3 cDNA and incubated in inducing conditions for 2 days. Cells were harvested and subjected to Western blot analysis by using anti-ATF3 and anti-actin antibodies. (B) Overexpression of ATF3 reduced mutant Htt (Htt-N63-148Q) induced toxicity using LDH-based toxicity assay. P =0.027 by Student's t-test.
Fig 5.
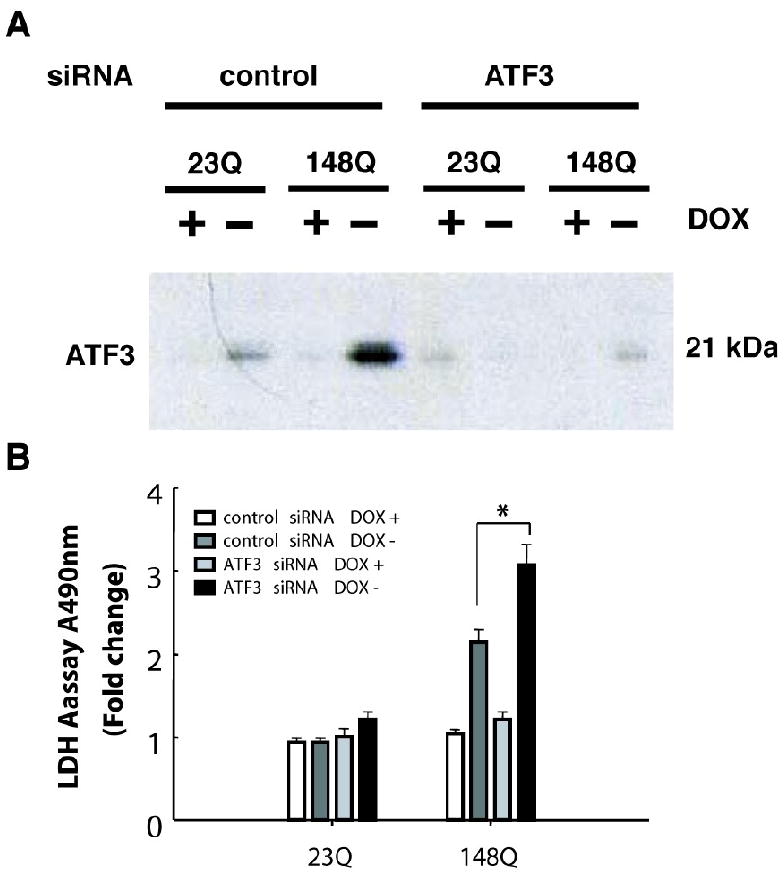
Knockdown of ATF3 expression enhances mutant Htt-N63-induced cell toxicity. (A) Htt N63-148Q stable cells were transfected with siRNA targeting ATF3 or non-targeting control RNA, and incubated in inducing conditions for 4 days. Cells were harvested and subjected to Western blot analysis by using anti-ATF3 and anti-actin antibodies. (B) Knockdown of ATF3 enhanced mutant Htt-N63 induced toxicity by using LDH-based toxicity assay. P < 0.05 by Student's t-test.
3. Discussion
In this study, we found that inducible expression of mutant Htt-148Q caused a change in gene expression profiles of transcription regulators, increased ATF3 at both mRNA and protein levels, and enhanced ATF3 transcriptional activity via ATF3/CRE DNA binding. Knockdown of ATF3 expression resulted in an increase in mutant Htt-induced cell toxicity, whereas overexpressing ATF3 rescued cell death, indicating a protective role for ATF3.
Mutant htt-148Q significantly increased ATF 3 mRNA and protein expression level. This result is consistent with previous findings in other systems (Apostol et al., 2006; Luthi-Carter et al., 2002). Importantly, we found that mutant Htt-148Q increased ATF3 promoter activity through the ATF/CRE binding site, suggesting that mutant Htt can facilitate transcription via a CRE-dependent mechanism, as previously described (Obrietan and Hoyt, 2004).
ATF3 functions as a stress response gene and is induced on exposure to a variety of physiological and pathological stimuli (Liang et al., 1996). The consequences of ATF3 induction that follows stress signals are not completely understood. Depending on the promoter and cellular context, ATF3 can activate transcription of some target genes and repress transcription of others (Hai et al., 1999; Miyazaki et al., 2009; Yin et al., 2007). Consequently, the role of ATF3 in cell death and cell cycle progression remains uncertain. In our study, overexpression of ATF3 reduced Htt-148Q-induced toxicity, and knockdown of ATF3 enhanced Htt-148Q toxicity, suggesting that ATF3 has a protective effect against mutant Htt-induced cell death. In line with this idea, a previous report showed that ATF3 enhances neurite outgrowth in adult rat dorsal root ganglion neurons (Seijffers et al., 2007). Francis et al. reported increased ATF3 and resistance to cell death in rat hippocampal neurons following in vivo injection of kainic acid (Francis et al., 2004). Another study found that ATF3 overexpression in PC12 cells and in rat superior cervical ganglion neurons inhibited c-Jun N-terminal kinase-induced apoptosis, induced neurite elongation via Akt activation, and increased HSP 27 (Nakagomi et al., 2003). Taken together, these findings support a pro-survival function for ATF3 in response to mutant Htt. However, this pro-survival effect may not overcome the cell death events that occur as a result of consistent expression of mutant Htt for 4-6 days.
Previous studies suggest that the primary site of mutant Htt toxicity is the nucleus (Peters et al., 2002; Saudou et al., 1998), although other reports indicate that htt can induce cytotoxicity in the cytosol as well. (Gervais et al., 2002; Panov et al., 2002). Increasing evidence indicates that many transcription factors or cofactors are involved in huntingtin toxicity in both sites, especially in the nucleus. Our results showed that expression of mutant Htt induced a change in the profile of transcription factors (Table 1) at the gene level. These changes included increases in several genes that control cell death [DNA-damage inducible transcript 3 (Cheng et al., 2008; Marciniak et al., 2004), v-jun (Shaulian et al., 2000), Egr-1 (de Belle et al., 1999; Huang et al., 1997)] and activator protein-1 family genes [Jun (c-jun, JunB and JunD) and Fos (c-Fos, Fos-related antigene-1fra-1 and fra2) (van Dam and Castellazzi, 2001)]. Forkhead box M1 gene (Foxm1), which has a dual DNA biding specificity (Major et al., 2004; Perez-Sanchez et al., 2000) and promotes cell proliferation (Kim et al., 2005), was significantly decreased in our cell line. These changes in genes that control cell death and regulate cell growth may converge on htt toxicity and overcome the ATF3 protective effect, thereby resulting in cell death.
In summary, we found that mutant Htt caused changes in the expression of genes that regulate transcription and increased ATF expression. These alterations may subsequently cause cells to have to fight for survival. These results provide insight into mutant Htt toxicity and may lead to a potential therapeutic target for ameliorating Htt-induced cell death at an early stage.
4. Experimental procedures
4.1 Constructs
Htt-N63-148Q-myc and Htt-N63-23Q-myc constructs were described by Cooper and collegues. (Cooper et al., 1998). Htt-N63-23 (wild type) and Htt-N63-148Q (mutant Htt); ATF3 in pCG vector (Hai et al., 1999) plasmids are used for transient transfection experiment.
ATF3 promoter constructs prepared in pGL3 vector (Promega) were a gift from Dr. Kitajima, Tokyo Medical and Dental University, Tokyo, Japan (Cai et al., 2000), and are described as follows: ATF WT, ATF3 wild type promoter; Delta ATF, pGL3 vector lacking the ATF3 promoter region; ATFm, ATF3 mutant promoter with two point mutations in ATF/CRE binding site known to block ATF/CRE binding. The pTK-Renilla vector was obtained from Promega.
4.2 Cell culture and transfection
A stable inducible PC12 cell model designed to express a myc-tagged N-terminal Htt fragment with either an expanded or normal polyQ region was previously described (Igarashi et al., 2003). To initiate differentiation, cells were incubated in Dulbecco's modified Eagle's medium (DMEM) containing 1% horse serum, 100 μg/ml G418, 200 μg/ml hygromycin, 200 ng/ml doxycycline, 100 units/ml penicillin, 100 units/ml streptomycin, and 50 ng/ml NGF (Roche). Mutant Htt expression was induced by washing the cells with PBS and eliminating doxycycline from the medium. Unless otherwise noted, all reagents were obtained from Invitrogen Life Sciences. PC12 tTA cells were used for transient transfection as described previously (Bae et al., 2005), Htt-N63-23Q or Htt-N63-148Q plasmids were transfected with lipofectamine 2000 (Invitrogen).
4.3 Cell viability studies
PC12 cells induced to express Htt-N63 were seeded on collagen-coated 24-well plates at a concentration of 104 cells/ml in either the presence or absence of doxyclycline. Cell viability was assessed by lactate dehydrogenase (LDH) assay (Roche Applied Science) according to the manufacturer's instructions. For each experiment, values from three wells were averaged; data are expressed as mean +/- SEM which collected from three separate experiment.
4.4 Western blot analysis
Protein extracts from differentiated Htt-N63 PC12 cells, grown for the indicated times, were prepared in M-Per lysis buffer (Pierce) supplemented with Complete Protease Inhibitor (Roche Applied Science). Protein concentration was determined in each lysate by the BCA protein assay (Pierce). Proteins were transferred onto nitrocellulose (Schleicher and Schuell) at 30 V for 2 hr. Blots were treated with blocking buffer (PBS + 0.1% Tween-20 + 5% nonfat dry milk) for 1 hr at room temperature and probed with one of the following primary antibodies overnight at 4°C: mouse anti-myc (9E10) (Roche) at 1:500, rabbit anti-ATF3 (Santa Cruz) at 1:200, mouse antibody to expanded polyQ (1C2) (Chemicon) at 1:3000, mouse anti-α-tubulin (Santa Cruz) at 1:500, and mouse anti-β-actin (Sigma) at 1:5000. Peroxidase-labeled secondary antibody (Boehringer Mannheim) for 1 hr at room temperature. Immunoreactive bands were detected by chemiluminescence (Amersham, Buckinghamshire U.K.). For densitometry measurements, autoradiograms were scanned with an Epson scanner and immunoreactive bands were quantified by using Scion Image beta version 4.0.2 (Scion Corporation).
4.5 RNA isolation and real-time quantitative PCR (RT-qPCR)
Differentiated PC12 cells induced to express Htt-N63-148Q were rinsed twice with PBS and collected. Total RNA extraction was carried out using an RNeasy Mini Kit (Qiagen) according to manufacturer's instructions. Cellular extracts were then treated with DNase (Qiagen) and total RNA was quantified on a Nanodrop spectrophotometer. Aliquots were prepared and either stored at −80 °C or used immediately. Two-step RT-PCR method were used (Ishigaki et al., 2002). First, after extraction of RNA, we immediately carried out reverse transcription to cDNA using a High Capacity Archive Kit (ABI) following the manufacturer's instructions; then we subjected the cDNA to PCR. Real-time quantitative polymerase chain reaction (qPCR) was carried out by the TaqMan Gene Expression Assay (ABI) according to manufacturer's instructions. In all experiments, actin (ID4352340E) was used as an endogenous control. PCR reactions were carried out in triplicate by using an ABI 7700 Prism Detection system (ABI) in a 50μl sample volume and 96-well plates under the following cycling conditions: 95°C for 10 min and 50°C for 2 min (1 cycle); 95°C for 15 sec and 60°C for 1 min (40 cycles). Each time for the q-PCR reaction, we used actin and ATF3 simultaneously. The data are expressed as the ratio of ATF3/actin, and the fold change of ATF3 was calculated as ATF3 of dox-/ATF3 of dox+. Negative control lacking the RT enzyme was routinely included.
4.6 Microarray data analysis
The mRNA samples were analyzed with Affymetrix GeneChip Rat genome 230 2.0 Arrays. Probe-level preprocessing was conducted with the statistical algorithm RMA (Robust Multi-array expression measure) (Irizarry et al., 2003). Using the bioconductor package Affy to produce probe set level signals. An empirical Bayes method with the lognormal-normal or Gamma-Gamma modeling, as implemented in the bioconductor package EBarrays, was then used to estimate the posterior probabilities of the differential expression of genes between the sample conditions (Kendziorski et al., 2003; Newton et al., 2001). The criterion of the posterior probability > 0.5, that is the posterior probability favors change, was used to produce the differentially expressed gene lists. All computation was performed under R environment (http://www.r-project.org) and all bioconductor packages are available at www.bioconductor.org.
4.7 Luciferase reporter assays
PC12 cells expressing either Htt N63-148Q-myc or Htt N63-23Q-myc were plated on 24-well plates at a density of 104 cells per well and grown for 24 hr. Cells were pre-incubated for 4 hr with DMEM supplemented with 1% horse serum and then transfected with pTK-Renilla vector and one of the promoter constructs described above by using Lipofectamine 2000 (Invitrogen) according to the manufacturer's protocol. Following transfection, the medium was replaced with serum-free medium and doxycycline was added to Dox+ cells. After 24 hr, cells were harvested in luciferase lysis buffer (Promega) and luciferase activity was determined using a dual luciferase assay kit (Promega). Data were collected in triplicate and normalized against pTK-renilla activity. The results shown represent the mean+/- SEM of at least three separate experiments with statistical analysis performed by Standard Student's t-test.
4.8 ATF3 siRNA knockdown experiments
To silence the expression of endogenous ATF3, an siRNA specific for ATF3 was designed and prepared by Dharmacon (Dharmacon, Inc. Chicago, IL) (Rat ATF3, Cat. M-080-117-00). Stable Htt N63-148Q PC12 cells were transiently transfected with either ATF3 or control siRNA (non-targeting control pool, Cat. D-001206-13-20) 1 day after induction of mutant Htt expression (Lipofectamine 2000, Invitrogen). The conditioned medium was collected 4 days after transfection and analyzed by the LDH assay. Simultaneously, cells were collected, lysed, and analyzed by Western blotting to determine the extent of ATF3 knockdown.
Acknowledgments
This work was supported by HDSA Coalition for the Cure, High Q / CHDI Foundation, NINDS 16375 (C.A.R), NINDS NS053679-03 (M.A.P.), and Hood College Board of Associates Grant (R.R.H.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Allen-Jennings AE, Hartman MG, Kociba GJ, Hai T. The roles of ATF3 in glucose homeostasis. A transgenic mouse model with liver dysfunction and defects in endocrine pancreas. J Biol Chem. 2001;276:29507–14. doi: 10.1074/jbc.M100986200. [DOI] [PubMed] [Google Scholar]
- Apostol BL, Illes K, Pallos J, Bodai L, Wu J, Strand A, Schweitzer ES, Olson JM, Kazantsev A, Marsh JL, Thompson LM. Mutant huntingtin alters MAPK signaling pathways in PC12 and striatal cells: ERK1/2 protects against mutant huntingtin-associated toxicity. Hum Mol Genet. 2006;15:273–85. doi: 10.1093/hmg/ddi443. [DOI] [PubMed] [Google Scholar]
- Bae BI, Xu H, Igarashi S, Fujimuro M, Agrawal N, Taya Y, Hayward SD, Moran TH, Montell C, Ross CA, Snyder SH, Sawa A. p53 mediates cellular dysfunction and behavioral abnormalities in Huntington's disease. Neuron. 2005;47:29–41. doi: 10.1016/j.neuron.2005.06.005. [DOI] [PubMed] [Google Scholar]
- Borrell-Pages M, Zala D, Humbert S, Saudou F. Huntington's disease: from huntingtin function and dysfunction to therapeutic strategies. Cell Mol Life Sci. 2006;63:2642–60. doi: 10.1007/s00018-006-6242-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai Y, Zhang C, Nawa T, Aso T, Tanaka M, Oshiro S, Ichijo H, Kitajima S. Homocysteine-responsive ATF3 gene expression in human vascular endothelial cells: activation of c-Jun NH(2)-terminal kinase and promoter response element. Blood. 2000;96:2140–8. [PubMed] [Google Scholar]
- Cheng WP, Hung HF, Wang BW, Shyu KG. The molecular regulation of GADD153 in apoptosis of cultured vascular smooth muscle cells by cyclic mechanical stretch. Cardiovasc Res. 2008;77:551–9. doi: 10.1093/cvr/cvm057. [DOI] [PubMed] [Google Scholar]
- Cooper JK, Schilling G, Peters MF, Herring WJ, Sharp AH, Kaminsky Z, Masone J, Khan FA, Delanoy M, Borchelt DR, Dawson VL, Dawson TM, Ross CA. Truncated N-terminal fragments of huntingtin with expanded glutamine repeats form nuclear and cytoplasmic aggregates in cell culture. Hum Mol Genet. 1998;7:783–90. doi: 10.1093/hmg/7.5.783. [DOI] [PubMed] [Google Scholar]
- Cornett J, Smith L, Friedman M, Shin JY, Li XJ, Li SH. Context-dependent dysregulation of transcription by mutant huntingtin. J Biol Chem. 2006;281:36198–204. doi: 10.1074/jbc.M607839200. [DOI] [PubMed] [Google Scholar]
- Cui L, Jeong H, Borovecki F, Parkhurst CN, Tanese N, Krainc D. Transcriptional repression of PGC-1alpha by mutant huntingtin leads to mitochondrial dysfunction and neurodegeneration. Cell. 2006;127:59–69. doi: 10.1016/j.cell.2006.09.015. [DOI] [PubMed] [Google Scholar]
- de Belle I, Huang RP, Fan Y, Liu C, Mercola D, Adamson ED. p53 and Egr-1 additively suppress transformed growth in HT1080 cells but Egr-1 counteracts p53-dependent apoptosis. Oncogene. 1999;18:3633–42. doi: 10.1038/sj.onc.1202696. [DOI] [PubMed] [Google Scholar]
- Francis JS, Dragunow M, During MJ. Over expression of ATF-3 protects rat hippocampal neurons from in vivo injection of kainic acid. Brain Res Mol Brain Res. 2004;124:199–203. doi: 10.1016/j.molbrainres.2003.10.027. [DOI] [PubMed] [Google Scholar]
- Gatchel JR, Zoghbi HY. Diseases of unstable repeat expansion: mechanisms and common principles. Nat Rev Genet. 2005;6:743–55. doi: 10.1038/nrg1691. [DOI] [PubMed] [Google Scholar]
- Gervais FG, Singaraja R, Xanthoudakis S, Gutekunst CA, Leavitt BR, Metzler M, Hackam AS, Tam J, Vaillancourt JP, Houtzager V, Rasper DM, Roy S, Hayden MR, Nicholson DW. Recruitment and activation of caspase-8 by the Huntingtin-interacting protein Hip-1 and a novel partner Hippi. Nat Cell Biol. 2002;4:95–105. doi: 10.1038/ncb735. [DOI] [PubMed] [Google Scholar]
- Hai T, Wolfgang CD, Marsee DK, Allen AE, Sivaprasad U. ATF3 and stress responses. Gene Expr. 1999;7:321–35. [PMC free article] [PubMed] [Google Scholar]
- Hai T, Hartman MG. The molecular biology and nomenclature of the activating transcription factor/cAMP responsive element binding family of transcription factors: activating transcription factor proteins and homeostasis. Gene. 2001;273:1–11. doi: 10.1016/s0378-1119(01)00551-0. [DOI] [PubMed] [Google Scholar]
- Huang RP, Fan Y, de Belle I, Niemeyer C, Gottardis MM, Mercola D, Adamson ED. Decreased Egr-1 expression in human, mouse and rat mammary cells and tissues correlates with tumor formation. Int J Cancer. 1997;72:102–9. doi: 10.1002/(sici)1097-0215(19970703)72:1<102::aid-ijc15>3.0.co;2-l. [DOI] [PubMed] [Google Scholar]
- Igarashi S, Morita H, Bennett KM, Tanaka Y, Engelender S, Peters MF, Cooper JK, Wood JD, Sawa A, Ross CA. Inducible PC12 cell model of Huntington's disease shows toxicity and decreased histone acetylation. Neuroreport. 2003;14:565–8. doi: 10.1097/00001756-200303240-00007. [DOI] [PubMed] [Google Scholar]
- Irizarry RA, Hobbs B, Collin F, Beazer-Barclay YD, Antonellis KJ, Scherf U, Speed TP. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics. 2003;4:249–64. doi: 10.1093/biostatistics/4.2.249. [DOI] [PubMed] [Google Scholar]
- Jiang H, Nucifora FC, Jr, Ross CA, DeFranco DB. Cell death triggered by polyglutamine-expanded huntingtin in a neuronal cell line is associated with degradation of CREB-binding protein. Hum Mol Genet. 2003;12:1–12. doi: 10.1093/hmg/ddg002. [DOI] [PubMed] [Google Scholar]
- Kendziorski CM, Newton MA, Lan H, Gould MN. On parametric empirical Bayes methods for comparing multiple groups using replicated gene expression profiles. Stat Med. 2003;22:3899–914. doi: 10.1002/sim.1548. [DOI] [PubMed] [Google Scholar]
- Kim IM, Ramakrishna S, Gusarova GA, Yoder HM, Costa RH, Kalinichenko VV. The forkhead box m1 transcription factor is essential for embryonic development of pulmonary vasculature. J Biol Chem. 2005;280:22278–86. doi: 10.1074/jbc.M500936200. [DOI] [PubMed] [Google Scholar]
- Liang G, Wolfgang CD, Chen BP, Chen TH, Hai T. ATF3 gene. Genomic organization, promoter, and regulation. J Biol Chem. 1996;271:1695–701. doi: 10.1074/jbc.271.3.1695. [DOI] [PubMed] [Google Scholar]
- Luthi-Carter R, Strand AD, Hanson SA, Kooperberg C, Schilling G, La Spada AR, Merry DE, Young AB, Ross CA, Borchelt DR, Olson JM. Polyglutamine and transcription: gene expression changes shared by DRPLA and Huntington's disease mouse models reveal context-independent effects. Hum Mol Genet. 2002;11:1927–37. doi: 10.1093/hmg/11.17.1927. [DOI] [PubMed] [Google Scholar]
- Major ML, Lepe R, Costa RH. Forkhead box M1B transcriptional activity requires binding of Cdk-cyclin complexes for phosphorylation-dependent recruitment of p300/CBP coactivators. Mol Cell Biol. 2004;24:2649–61. doi: 10.1128/MCB.24.7.2649-2661.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marciniak SJ, Yun CY, Oyadomari S, Novoa I, Zhang Y, Jungreis R, Nagata K, Harding HP, Ron D. CHOP induces death by promoting protein synthesis and oxidation in the stressed endoplasmic reticulum. Genes Dev. 2004;18:3066–77. doi: 10.1101/gad.1250704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyazaki K, Inoue S, Yamada K, Watanabe M, Liu Q, Watanabe T, Adachi MT, Tanaka Y, Kitajima S. Differential usage of alternate promoters of the human stress response gene ATF3 in stress response and cancer cells. Nucleic Acids Res. 2009 doi: 10.1093/nar/gkn1082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakagomi S, Suzuki Y, Namikawa K, Kiryu-Seo S, Kiyama H. Expression of the activating transcription factor 3 prevents c-Jun N-terminal kinase-induced neuronal death by promoting heat shock protein 27 expression and Akt activation. J Neurosci. 2003;23:5187–96. doi: 10.1523/JNEUROSCI.23-12-05187.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newton MA, Kendziorski CM, Richmond CS, Blattner FR, Tsui KW. On differential variability of expression ratios: improving statistical inference about gene expression changes from microarray data. J Comput Biol. 2001;8:37–52. doi: 10.1089/106652701300099074. [DOI] [PubMed] [Google Scholar]
- Nucifora FC, Jr, Sasaki M, Peters MF, Huang H, Cooper JK, Yamada M, Takahashi H, Tsuji S, Troncoso J, Dawson VL, Dawson TM, Ross CA. Interference by huntingtin and atrophin-1 with cbp-mediated transcription leading to cellular toxicity. Science. 2001;291:2423–8. doi: 10.1126/science.1056784. [DOI] [PubMed] [Google Scholar]
- Obrietan K, Hoyt KR. CRE-mediated transcription is increased in Huntington's disease transgenic mice. J Neurosci. 2004;24:791–6. doi: 10.1523/JNEUROSCI.3493-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panov AV, Gutekunst CA, Leavitt BR, Hayden MR, Burke JR, Strittmatter WJ, Greenamyre JT. Early mitochondrial calcium defects in Huntington's disease are a direct effect of polyglutamines. Nat Neurosci. 2002;5:731–6. doi: 10.1038/nn884. [DOI] [PubMed] [Google Scholar]
- Perez-Sanchez C, Gomez-Ferreria MA, de La Fuente CA, Granadino B, Velasco G, Esteban-Gamboa A, Rey-Campos J. FHX, a novel fork head factor with a dual DNA binding specificity. J Biol Chem. 2000;275:12909–16. doi: 10.1074/jbc.275.17.12909. [DOI] [PubMed] [Google Scholar]
- Peters PJ, Ning K, Palacios F, Boshans RL, Kazantsev A, Thompson LM, Woodman B, Bates GP, D'Souza-Schorey C. Arfaptin 2 regulates the aggregation of mutant huntingtin protein. Nat Cell Biol. 2002;4:240–5. doi: 10.1038/ncb761. [DOI] [PubMed] [Google Scholar]
- Ross CA. Polyglutamine pathogenesis: emergence of unifying mechanisms for Huntington's disease and related disorders. Neuron. 2002;35:819–22. doi: 10.1016/s0896-6273(02)00872-3. [DOI] [PubMed] [Google Scholar]
- Ross CA, Thompson LM. Transcription meets metabolism in neurodegeneration. Nat Med. 2006;12:1239–41. doi: 10.1038/nm1106-1239. [DOI] [PubMed] [Google Scholar]
- Saudou F, Finkbeiner S, Devys D, Greenberg ME. Huntingtin acts in the nucleus to induce apoptosis but death does not correlate with the formation of intranuclear inclusions. Cell. 1998;95:55–66. doi: 10.1016/s0092-8674(00)81782-1. [DOI] [PubMed] [Google Scholar]
- Seijffers R, Mills CD, Woolf CJ. ATF3 increases the intrinsic growth state of DRG neurons to enhance peripheral nerve regeneration. J Neurosci. 2007;27:7911–20. doi: 10.1523/JNEUROSCI.5313-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaulian E, Schreiber M, Piu F, Beeche M, Wagner EF, Karin M. The mammalian UV response: c-Jun induction is required for exit from p53-imposed growth arrest. Cell. 2000;103:897–907. doi: 10.1016/s0092-8674(00)00193-8. [DOI] [PubMed] [Google Scholar]
- Steffan JS, Kazantsev A, Spasic-Boskovic O, Greenwald M, Zhu YZ, Gohler H, Wanker EE, Bates GP, Housman DE, Thompson LM. The Huntington's disease protein interacts with p53 and CREB-binding protein and represses transcription. Proc Natl Acad Sci U S A. 2000;97:6763–8. doi: 10.1073/pnas.100110097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugars KL, Rubinsztein DC. Transcriptional abnormalities in Huntington disease. Trends Genet. 2003;19:233–8. doi: 10.1016/S0168-9525(03)00074-X. [DOI] [PubMed] [Google Scholar]
- van Dam H, Castellazzi M. Distinct roles of Jun : Fos and Jun : ATF dimers in oncogenesis. Oncogene. 2001;20:2453–64. doi: 10.1038/sj.onc.1204239. [DOI] [PubMed] [Google Scholar]
- Yin X, Dewille JW, Hai T. A potential dichotomous role of ATF3, an adaptive-response gene, in cancer development. Oncogene. 2007 doi: 10.1038/sj.onc.1210861. [DOI] [PubMed] [Google Scholar]
- Yoshida T, Sugiura H, Mitobe M, Tsuchiya K, Shirota S, Nishimura S, Shiohira S, Ito H, Nobori K, Gullans SR, Akiba T, Nitta K. ATF3 protects against renal ischemia-reperfusion injury. J Am Soc Nephrol. 2008;19:217–24. doi: 10.1681/ASN.2005111155. [DOI] [PMC free article] [PubMed] [Google Scholar]