

Abstract
Recently, evidence has emerged indicating that assessment of KRAS mutations before anti-epidermal growth factor receptor therapy improves outcome in patients with metastatic colorectal cancer (CRC). We report here a novel reverse-hybridization (RH) assay to screen for KRAS mutations in formalin-fixed paraffin-embedded colorectal tissue samples. We combined mutant-enriched PCR based on peptide nucleic acid clamping and RH of amplification products to nitrocellulose test strips that contained a parallel array of oligonucleotide probes targeting 10 frequent mutations in codons 12 and 13 of the KRAS gene. DNA mixing experiments, which included eight different tumor cell lines with known KRAS mutations, were performed to examine the sensitivity of mutation detection. All KRAS mutations present in tumor cell lines were unambiguously identified by the RH assay with 1% of each cell line DNA diluted in normal DNA. RH was then used to screen for KRAS mutations in 74 colorectal tumor and 4 normal control samples. Twenty-six (35%) of the 74 tumor samples showed KRAS mutations. No mutation was found in the four samples of normal colorectal tissue. DNA sequencing without previous mutant enrichment, however, failed to detect four (15%) out of 26 KRAS-positive formalin-fixed paraffin-embedded samples (FFPE). This finding suggests that even after microdissection, mutant sequences in a given DNA isolate can be rare and more sensitive methods are needed for mutation analysis.
The KRAS proto-oncogene (v-Ki-ras2 Kirsten rat sarcoma viral oncogene homolog: Gene Bank Accession Number NM_033360) is one of the most prominent and most commonly mutated RAS family members in colorectal cancer (CRC). Oncogenic mutations of KRAS disrupt binding to GTP and allow it to remain in an active state.1 The most common mutations in CRC and other cancer types affect codons 12 and 13, and to a lesser extend codon 61.2,3,4 A link between the KRAS gene and an underlying epigenetic disorder in CRC was shown recently; KRAS mutations were associated with a subset of CRCs that exhibit methylation in multiple sets of genes and are referred to as CpG methylator phenotype. These tumors have distinct clinical, molecular and pathological features, and KRAS mutations in codon 12 and 13 were associated with so-called CpG methylator phenotype-low cases.5,6 In another recent study, KRAS mutational status and methylation status were associated with decreased survival.7 The clinical implication of assessment of the KRAS mutation status in metastatic CRC was demonstrated very recently. The effect of antibody treatment of patients suffering from metastatic CRC with cetuximab was significantly enhanced as compared with standard chemotherapy alone when genomic DNA isolated from tumor tissue did not contain a KRAS mutation, whereas patients with KRAS mutations could not be shown to benefit from cetuximab treatment.8 Monoclonal antibodies such as cetuximab or panitumumab target the extracellular domain of the epidermal growth factor receptor thereby blocking ligand-induced epidermal growth factor receptor activation and subsequent signal transduction through pathways like the RAS/RAF/MAPK and P13/AKT cascades.9,10 Results from recent randomized controlled trials suggest that patients with KRAS mutations in codons 12 and 13 do not benefit from these anti-epidermal growth factor receptor monoclonal antibody therapies,11 and accurate treatment response prediction will spare the patient unnecessary treatment thereby focusing on more individualized therapy. Thus, reliable and sensitive determination of the KRAS mutation status becomes increasingly important in individual treatment decisions. Archival tissue of the primary tumor is easily accessible and is an important source for KRAS mutation testing. We developed a novel biomarker assay to detect KRAS mutations in archived formalin-fixed paraffin-embedded (FFPE) tissue. The test combines mutant-enriched PCR based on peptide nucleic acid clamping and reverse-hybridization (RH) to nitrocellulose test strips containing a parallel array of oligonucleotide probes targeting 10 mutations in codons 12 and 13 of the KRAS gene. Because mutations in codon 61 are extremely rare in CRC cases, these were not included in the assay.2,3,4
We then used the novel RH assay to screen for KRAS mutations in DNA extracted from FFPE tissue samples obtained from patients operated because of CRC.
Materials and Methods
Patients
Patients were treated at the Departments of Surgery and Medicine, Danube Hospital SMZ Ost, Vienna, Austria between 2002 and 2005. Surgically resected tissues were collected from 78 patients. All 73 cancer cases (51 male and 22 female patients) were adenocarcinomas, one benign tumor was an adenoma with high grade dysplasia. Cases regarded as normal controls (two male and two female patients) included one lipoma, two cases of diverticulosis and one case without any pathological diagnosis. Median age at surgery was 63 (range, 30 to 87 years). The cancers were classified according to the International Union Against Cancer (UICC) TNM guidelines.12 Two patients (1.5%) had a carcinoma in situ. 12 (9%) patients were UICC stage I, 16 (12%) patients were UICC stage II, 25 (18%) UICC stage III, 12 (9%) patients were UICC stage IV, and 6 (4.5%) patients had local relapses. The study was approved by the local ethics committee and reviewed by the institutional review board.
DNA Extraction
An appropriate paraffin block containing tumor tissue was selected for analysis after reviewing the H&E-stained slides. An area of tumor on the H&E-stained slide was identified by an experienced pathologist and was marked and microdissected on a corresponding unstained slide for subsequent DNA isolation as described previously.13 DNA was extracted using a commercially available DNA extraction kit (QIAmp DNA Mini Kit; Qiagen, Hilden, Germany) and quantified by fluorometry (Quant-iT dsDNA HS Assay; Invitrogen, Carlsbad, CA).
Mutant–Enriched PCR
Mutant-enriched duplex PCR was done as described by Prix et al14 except for the fact that all primers were 5′-biotinylated. PCR was performed in a 25 μl reaction, containing 1 × PCR Buffer (Qiagen), 100 μmol/L each deoxyribonucleoside triphosphate, 0.1 μmol/L HLA-DRA primers, 0.25 μmol/L KRAS primers, 2.84 μmol/L peptide nucleic acid, 1 U Hot Star Taq Polymerase (Qiagen), and up to 50 ng DNA template. The amplification was performed on a PE 9700 cycler (Applied Biosystems, Foster City, CA) starting with an initial denaturation step at 94°C for 15 minutes, then running for 35 cycles as follows: 94°C for 1 minute, 70°C for 50 seconds, 58°C for 50 seconds, 72°C for 50 seconds, and a final extension at 72°C for 7 minutes.
Real-Time PCR
Real-time PCR was used to quantitate genomic DNA isolated from tumor cell lines as well as mutant PCR products generated by site-directed mutagenesis. SYBR green PCR amplification was performed using the StepOne Real-time PCR System (Applied Biosystems). Amplification was performed in a 25-μl (final volume) mixture containing 3 μl DNA template, each of the two KRAS-specific primers 5′-TTATAAGGCCTGCTGAAAATGACTGAA-3′ (forward) and 5′-TGAATTAGCTGTATCGTCAAGGCACT-3′ (reverse)23 at 0.5 μmol/L, and 12.5 μl Maxima SYBR Green qPCR Master Mix (Fermentas, Vilnius, Lithuania). PCR conditions comprised an initial denaturation step at 95°C for 10 minutes followed by 40 cycles of 95°C 10 seconds and a combined annealing/elongation step at 65°C for 30 seconds. Threshold cycle (CT) values were determined with the StepOne software (version 2.0), using the second derivative method. Standard curves using DNA isolated from human blood (Human genomic DNA; Roche Diagnostics, Mannheim, Germany) were generated by plotting the CT values as a function of the log of the initial DNA concentration.
DNA Sequencing
Reactions were set up in 25-μl volumes as follows: 10 pmol primer 5′-GTGTATTAACCTTATGTGTGAC-3′ (forward) and 5′-GTCAGAGAAACCTTTATCTG-3′ (reverse), 12.5 μl JumpStart REDTaq ReadyMix 2 × PCR Reaction Mix (Sigma-Aldrich, St. Louis, MO) and 20 ng genomic DNA. PCR was performed on a GeneAmp PCR system 2700 (Applied Biosystems). PCR conditions comprised an initial denaturation step at 94°C for 3 minutes, followed by 35 cycles of 94°C 30 seconds, 58°C 30 seconds, 72°C 30 seconds, and a final extension at 72°C for 5 minutes. Sequence analysis of PCR products was performed on the ABI 310 automatic sequencer (Applied Biosystems) to the manufacturers instructions (BigDye Terminator v1.1 Cycle Sequencing Kit; Applied Biosystems).
Reverse-Hybridization
For RH, oligonucleotides were synthesized as probes targeting 10 mutations in codons 12 and 13 of the KRAS gene, namely Val12 (GGT→GTT), Asp12 (GGT→GAT), Leu12 (GGT→CTT), Ser12 (GGT→AGT), Ala12 (GGT→GCT), Ile12 (GGT→ATT), Cys12 (GGT→TGT), Arg12 (GGT→ CGT), Cys13 (GGC→TGC), and Asp13 (GGC→GAC). Additionally, an oligonucleotide probe specific for the human HLA-DRA locus and a 5′-biotinylated oligonucleotide were used to control PCR performance and detection reagents, respectively. Terminal deoxynucleotidyl transferase (Fermentas) was used to endue each oligonucleotide probe with a 3′-poly(dT) tail according to the manufacturer′s instruction. Tailed oligonucleotide probes were immobilized on nitrocellulose membrane as an array of parallel lines and membrane sheets were then cut to define test strips 3 mm wide. Following mutant-enriched duplex PCR, biotinylated amplification products were hybridized to nitrocellulose test strips strictly controlling temperature (45 ± 0.5°C), and bound sequences were visualized using a streptavidin-alkaline phosphatase conjungate and color substrates.15 RH was performed either manually, using a shaking waterbath (GFL, Burgwedel, Germany) set to 45°C, or essentially automated (profiBlot T48; TECAN, Grödig, Austria). A schematic outline of the RH protocol is shown in Figure 1. To validate oligonucleotide probes being specific for the mutations targeted, tumor cell lines SW480 (Val12), LS174T (Asp12), A549 (Ser12), SW1116 (Ala12), MIA Paca2 (Cys12), H157 (Arg12), H1355 (Cys13), DLD1 (Asp13), and Colo320 (wild-type) were available.14 Genomic DNA isolated from tumor cell lines containing a KRAS mutation was used to generate recombinant plasmid clones (TOPO TA Cloning Kit; Invitrogen). After confirming the presence of mutations by DNA sequencing, plasmid clones served as reference DNA templates in the PCR. Because no tumor cell lines for KRAS mutations Leu12 and Ile12 were obtainable, mutant PCR products generated by site-directed mutagenesis served as controls.14
Figure 1.
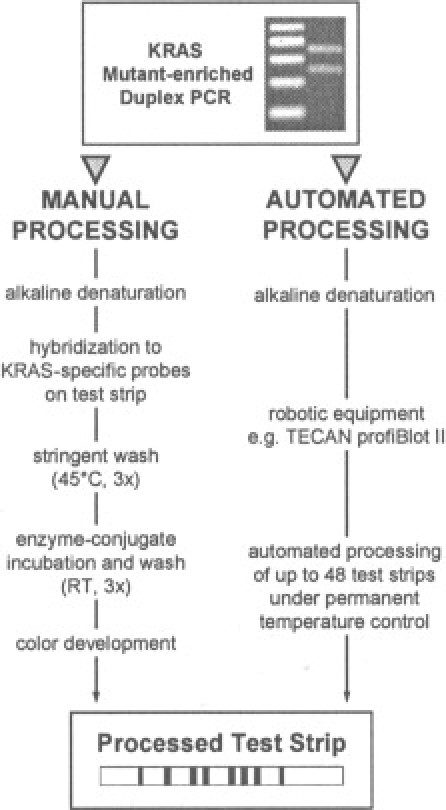
Schematic pictorial of our RH protocol. After mutant-enriched duplex PCR, RH of biotinylated amplification products to test strips was carried out manually (left) or, for increased sample throughput, essentially automated using commercially available instrumentation (right).
Results
The specificity of the RH assay reported here was verified by hybridizing reference PCR products obtained from plasmid clones to individual test strips. RH and sequencing results concurred for each KRAS mutation covered by the RH assay (data not shown). The RH assay's limit for detecting KRAS mutations was determined using different amounts of tumor cell line DNA or mutant PCR products mixed with 50 ng of wild-type DNA as templates. Figure 2 shows the RH results obtained for a mixture of 50 ng wild-type and 0.5 ng mutant genomic DNA demonstrating an analytical sensitivity of 1% for each KRAS mutation under investigation. Test strip signals obtained for DNA mixtures containing rising amounts of mutant DNA (eg, 1%, 5%, and 10%) did not show a linear increase in intensity (data not shown). Cross-reactivity caused by nonspecific interactions between oligonucleotide probes and amplification products could not be observed. Suppression of wild-type amplification by peptide nucleic acid clamping using 50 ng of wild-type DNA template was found to be complete as judged by agarose gel electrophoresis (data not shown). Accordingly, test strips hybridized to PCR products obtained for 50 ng wild-type DNA remained negative (Figure 2).
Figure 2.
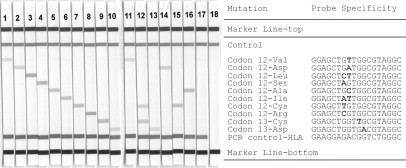
Identification of KRAS mutations by RH. Test trips obtained after hybridization and enzymatic color development are shown. Strips 1–10: Mutant-enriched PCR was perfomed using a mix of 50 ng wild-type and 0.5 ng genomic DNA derived from tumor cell lines (strips 1, 2, 4, 5, 7–10) and mutant PCR product generated by site-directed mutagenesis (strips 3, 6), respectively. Strips 11–18: Mutant-enriched PCR was performed on genomic DNA isolated from selected FFPE tissue samples (strips 11–16). The following genotypes were identified: Val12 (strip 11), Cys12/Cys13 (strip 12), Asp13 (strip 13), Asp12 (strip 14), Cys12 (strip 15), Ala12 (strip 16), 50 ng Colo320 (strip 17), and negative PCR control (strip 18).
Of 73 FFPE samples from CRC patients, we identified 26 (35%) samples to carry a KRAS mutation. Also, the single adenoma case contained a KRAS mutation. No mutation was detected in the four normal control samples. All FFPE samples were analyzed in duplicate, and results concurred within each sample, indicating good assay reproducibility (data not shown). We used DNA sequencing as a confirmatory method to verify the results obtained with the RH assay. DNA sequencing results concurred for 22 (85%) KRAS positive and all (100%) KRAS negative samples (data not shown). Thus, four FFPE samples identified KRAS positive by RH were wild-type by sequencing. However, mutations in all four samples were detectable when mutant enrichment was performed before DNA sequencing.
Twenty two (16%) samples were positive for codon 12 mutations, and 1 (0.7%) was positive for a codon 13 mutation. One sample contained 2 codon 12 mutations and one a codon 12 and codon 13 mutation. 11 (47%) of 27 mutations found in our study were Asp12, followed by 8 tumors containing the Val12 mutation, 2 tumors containing the Cys12 mutation, 2 tumors containing a Ser12 mutation, 1 tumor containing the Asp13 mutation, and 1 containing both the Asp12 and Ser12 mutation and 1 containing the Cys12 and Cys13 mutation. The distribution of codons 12 and 13 mutations with respect to histology, tumor stage and grading is shown in Table 1. Individual KRAS mutations with respective tumor stages and gradings are shown in Table 2. In summary, 1 (50%) of the 2 carcinoma in situ cases, 4 (33%) of the 12 patients with stage I, 4 (25%) of the 16 patients with stage II, 8 (32%) of the 25 patients with stage III, 6 (50%) of the 12 patients with UICC stage IV, and 2 of the 6 patients with local relapse were positive for KRAS mutations (Table 2).
Table 1.
Characteristics of 78 Colorectal Tissue Samples
|
KRAS Mutations |
||||
|---|---|---|---|---|
| n | n (%) | Codon 12 | Codon 13 | |
| Colorectal tumors | 74 | 26 (35) | 25 | 2 |
| Colon | 49 | 17 (34) | 17* | 0 |
| Rectum | 24 | 8 (33) | 6 | 2† |
| Histologic type | ||||
| Adenocarcinoma | 73 | 25 (34) | 24* | 2† |
| Rectal adenoma | 1 | 1 (0.7) | 1 | 0 |
| Normal tissue | 4 | 0 (0) | 0 | 0 |
| Differentiation grade | ||||
| G1 | 6 | 0 (0) | 0 | 0 |
| G2 | 35 | 14 (40) | 14 | 1 |
| G3 | 31 | 11 (35) | 11* | 1† |
| ND | 1 | 0 (0) | 0 | 0 |
| Staging | ||||
| pTis | 2 | 1 (50) | 1 | 0 |
| UICC I | 12 | 4 (33) | 4 | 0 |
| UICC IIA | 11 | 2 (18) | 2 | 0 |
| UICC IIB | 5 | 2 (40) | 2 | 0 |
| UICC IIIA | 5 | 1 (20) | 0 | 1 |
| UICC IIIB | 14 | 6 (43) | 6* | 1† |
| UICC IIIC | 6 | 1 (17) | 1 | 0 |
| UICC IV | 12 | 6 (50) | 6 | 0 |
| Local relapse | 6 | 2 (33) | 2 | 0 |
ND: Not determined in a case with local relapse of an adenocarcinoma of the rectum.
One tumor combined both Asp 12 and Ser 12 mutations.
One tumor combined both Cys 12 and Cys 13 mutations.
Table 2.
Individual KRAS Mutations Detected in 26 Colorectal Tumor Samples Using the RH Assay
| Sample ID | Histologic finding | WHO grading | Mutation |
|---|---|---|---|
| 69 | Rectal polyp | − | Val12 |
| 7 | I | 2 | Asp12 |
| 10 | I | 2 | Asp12 |
| 12 | I | 2 | Cys12 |
| 41 | I | 3 | Ser12 |
| 9 | IIA | 3 | Asp12 |
| 16 | IIA | 2 | Ser12 |
| 3 | IIB | 3 | Asp12 |
| 48 | IIB | 2 | Val12 |
| 19 | IIIA | 2 | Asp13 |
| 26 | IIIB | 2 | Val12 |
| 28 | IIIB | 2 | Val12 |
| 34 | IIIB | 2 | Val12 |
| 37 | IIIB | 3 | Asp12/Ser12 |
| 45 | IIIB | 3 | Cys12 |
| 71 | IIIB | 3 | Cys12/Cys13 |
| 77 | IIIB | 2 | Asp12 |
| 33 | IIIC | 2 | Val12 |
| 8 | IV | 3 | Asp12 |
| 35 | IV | 3 | Val12 |
| 43 | IV | 2 | Asp12 |
| 57 | IV | 3 | Asp12 |
| 61 | IV | 3 | Asp12 |
| 68 | IV | 3 | Asp12 |
| 47 | L.R. | − | Val12 |
| 60 | L.R. | − | Asp12 |
L.R.: local relapse.
Discussion
This study reports a sensitive, nonquantitative assay for the detection of KRAS mutations in FFPE tissue, combining mutant-enriched PCR and RH. The RH assay′s limit for detecting KRAS mutations was determined to be 1% based on DNA mixing experiments using genomic DNA isolated from various tumor cell lines. Test strip signals did not increase correspondingly when higher amounts of mutant DNA (eg, 5% or 10%) were analyzed, indicating that the RH assay cannot be used to estimate the quota of mutant DNA present in a sample. In this study, 26 (35%) of 73 FFPE samples obtained from CRC patients contained a KRAS mutation as determined by RH. This finding is consistent with published studies that detected oncogenic KRAS mutations in 20% to 41% of CRC cases2,16,17 and in approximately 20% and 50% of small and more advanced adenomas, respectively.2,16 Moreover, KRAS mutations occurred predominantly in codon 12, which is well in line with previous reports.2,3,4
A wide variety of methods, such as enriched PCR-restriction fragment length polymorphism,18,19 single-strand conformational polymorphism,20 denaturing gradient gel electrophoresis,21 high performance liquid chromatography,22 amplification refractory mutation system,23 high resolution melting analysis,24 and DNA sequencing, have been applied to KRAS mutation analysis. While simpler methods like PCR-restriction fragment length polymorphism or amplification refractory mutation system-PCR can be used for the sensitive detection of KRAS mutations in a background of wild-type DNA (analytical sensitivity ≤1%), they are hardly capable of identifying more than one mutation in a single tube, thus limiting their applicability to diagnostic settings with finite sample throughput only. High resolution melting analysis is a rapid in-tube methodology that enables high-throughput screening of KRAS mutations with a moderate analytical sensitivity of 5% to 6%. Although high resolution melting analysis, single-strand conformational polymorphism, denaturing gradient gel electrophoresis, and high performance liquid chromatography are useful screening tools, DNA sequencing remains the gold standard for the confirmation and identification of specific mutations. Dideoxy DNA sequencing is the most commonly used sequencing method, however, sensitivity for detecting mutant DNA is low (ie, detection limit around 20%) and sample throughput is limited. In this study, DNA sequencing without previous enrichment of mutants failed to detect 4 (15%) out of 26 KRAS positive FFPE samples. This finding suggests that even after microdissection, mutant sequences in a given DNA isolate can be rare and more sensitive methods are needed for mutation analysis. Recently, a KRAS pyrosequencing assay (ie, a real-time, non electrophoretic, nucleotide extension sequencing method with an allele quantification capability) has demonstrated a superior analytical sensitivity of approximately 5% on mixed DNA samples containing various amounts of mutant DNA.25
The RH assay described here is a sensitive tool (analytical sensitivity 1%) for the simultaneous detection of 10 mutations in codons 12 and 13 of the KRAS gene. The procedure is relatively fast (<6 hours excluding DNA isolation) and follows a simple protocol using ready to use test strips and visible color detection. The method requires standard laboratory equipment only and, for increased sample throughput, hybridization/detection may be fully automated by means of commercially available instrumentation. Therefore, this approach appears to be a good alternative to methods currently in use for the detection of KRAS mutations in DNA isolated from archived FFPE tissue.
Acknowledgements
We acknowledge the excellent technical assistance of Bettina Rauscher. We are grateful to Lothar Prix and Andreas Schütz for providing cell lines and mutant PCR products.
Footnotes
Supported by the Fund for Innovative Cancer Research, City of Vienna.
Disclosures: G.K. and C.O. are employees of Vienna Lab Diagnostics and R.Z. is the Managing Director and a shareholder of Vienna Lab Diagnostics.
References
- 1.Barbacid M. Ras Genes. Annu Rev Biochem. 1987;56:779–827. doi: 10.1146/annurev.bi.56.070187.004023. [DOI] [PubMed] [Google Scholar]
- 2.Fang JY, Richardson BC. The MAPK signalling pathways and colorectal cancer. Lancet Oncol. 2005;6:322–327. doi: 10.1016/S1470-2045(05)70168-6. [DOI] [PubMed] [Google Scholar]
- 3.Grady W, Markowitz S. Genetic and epigenetic alterations in colon cancer. Annu Rev Genomics Hum Genet. 2002;3:101–128. doi: 10.1146/annurev.genom.3.022502.103043. [DOI] [PubMed] [Google Scholar]
- 4.Toyooka S, Tsukuda K, Ouchida M, Tanino M, Inaki Y, Kobayashi K, Yano M, Soh J, Kobatake T, Shimizu N, Shimizu K. Detection of codon 61 point mutations of the K-ras gene in lung and colorectal cancers by enriched PCR. Oncol Rep. 2003;10:1455–1459. doi: 10.3892/or.10.5.1455. [DOI] [PubMed] [Google Scholar]
- 5.Ogino S, Kawasaki T, Kirkner GJ, Loda M, Fuchs CS. CpG island methylator phenotype-low (CIMP-low) in colorectal cancer: possible associations with male sex and KRAS mutations. J Mol Diagn. 2006 Nov;8:582–588. doi: 10.2353/jmoldx.2006.060082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nosho K, Irahara N, Shima K, Kure S, Kirkner GJ, Schernhammer ES, Hazra A, Hunter DJ, Quackenbush J, Spiegelman D, Giovannucci EL, Fuchs CS, Ogino S. Comprehensive biostatistical analysis of CpG island methylator phenotype in colorectal cancer using a large population-based sample. PLoS ONE. 2008;3:e3698. doi: 10.1371/journal.pone.0003698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Barault L, Charon-Barra C, Jooste V, de la Vega MF, Martin L, Roignot P, Rat P, Bouvier AM, Laurent-Puig P, Faivre J, Chapusot C, Piard F. Hypermethylator phenotype in sporadic colon cancer: study on a population-based series of 582 cases. Cancer Res. 2008;68:8541–8546. doi: 10.1158/0008-5472.CAN-08-1171. [DOI] [PubMed] [Google Scholar]
- 8.Van Cutsem E, Lang I, D' haens G, Moiseyenko V, Zaluski J, Folprecht G, Tejpar S, Kisker O, Stroh C, Rougier P. KRAS status and efficacy in the first-line treatment of patients with metastatic colorectal cancer (mCRC) treated with FOLFIRI with or without cetuximab. J Clin Oncol. 2008;26:A2. [Google Scholar]
- 9.Mendelsohn J, Baselga J. Epidermal growth factor receptor targeting in cancer. Semin Oncol. 2006;33:369–385. doi: 10.1053/j.seminoncol.2006.04.003. [DOI] [PubMed] [Google Scholar]
- 10.Hynes NE, Lane HA. ERBB receptors and cancer: the complexity of targeted inhibitors. Nat Rev Cancer. 2005;5:341–354. doi: 10.1038/nrc1609. [DOI] [PubMed] [Google Scholar]
- 11.Allegra CJ, Jessup JM, Somerfield MR, Hamilton SR, Hammond EH, Hayes DF, McAllister PK, Morton RF, Schilsky RL. American Society of Clinical Oncology Provisional Clinical Opinion: testing for kras gene mutations in patients with metastatic colorectal carcinoma to predict response to anti-epidermal growth factor receptor monoclonal antibody therapy. J Clin Oncol. 2009;20:2091–2096. doi: 10.1200/JCO.2009.21.9170. [DOI] [PubMed] [Google Scholar]
- 12.TNM Atlas . In: Illustrated Guide to the TNM/pTNM Classification of Malignant Tumours. Wittekind C, Greene FL, Hutter RVP, Klimpfinger M, Sobin LH, editors. Springer; New York: 2007. pp. 101–113. [Google Scholar]
- 13.Fabjani G, Kucera E, Schuster E, Minai-Pour M, Czerwenka K, Sliutz G, Leodolter S, Reiner A, Zeillinger R. Genetic alterations in endometrial hyperplasia and cancer. Cancer Lett. 2002;175:205–211. doi: 10.1016/s0304-3835(01)00714-5. [DOI] [PubMed] [Google Scholar]
- 14.Prix L, Uciechowski P, Böckmann B, Giesing M, Schuetz AJ. Diagnostic biochip array for fast and sensitive detection of K-ras mutations in stool. Clin Chem. 2002;48:428–435. [PubMed] [Google Scholar]
- 15.Kriegshaeuser G, Krugluger W, Halsall D, Kury F, Oberkanins C. Reverse-Hybridization assay for mutations associated with hereditary sugar intolerance. Eur J Hum Genet. 2004;(12 Suppl):S1–S250. [Google Scholar]
- 16.Boughdady IS, Kinsella AR, Haboubi NY, Schofield PF. K-ras gene mutations in adenomas and carcinomas of the colon. Surg Oncol. 1992;1:275–282. doi: 10.1016/0960-7404(92)90088-3. [DOI] [PubMed] [Google Scholar]
- 17.Russo A, Bazan V, Agnese V, Rodolico V, Gebbia N. Prognostic and predictive factors in colorectal cancer: Kirsten Ras in CRC (RASCAL) and TP53CRC collaborative studies. Ann Oncol. 2005;16(Suppl):S44–S49. doi: 10.1093/annonc/mdi907. [DOI] [PubMed] [Google Scholar]
- 18.Hardingham JE, Kotasek D, Farmer B, Butler RN, Mi JX, Sage RE, Dobrovic A. Immunobead-PCR: a technique for the detection of circulating tumor cells using immunomagnetic beads and the polymerase chain reaction. Cancer Res. 1993;53:3455–3458. [PubMed] [Google Scholar]
- 19.Behn M, Thiede C, Neuberger A, Pankow W, Schuermann M. Facilitated detection of oncogene mutations from exfoliated tissue material by a PNA-mediated ‘enriched PCR’ protocol. J Pathol. 2000;190:69–75. doi: 10.1002/(SICI)1096-9896(200001)190:1<69::AID-PATH503>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- 20.Kalra R, Paderanga DC, Olson K, Shannon KM. Genetic analysis is consistent with the hypothesis that NF1 limits myeloid cell growth through p21ras. Blood. 1994;84:3435–3439. [PubMed] [Google Scholar]
- 21.Keohavong P, Zhu D, Whiteside TL, Swalsky P, Bakker A, Elder EM, Siegfried JM, Srivastava S, Finkelstein SD. Detection of infrequent and multiple K-ras mutations in human tumors and tumor-adjacent tissues. Anal Biochem. 1997;247:394–403. doi: 10.1006/abio.1997.2100. [DOI] [PubMed] [Google Scholar]
- 22.Lilleberg SL, Durocher J, Sanders C, Walters K, Culver K. High sensitivity scanning of colorectal tumors and matched plasma DNA for mutations in APC,TP53,K-RAS, and BRAF genes with a novel DHPLC fluorescence detection platform. Ann NY Acad Sci. 2004;1022:250–256. doi: 10.1196/annals.1318.039. [DOI] [PubMed] [Google Scholar]
- 23.Amado RG, Wolf M, Peeters M, Van Cutsem E, Siena S, Freeman DJ, Juan T, Sikorski R, Suggs S, Radinsky R, Patterson SD, Chang DD. Wild-type KRAS is required for panitumumab efficacy in patients with metastatic colorectal cancer. J Clin Oncol. 2008;26:1626–1634. doi: 10.1200/JCO.2007.14.7116. [DOI] [PubMed] [Google Scholar]
- 24.Krypuy M, Newnham GM, Thomas DM, Conron M, Dobrovic A. High resolution melting analysis for the rapid and sensitive detection of mutations in clinical samples: KRAS codon 12 and 13 mutations in non-small cell lung cancer. BMC Cancer. 2006;6:295. doi: 10.1186/1471-2407-6-295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ogino S, Kawasaki T, Brahmandam M, Yan L, Cantor M, Namgyal C, Mino-Kenudson M, Lauwers GY, Loda M, Fuchs CS. Sensitive sequencing method for KRAS mutation detection by pyrosequencing. J Mol Diagn. 2005;7:413–421. doi: 10.1016/S1525-1578(10)60571-5. [DOI] [PMC free article] [PubMed] [Google Scholar]