

Abstract
In 5% to 10% of cases, neurofibromatosis type 1 is caused by microdeletions scattered across the entire NF1 gene and various neighboring genes. The phenotype appears to be more severe in patients with NF1 microdeletions than in patients with NF1 single point mutations. We have developed a new method for detecting and characterizing NF1 microdeletions based on a custom high-resolution oligonucleotide array comparative genomic hybridization by using the custom 8x15K Agilent array format. The array comprised a total of 14,207 oligonucleotide probes spanning the whole of chromosome 17, including 12,314 probes spanning an ∼8 Mb interval surrounding the NF1 locus. We validated this approach by testing NF1 microdeleted DNA samples previously characterized by means of microsatellites and real-time PCR methods. Our array comparative genomic hybridization provided enough information for subsequent long-range PCR and nucleotide sequencing of the microdeletion endpoints. Unlike previously described methods, our array comparative genomic hybridization was able to unambiguously differentiate between the three types of microdeletions (type I, type II, and atypical) and to characterize atypical microdeletions. Further comparative studies of patients with well-characterized genotypes and phenotypes and different microdeletions sizes and breakpoints will help determine whether haploinsufficiency of deleted genes and/or genes rearrangements influence clinical outcomes.
Neurofibromatosis 1 (NF1, OMIM 162200) is an autosomal disorder with an estimated incidence of 1 in 3500 live births.1 The main features of NF1 are multiple neurofibromas, café-au-lait spots, axillary freckling, Lisch nodules, tibial pseudarthrosis, and a predisposition to develop certain benign and malignant nervous system tumors.2 NF1 is due to autosomal dominant loss-of-function mutations of the NF1 gene (neurofibromin 1; NM_000267).3 NF1 is located at 17q11.2 and contains 60 translated exons distributed over ∼300 kb.
Most germline mutations identified so far in NF1 patients are intragenic single point mutations that cause truncation or loss of the encoded protein. However, in approximately 5% to 10% of cases, NF1 is caused by microdeletions scattered along the entire NF1 gene and neighboring genes.4 Most NF1 patients have one of two recurrent (typical) microdeletions.5 The type I microdeletion is 1.4 Mb long and is caused by non allelic homologous recombination between the NF1 proximal and distal low-copy repeats (NF1-REP-a and –c) flanking the NF1 gene.6 The type II microdeletion is smaller (1.2 Mb) and its breakpoints are located in the SUZ12 gene (suppressor of zeste 12 homolog; NM_015355) and its pseudogene SUZ12P.5,7 Less frequent than these two types of microdeletion are so-called atypical NF1 microdeletions with non-recurring breakpoints.8
NF1 patients with microdeletions that encompass the entire NF1 coding region and various contiguous genes often have a more severe phenotype than patients with intragenic NF1 mutations—including dysmorphic features, learning disability, an excessive number and earlier onset of benign neurofibromas, developmental delay, and possibly a higher incidence of malignant peripheral nerve sheath tumors—supporting the existence of a contiguous gene syndrome.8,9,10,11 However, the high phenotypic variability of the NF1 microdeletion syndrome, the small number of microdeletions so far characterized (for example fewer than 20 atypical NF1 microdeletions), and the imprecise definition of their boundaries make it difficult to establish reliable genotype/phenotype correlations.8,10,12,13 To improve the identification of NF1 microdeletions, we developed a custom microarray and validated it by testing well-characterized NF1 microdeletions with typical/atypical microdeletions distinction using microsatellite markers and PCR-based gene dosage.
Materials and Methods
Patients
A neurofibromatosis 1 database (NF-France) has been established in France, thanks to a grant from the French Clinical Research Program (coordinator: Pr. P. Wolkenstein, Henri Mondor Hospital, Créteil, France). The collection consists of 561 families with 1083 NF1 patients. The performance of our array CGH was validated on eight well-characterized NF1 microdeletions (by microsatellites polymorphisms studies and real-time PCR-based gene dosage) included in the database: patients NF00028, NF00085, NF00358, NF00437, NF01465, NF01470, NF01507, and NF01585. Six normal control DNAs were also collected from six healthy donors. All blood samples used for DNA extraction were collected with the patients' informed consent.
DNA Extraction
High-molecular-weight DNAs from patients and healthy donors were prepared by standard proteinase K digestion followed by phenol-chloroform extraction from whole-blood leukocytes. The quantity and quality of the DNA samples were assessed with Nanodrop technology (Coleman Technologies, Orlando, FL) and verified by electrophoresis through agarose gels and staining with ethidium bromide.
Microsatellite Typing
A series of 17q11.2-linked microsatellite markers were characterized, consisting of four NF1 intragenic (D17S1307, D17S2163, D17S1166, and IVS38-GT53.0) and three NF1 extragenic (D17S841, D17S1800, and D17S798) polymorphic microsatellites (primer sequences are given in Table 1). After amplification with a 5′-end-labeled primer, followed by the addition of an internal size standard (Genescan 2500-ROX: Applied Biosystems), the PCR products were separated on 6% polyacrylamide/7 M/L urea gel, using an ABI PRISM 310 DNA sequencer (Applied Biosystems). The results were analyzed with the Genescan 672 (version 1.2) software package.
Table 1.
Primers Designed to Analyze Four NF1 Intragenic (D17S1307, D17S2163, D17S1166, and IVS38-GT53.0) and Three NF1 Extragenic (D17S841, D17S1800, and D17S798) Polymorphic Microsatellites
| Target | Primer | Sequence |
|---|---|---|
| D17S1307 | IVS1-R | 5′-Fam-TGATCTGGACGTAGGAGAC-3′ |
| IVS1-F | 5′-GATTATCGCCCATTTATTACACTT-3′ | |
| D17S2163 | STR27III-F | 5′-Fam-TGAAGTATGCAGTTTTCCAG-3′ |
| STR27III-R | 5′-GGCTAAGTGTAAACGCAAAG-3′ | |
| D17S1166 | IVS27IV-1 | 5′-Fam-TAGATTATATGGGACAGAAAATG-3′ |
| IVS27IV-2 | 5′-CTTGAGGTGATGACAGGATG-3′ | |
| IVS38-GT53.0 | IVS38-F | 5′-Fam-GAGCAAGACCCTGTCTCCA-3′ |
| IVS38-R | 5′-CTCCTAACATTTATTAACCTTA-3′ | |
| D17S841 | D17S841-F | 5′-Fam-TGGACTTTCTTACATGGCAG-3′ |
| D17S841-R | 5′-AGGTTAGTAGTCTATGTCACAGCG-3′ | |
| D17S635 | D17S635-L1 | 5′-Fam-TTGTGCCACTGCACTCCAG-3′ |
| D17S635-U1 | 5′-GACTCTTGATTGTAAGCNACAGAA-3′ | |
| D17S1800 | D17S1800-F | 5′-Fam-CTAAACTAGGTTGGGTTGAAATCTC-3′ |
| D17S1800-R | 5′-TCTGGCACAAAGACCTGAG-3′ | |
| D17S798 | D17S798-F | 5′-Fam-CCATGAGAAAGTTGTTTAGTAGT-3′ |
| D17S798-R | 5′-TGTTCTTGGGAGTGCAG-3′ |
Sequence Tagged Site Real-Time PCR-Based Gene Dose Mapping
The theoretical and practical aspects of sequence tagged site (STS) real-time PCR-based gene dose mapping have been described in detail elsewhere.13 In brief, quantitative values are obtained from the threshold cycle number at which the increase in the signal associated with exponential growth of PCR products begins to be detected by the Applied Biosystems analysis software. The precise amount of genomic DNA added to each reaction mix (based on optical density) and its quality (ie, lack of extensive degradation) are both difficult to assess. We therefore also quantified the ALB gene (encoding albumin and mapping to chromosome region 4q11-q13) as an endogenous DNA control, and each sample was normalized on the basis of its ALB content. The relative copy number of the target STS marker was also normalized to a calibrator, or 1× sample, consisting of genomic DNA from a normal subject. Final results, expressed as N-fold differences in the target STS marker copy number relative to the ALB gene and the calibrator, and termed “NSTS” were determined as follows: NSTS = 2(ΔCtsample − ΔCtcalibrator), where ΔCt values of the sample and calibrator are determined by subtracting the average Ct value of the target STS marker from the average Ct value of the ALB gene. Given the target STS marker, samples with NSTS values of 0.5 and 1.0 were considered deleted and normal, respectively. Primers for ALB and the forty-five 17q11.2-linked STS markers were chosen with the assistance of the computer program Oligo 4.0 (National Biosciences). We conducted BLASTN searches against nr (the non-redundant set of the GenBank, European Molecular Biology Laboratory, and DNA Data Bank of Japan database sequences) to confirm the total STS specificity of the nucleotide sequences chosen for the primers. All PCR reactions were performed with an ABI Prism 7900 Sequence Detection System (Applied Biosystems) and the SYBR Green PCR Core Reagents kit (Applied Biosystems). Experiments were performed in triplicates for each data point.
Microarray Design
This high-resolution oligonucleotide-based microarray contained 15,689 60-mer probes (including 1482 manufacturer control probes) spanning both coding and non-coding genomic sequences of chromosome 17 (Figure 1). The multistep design of our custom array CGH included 14,207 probes distributed among four groups with various probe densities. Group I comprised 1893 probes spread along chromosome 17 with a ∼40-kb average probe spacing, except in an 8-Mb interval. This 8-Mb interval spanned the NF1 gene region and included the largest NF1 atypical microdeletions described in the literature.8,10,12,13 Group II spanned 1.3 Mb and 2.7 Mb on centromeric and telomeric sides of this 8-Mb interval respectively, and comprised 4000 probes with a 1-kb average probe spacing. Group III spanned the central 4 Mb of the 8 Mb interval and had a probe spacing of ∼500 bp and 8003 probes. Group III covered the NF1 gene region and included type I, type II and the majority of atypical NF1 microdeletions described in the literature.8,10,12 Finally, group IV was designed to further differentiate between type I and type II microdeletions. It included 311 additional probes designed in the least similar sequences (with maximum mismatches) of the paralogous regions flanking the NF1 gene (NF1-REP-a and –c and SUZ12 and SUZ12P). Group I probes were selected from catalog probes of Agilent's 44K commercial array CGH. High-density probes in groups II, III, and IV were selected from Agilent's eArray library (Agilent Technology, Inc. Santa Clara, CA), which contains a panel of four million validated synthetic probes that genomically tile specific regions avoiding all common repeats and other redundant sequences. The 14,207 oligonucleotide probe sequences and locations are given in Supplemental Table S1 at http://jmd.amjpathol.org. DNA sequence information referred to the public University of California Santa Cruz database [Human Genome Browser, March 2006 assembly: hg 18, National Center for Biotechnology Information Build 36.1].
Figure 1.
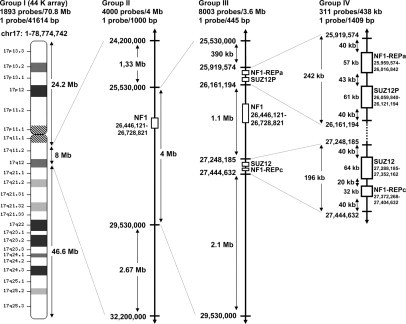
Multistep design of the high-resolution 15K NF1 array CGH. The array comprised 15,689 60-mer probes (with 1482 manufacturer control probes) spanning coding and non-coding genomic sequences of chromosome 17 with regions of various probe densities. DNA sequence information referred to the public UCSC database (Human Genome Browser, March 2006 assembly: hg 18, NCBI Build 36.1).
Microarray Procedure and Data Analysis
CGH labeling and hybridization were performed as recommended in the Agilent manual (Protocol v5.0, June 2007, Agilent technologies). Briefly, genomic DNA (300 ng) from the reference sample (pool of six normal control DNAs) and from each experimental sample was double-digested with AluI and RsaI (Promega, Madison, WI) for 2 hours at 37°C. The digested DNAs were labeled by random priming with the Agilent Genomic DNA Labeling Kit Plus (Agilent technologies) for 2 hours at 37°C, according to the manufacturer's instructions. Patients' DNA and pooled normal control DNAs (reference) were labeled with Cy5-dUTP and Cy3-dUTP, respectively. Labeled products were purified with Microcon YM-30 filters (Millipore, Billerica, MA). Patient and normal control DNAs (reference) were mixed and hybridized with Human Cot I DNA (Invitrogen) at 65°C for 24 hours. The sandwiched slides were hybridized for 24 hours at 65°C with 15 rotations per minute and washed according to the Agilent protocol. A reference versus reference control hybridization step was used to assess array hybridization quality. Arrays were scanned with an Agilent DNA Microarray Scanner (G2565BA). Log2 ratios were determined with Agilent Feature Extraction software (v 9.1.3.1) and the global quality of the individual microarrays used in the experiment was validated against the quality metrics (QC metrics) of this software. Results were visualized and analyzed with Agilent's CGH Analytics v3.5 software, and copy number aberrations were detected with the Aberration Detection Method 2 (ADM-2) algorithm using a threshold value of 6.0. This threshold value of the altered copy number was determined from the normal variation in the control hybridization.
Fine Characterization of Two Atypical Microdeletion Breakpoints
Long-range PCR was performed with the Expand 20 kb Plus PCR kit as recommended by the manufacturer (Roche Applied Science). Forward and reverse long-range PCR primers used to characterize the NF00028 (NF00028F1: 5′-TTATTGACGTGACCCTTCTCCTGACT-3′ and NF00028R1: 5′-GTATCTCCCGACCTCACTGCAGAA-3′) and NF00358 (NF00358F1: 5′-ATGTTTGGCGAGCTATTTTGTCTGTT-3′ and NF00358R1: 5′-AAAGCCCCTTCTGAATATGAGCAAA-3′) atypical microdeletions were designed in the last proximal and distal non deleted 60-mer probe sequences. The primer pairs were selected to be unique in the human genome and to be able to amplify only a 712-bp and 905-bp deleted genomic fragment from patients NF00358 and NF00028, respectively, the normal fragments being too large (>800 kb) to be amplified. The 712-bp and 905-bp PCR products were sequenced with the ABI BigDye terminator sequencing kit (Applied Biosystems) on an ABI Prism 3130 automatic DNA sequencer (Applied Biosystems).
Results
The performance of our array CGH was validated on eight well-characterized NF1 microdeletions from the NF-France database. These eight microdeletions had been initially characterized with another method.13 Briefly, a series of four NF1 intragenic and three extragenic microsatellite markers were characterized to identify NF1 microdeletions. To further determine the size of the microdeletions, we estimated the number of copies (one or two) of a large panel of sequence tagged sites (STS) regularly spaced within breakpoints, by using a multistep real-time PCR-based dosage assay. Six typical and two atypical deletions were identified using this combined approach.
The high-quality array CGH gave an excellent mean DLRSpread value of 0.17 ± 0.02 (range from 0.13 to 0.19) and high signal-to-noise ratios (84.5 ± 7.1 and 92.4 ± 11.1 for the green and red channel signals, respectively). Excellent quality metric values were also obtained with the reference versus reference control hybridization (DLRSpread = 0.14; SNR_green = 84; SNR_red = 111).
A panoramic view of the region, as displayed by CGH Analytics software, gave the precise location of the deleted sequences (Figure 2A). The eight NF1 microdeletions were accurately characterized with our custom array CGH, and comprised four type I (NF00437, NF01470, NF01507, and NF01585), two type II (NF00085 and NF01465), and two atypical deletions (NF00028 and NF00358). Figures 2A and 2B show characteristic array CGH profiles for four of the eight deletions. Among the two type II microdeletions herein characterized, patient NF01465 has a de novo microdeletion whereas NF00085 showed an inherited microdeletion. Patients with type II microdeletions have often been shown to be somatic mosaicism.5,7 However, mosaicism was not clearly observed in NF01465 with both our custom array CGH and NF1 microsatellite markers typing, but it could not be excluded in this patient in particular because of the very high percentage of cells with microdeletion often reported in NF1 type II microdeleted patients peripheral blood.5,7 The breakpoints of the two atypical deletions were precisely determined by sequencing the junction long-range PCR products (Figure 2C). The NF00028 deletion was exactly 836,791 bp long and was located on chromosome arm 17q between nucleotides 25,943,375 and 26,780,167 (numbered as in build 36.1 of the NCBI). The NF00358 deletion was exactly 1185,175 bp long and was located between nucleotides 25,609,630 and 26,794,806.
Figure 2.

A: Array CGH profiles of four deletions of the eight characterized deletions: one type I (NF00437), one type II (NF00085), and two atypical (NF00028, NF00358) deletions were easily identified. B: Zoom on telomeric breakpoints: SUZ12 is entirely deleted in the type I deletion (NF01470) and partially deleted in type II (NF00085). C: Electrophoregram of part of the specific junction sequence generated by long-range PCR from atypical deletions NF00028 and NF00358.
It is noteworthy that the NF00028 centromeric and telomeric breakpoints were located within a 116-bp LINE (Long Interspersed Nucleotide Elements) and within a 96-bp AluSq, respectively. The NF00358 telomeric breakpoint was located within a 307-bp AluSx. No significant overlap was found between the centromeric and telomeric sequences of both NF00028 and NF00358 microdeletions, suggesting non-homologous end joining (NHEJ) as the likely involved mechanism.14
Interestingly, both NF00028 and NF00358 telomeric breakpoints were located in the RAB11FIP4 gene (RAB11 family interacting protein 4; NM_032932) in introns 1 and 3, respectively, suggesting a new NF1 microdeletion recurrent breakpoint. The NF00028 centromeric breakpoint was located in intron 1 of the LRRC37B2 gene (Leucine rich repeat containing 37, member B2; NM_207323) and the NF00358 centromeric breakpoint was located in intron 1 of the BLMH gene (Bleomycin hydrolase; NM_000386).
Discussion
Until recently it was notoriously laborious to detect NF1 microdeletions and the characterization of such alterations is not yet fully optimized. Fluorescence in situ hybridization (FISH) has been widely used and a combined approach has been developed to improve resolution.4,10,12,15 The stepwise FISH procedure consists of YAC (Yeast Artificial Chromosome) clones of a NF1 region contig FISH followed by high-resolution FISH technique with locus-specific cloned PCR product probes.10,12 A set of FISH probes has been specifically designed to distinguish between type I and II microdeletions.15
However, many laboratories are not equipped for FISH analysis, and multiplex ligation-dependent probe amplification (MLPA) may be considered as an alternative. Two MLPA assays have been specifically designed to detect NF1 whole-gene deletions, namely SALSA P081/082 NF1 MLPA assay and SALSA P122 NF1 area (MRC-Holland, Amsterdam, The Netherlands).15,16,17 The SALSA P081/082 NF1 MLPA assay version 04 included a total of 81 probes and tested only 51 of the 60 NF1 exons. The SALSA P122 NF1 area assay version 01 (V.01), with five probes in NF1 and seven additional probes in NF1 flanking genes, distinguished between type II and the larger type I microdeletions with only two probes.15,16 False-positive results have been reported with the MLPA technique, due to subtle point mutations close to the ligation sites at the ends of MLPA hemiprobes.15,16 However, MLPA allows a fast and inexpensive fist-line screening for NF1 microdeletion, complementary to a high-resolution technique such as array CGH.
Two specific microarrays derived from pooled PCR products for CGH analysis of the NF1 locus have previously been developed.18,19 The first one, developed by Mantripragada et al contained 183 probes.18 The PCR products had an average size of 637 bp and covered 2.24 Mb of region 17q11.2, with an average resolution of ∼12 kb. The second PCR products array from Shen et al had an average resolution of 4.5 kb with 493 pooled probes measuring 200 to 998 bp.19 These two arrays were reliable tools for molecular diagnosis of NF1 microdeletions. However, neither array CGH reached exon-level resolution in NF1, as they did not cover all 60 exons. Another limitation was that neither array CGH could differentiate between type I and type II microdeletions.18,19 Moreover, the breakpoints of two atypical deletions could not be determined because their size was greater than the span of the microarray from Mantripragada et al18 As these arrays had limited resolution, we developed a high-resolution oligonucleotide array CGH-based approach for the detection of large NF1 rearrangements.
Contrary to previously developed techniques, our high-resolution array CGH clearly distinguishes between type I and type II microdeletions (Figure 2B), provides enough information for subsequent breakpoint sequencing, and has high density coverage of the NF1 gene (with 821 probes in NF1).
In conclusion, we have developed a sensitive, cost effective (∼100 є per patient) and rapid approach (eight patients can be analyzed simultaneously per 8x15K slide) suitable for accurate characterization of NF1 microdeletions. Further studies comparing NF1 patients with well-characterized genotypes and phenotypes and different microdeletion sizes and breakpoints will help determine whether haploinsufficiency of deleted genes and/or gene rearrangements influence the nature and severity of clinical manifestations, and to identify genes or mechanisms contributing to the specific NF1-microdeletion phenotype. Large series of NF1 microdeletion samples have now to be collected in a prospective study to assess this clinical issue.
Acknowledgements
We thank the patients and their parents for their participation. We are grateful to Lucie Hernandez and Samuel Quentin from Plate-Forme Génomique de l'Institut Universitaire d'Hématologie de l'Hopital Saint-Louis for expert technical assistance.
Footnotes
Supported in part by grants from Association Neurofibromatoses et Recklinghausen, Ligue Française Contre les Neurofibromatoses, the Clinical Research programme (PHRC 2002), INSERM Projet NF1GeneModif and Ministère de l'Enseignement Supérieur et de la Recherche.
Supplemental material for this article can be found on http://jmd.amjpathol.org.
Supplementary data
References
- 1.Carey JC, Baty BJ, Johnson JP, Morrison T, Skolnick M, Kivlin J. The genetic aspects of neurofibromatosis. Ann NY Acad Sci. 1986;486:45–56. doi: 10.1111/j.1749-6632.1986.tb48061.x. [DOI] [PubMed] [Google Scholar]
- 2.Ferner RE, Huson SM, Thomas N, Moss C, Willshaw H, Evans DG, Upadhyaya M, Towers R, Gleeson M, Steiger C, Kirby A. Guidelines for the diagnosis and management of individuals with neurofibromatosis 1. J Med Genet. 2007;44:81–88. doi: 10.1136/jmg.2006.045906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Radtke HB, Sebold CD, Allison C, Haidle JL, Schneider G. Neurofibromatosis type 1 in genetic counseling practice: recommendations of the National Society of Genetic Counselors. J Genet Couns. 2007;16:387–407. doi: 10.1007/s10897-007-9101-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kluwe L, Siebert R, Gesk S, Friedrich RE, Tinschert S, Kehrer-Sawatzki H, Mautner VF. Screening 500 unselected neurofibromatosis 1 patients for deletions of the NF1 gene. Hum Mutat. 2004;23:111–116. doi: 10.1002/humu.10299. [DOI] [PubMed] [Google Scholar]
- 5.Steinmann K, Cooper DN, Kluwe L, Chuzhanova NA, Senger C, Serra E, Lazaro C, Gilaberte M, Wimmer K, Mautner VF, Kehrer-Sawatzki H. Type 2 NF1 deletions are highly unusual by virtue of the absence of nonallelic homologous recombination hotspots and an apparent preference for female mitotic recombination. Am J Hum Genet. 2007;81:1201–1220. doi: 10.1086/522089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Raedt TD, Stephens M, Heyns I, Brems H, Thijs D, Messiaen L, Stephens K, Lazaro C, Wimmer K, Kehrer-Sawatzki H, Vidaud D, Kluwe L, Marynen P, Legius E. Conservation of hotspots for recombination in low-copy repeats associated with the NF1 microdeletion. Nat Genet. 2006;38:1419–1423. doi: 10.1038/ng1920. [DOI] [PubMed] [Google Scholar]
- 7.Kehrer-Sawatzki H, Kluwe L, Sandig C, Kohn M, Wimmer K, Krammer U, Peyrl A, Jenne DE, Hansmann I, Mautner VF. High frequency of mosaicism among patients with neurofibromatosis type 1 (NF1) with microdeletions caused by somatic recombination of the JJAZ1 gene. Am J Hum Genet. 2004;75:410–423. doi: 10.1086/423624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Venturin M, Guarnieri P, Natacci F, Stabile M, Tenconi R, Clementi M, Hernandez C, Thompson P, Upadhyaya M, Larizza L, Riva P. Mental retardation and cardiovascular malformations in NF1 microdeleted patients point to candidate genes in 17q11.2. J Med Genet. 2004;41:35–41. doi: 10.1136/jmg.2003.014761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Upadhyaya M, Ruggieri M, Maynard J, Osborn M, Hartog C, Mudd S, Penttinen M, Cordeiro I, Ponder M, Ponder BA, Krawczak M, Cooper DN. Gross deletions of the neurofibromatosis type 1 (NF1) gene are predominantly of maternal origin and commonly associated with a learning disability, dysmorphic features and developmental delay. Hum Genet. 1998;102:591–597. doi: 10.1007/s004390050746. [DOI] [PubMed] [Google Scholar]
- 10.Riva P, Corrado L, Natacci F, Castorina P, Wu BL, Schneider GH, Clementi M, Tenconi R, Korf BR, Larizza L. NF1 microdeletion syndrome: refined FISH characterization of sporadic and familial deletions with locus-specific probes. Am J Hum Genet. 2000;66:100–109. doi: 10.1086/302709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.De Raedt T, Brems H, Wolkenstein P, Vidaud D, Pilotti S, Perrone F, Mautner V, Frahm S, Sciot R, Legius E. Elevated risk for MPNST in NF1 microdeletion patients. Am J Hum Genet. 2003;72:1288–1292. doi: 10.1086/374821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kehrer-Sawatzki H, Tinschert S, Jenne DE. Heterogeneity of breakpoints in non-LCR-mediated large constitutional deletions of the 17q11.2 NF1 tumour suppressor region. J Med Genet. 2003;40:e116. doi: 10.1136/jmg.40.10.e116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pasmant E, de Saint-Trivier A, Laurendeau I, Dieux-Coeslier A, Parfait B, Vidaud M, Vidaud D, Bièche I. Characterization of a 7.6-Mb germline deletion encompassing the NF1 locus and about a hundred genes in an NF1 contiguous gene syndrome patient. Eur J Hum Genet. 2008;16:1459–1466. doi: 10.1038/ejhg.2008.134. [DOI] [PubMed] [Google Scholar]
- 14.Venturin M, Gervasini C, Orzan F, Bentivegna A, Corrado L, Colapietro P, Friso A, Tenconi R, Upadhyaya M, Larizza L, Riva P. Evidence for non-homologous end joining and non-allelic homologous recombination in atypical NF1 microdeletions. Hum Genet. 2004;115:69–80. doi: 10.1007/s00439-004-1101-2. [DOI] [PubMed] [Google Scholar]
- 15.De Luca A, Bottillo I, Dasdia MC, Morella A, Lanari V, Bernardini L, Divona L, Giustini S, Sinibaldi L, Novelli A, Torrente I, Schirinzi A, Dallapiccola B. Deletions of NF1 gene and exons detected by multiplex ligation-dependent probe amplification. J Med Genet. 2007;44:800–808. doi: 10.1136/jmg.2007.053785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wimmer K, Yao S, Claes K, Kehrer-Sawatzki H, Tinschert S, De Raedt T, Legius E, Callens T, Beiglböck H, Maertens O, Messiaen L. Spectrum of single- and multiexon NF1 copy number changes in a cohort of 1,100 unselected NF1 patients. Genes Chromosomes Cancer. 2006;45:265–276. doi: 10.1002/gcc.20289. [DOI] [PubMed] [Google Scholar]
- 17.Upadhyaya M, Spurlock G, Monem B, Thomas N, Friedrich RE, Kluwe L, Mautner V. Germline and somatic NF1 gene mutation spectrum in NF1-associated malignant peripheral nerve sheath tumors (MPNSTs) Hum Mutat. 2008;29:74–82. doi: 10.1002/humu.20601. [DOI] [PubMed] [Google Scholar]
- 18.Mantripragada KK, Thuresson AC, Piotrowski A, Díaz de Stahl T, Menzel U, Grigelionis G, Ferner RE, Griffiths S, Bolund L, Mautner V, Nordling M, Legius E, Vetrie D, Dahl N, Messiaen L, Upadhyaya M, Bruder CE, Dumanski JP. Identification of novel deletion breakpoints bordered by segmental duplications in the NF1 locus using high resolution array-CGH. J Med Genet. 2006;43:28–38. doi: 10.1136/jmg.2005.033795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shen MH, Mantripragada K, Dumanski JP, Frayling I, Upadhyaya M. Detection of copy number changes at the NF1 locus with improved high-resolution array CGH. Clin Genet. 2007;72:238–244. doi: 10.1111/j.1399-0004.2007.00858.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.