

Abstract
Hereditary hemorrhagic telangiectasia is an autosomal dominant disease caused by mutations in the ACVRL1 and ENG genes characterized by arterio-venous malformations and telangiectases. Over 700 mutations have been described in these two genes, and missense mutations are common. We describe 10 cases in which more than one potentially pathogenic mutation was identified. We report that 8 novel missense mutations, as well as previously reported pathogenic missense mutations, were seen in combination with a second mutation, which raises questions with regards to their respective pathogenicity. Our data and discussion indicate the challenges of classifying missense mutations as pathogenic or benign and the value of co-segregation studies, as well as suggest that there may be hereditary hemorrhagic telangiectasia gene mutations that have only mild phenotypic effects. We present evidence to suggest that four missense mutations (ENG p.G331S, ENG p.L8P, ENG p.P452L and ACVRL1 p.C344R) are pathogenic, two novel mutations (ACVRL1 p.A311T and ENG p.S576G) are neutral, and two previously reported disease-causing mutations are benign or have suspected benign variants (ACVRL1 p.A482V and ENG p.V504M). We conclude that for the purpose of establishing a causative hereditary hemorrhagic telangiectasia mutation in a family proband, all exons and intron/exon borders of both genes should be sequenced and deletion/duplication analysis should be performed unless a mutation that is well-proven to be pathogenic is identified.
Telangiectases and arteriovenous malformations (AVMs) are the characteristic phenotypic manifestations of hereditary hemorrhagic telangiectasia (HHT), a heterogeneous autosomal dominant disorder. Recurring nosebleeds are the most common complication of telangiectases in patients with HHT, but can be mild and infrequent when they begin, typically in late childhood or early teens.1 AVMs that occur in the lungs, brain, or gastrointestinal tract are usually congenital lesions and can cause sudden, life-threatening complications secondary to either hemorrhage or the shunting of blood through these abnormal blood vessels. Oral and dermal telangiectasia, which can be seen by careful examination, often do not appear until the third or fourth decade of life. Although the penetrance approaches 100% by age 40, clinical expression is extremely variable and age-dependent, making HHT often a difficult diagnosis to make based on clinical examination alone.2 Yet all patients with HHT should be screened for internal AVMs, which can usually be effectively removed or permanently occluded. To allow for early diagnosis and prevention of severe complications in HHT families, determination of the family's disease causing mutation is important.
Five molecular subtypes of HHT have been described to date and cannot be distinguished on a clinical basis. Although AVMs in certain organs have been shown to be relatively more common in one molecular type versus another (i.e., pulmonary AVMs are more common in HHT1 and hepatic AVMs more common in HHT2), the gene involved cannot be reliable predicted based on clinical presentation.3 HHT1 is caused by mutations in the Endoglin gene (9q33-q34.1)4 and HHT2 by mutations in the ACVRL1 gene (12q11-q14).5 Detectable mutations in these two genes account for approximately 85% of the HHT cases reported.6,7 Two HHT families have been published that show linkage to 5q31.3–32 (HHT3)8 and chromosome 7p14 (HHT4).9 Mutations in SMAD4 cause a combined syndrome of HHT and juvenile polyposis, but probably represent less than 3% of families diagnosed with HHT.10 It is unknown whether additional loci for HHT exist and there is little data regarding possible regulatory region mutations for the ACVRL1 and ENG genes.
Clinical molecular genetic testing for SMAD4 has only recently become available and mutation analysis for HHT3 and HHT4 is not possible until specific genes are identified. Sequence analysis to look for mutations in the ENG and ACVRL1 has been clinically available since 2003. There are no common mutations in these two genes, the majority of mutations identified in family probands are novel, and de novo mutations are rare. Although a dominant-negative mechanism is not ruled out for all mutations, evidence suggests that haploinsufficiency is the main mechanism leading to HHT.2 No affected individual has been reported to date with two deleterious mutations. In ENG, approximately 15% of sequence variants are nonsense, 38% are small deletions/duplications, 7% are large deletions/duplications, 12% are splice site mutations, and 27% are missense mutations. In ACVRL1 approximately 15% are nonsense, 14% are small deletions/duplications, 7% are large deletions/duplications, 2% are splice site mutations, and 62% are missense mutations.7 Thus, a significant percentage of the sequence variants/mutations identified in affected family probands are novel and missense.6,7,11
Here we describe ten cases in which two possibly pathogenic mutations were detected in either ACVRL1 or ENG in families with HHT clinically confirmed according to published criteria.12 These cases demonstrate the challenge clinical labs face when interpreting the significance of missense mutations, including the limitations in the clinical laboratory setting of prediction programs, such as SIFT and PolyPhen. Our analysis suggests that previously published assumptions regarding the effect of some missense mutations are incorrect or uncertain.
Materials and Methods
Since 2004 individuals from approximately 400 families have had molecular diagnostic testing for HHT at Associated Regional and University Pathologists Laboratories. Coding regions, exon-intron boundaries, and parts of the untranslated regions of both ENG and ACVRL1 genes were sequenced bidirectionally. Our general approach to the evaluation of the clinical significance of missense mutations has been previously reported13 and is in accordance with recently published American College of Medical Genetics guidelines.14 Coding regions and exon/intron boundaries of both genes were sequenced in 100 healthy individuals to help interpret novel sequence changes found in our laboratory. None of the mutations listed in this study were found in 100 healthy controls. A missense mutation is initially classified as of uncertain significance if it has not been previously reported in more than one HHT family in our laboratory, the published literature and/or the International HHT Mutation Database (hhtmutation.org). Multiplex ligation-dependent probe amplification analysis of both genes is performed using the MRC-Holland (Amsterdam, the Netherlands) kit according to manufacturer's instructions to rule out a large deletion or duplication. As is recommended by the American College of Medical Genetics, family co-segregation studies are offered at no charge to families in which novel missense mutations, or other mutations considered to be of uncertain clinical significance, are identified. Family co-segregation studies are accomplished using an Institutional Review Board approved protocol (#12259) at the University of Utah. Effort is made to include the most distant affected relative(s) possible to increase the power of the co-segregation study. Lastly, prediction programs, SIFT, and PolyPhen,15,16 are consulted to aide in the assessment of the amino acid substitution effect. Exon Splice Enhancer (ESE 3.0) Finder was used to evaluate splice site alterations.
Results
Mutation results for ten cases and our interpretation of the clinical significance are shown in Table 1. Figure 1 shows mutation results with clinical findings in the ten cases and family members. Co-segregation information is detailed in the text below.
Table 1.
Mutation Results for Ten Cases and Our Interpretation of the Clinical Significance
| Case | Gene | Nucleotide change | Amino acid change | Effect | As previously reported | References | Cis/trans | SIFT result (P value) | Polyphen result (PSIC score) | Co-segregation (variant found in) | Our interpretation |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | ENG | c.991G>A | p.G331S | Splice, missense | Pathogenic | [19] | N/A | NT (0.00) | B (1.192) | Affected father | Pathogenic |
| ACVRL1 | c.931G>A | p.A311T* | Missense | N/A (novel) | Novel | N/A | T (0.14) | B (0.477) | Unaffected mother | Benign | |
| 2 | ENG | c.991G>A | p.G331S | Splice, missense | Pathogenic | [19] | N/A | NT (0.00) | B (1.192) | Not performed | Pathogenic |
| ACVRL1 | c.1445C>T | p.A482V | Missense | Pathogenic | [19, 20, 21, 22] | N/A | NT (0.00) | B (0.853) | Benign | ||
| 3 | ACVRL1 | c.259C>G | p.H87D | Missense | N/A (novel) | Novel | N/A | T (0.18) | Prob D (2.170) | Affected grandparent | VUS |
| ENG | c.1712G>A | p.R571H | Missense | N/A (novel) | Novel | N/A | NT (0.00) | Poss D (1.852) | Unaffected grandparent and sib with recurrent epistaxis | VUS | |
| 4 | ENG | c.23T>C | p.L8P | Missense | Pathogenic | [21] | Cis | T (0.07) | B (0.225) | Affected father | Pathogenic |
| ENG | c.1712G>A | p.R571H | Missense | N/A (novel) | Novel | Cis | NT (0.00) | Poss D (1.852) | Affected father | VUS | |
| 5 | ENG | c.23T>C | p.L8P | Missense | Pathogenic | [21] | Cis | T (0.07) | B (0.225) | Affected father and sibling | Pathogenic |
| ENG | c.1712G>A | p.R571H | Missense | N/A (novel) | Novel | Cis | NT (0.00) | Poss D (1.852) | Affected father and sibling | VUS | |
| 6 | ENG | c.23T>C | p.L8P | Missense | Pathogenic | [21] | Trans | T (0.07) | B (0.225) | Affected nephew, symptomatic mother | Pathogenic |
| ENG | c.1510G>A | p.V504M | Missense | Pathogenic | [21, 23] | Trans | NT (0.00) | B (1.154) | Unaffected brother | Suspected benign | |
| 7 | ENG | c.726C>A | p.C242X | Nonsense | N/A (novel) | Novel | Trans | N/A | N/A | Affected pat uncle | Pathogenic |
| ENG | c.674C>T | p.P225L | Missense | N/A (novel) | Novel | Trans | NT (0.03) | Prob D (2.130) | Mother with recurrent epistaxis | Possible mild effect | |
| 8 | ACVRL1 | c.1355C>T | p.P452L | Missense | Pathogenic | [6, 17] | N/A | T (0.17) | Poss D (1.886) | Proband | Pathogenic |
| ENG | c.1726A>G | p.S576G | Missense | N/A (novel) | Novel | N/A | T (0.4) | Poss D (1.528) | Unaffected father | Benign | |
| 9 | ACVRL1 | c.698C>T | p.S233L | Missense | Pathogenic | [24] | N/A | NT (0.00) | Prob D (2.326) | Not performed | Suspected Pathogenic |
| ENG | c.593C>T | p.P198L | Missense | N/A (novel) | Novel | N/A | NT (0.03) | Poss D (1.858) | VUS | ||
| 10 | ACVRL1 | c.890A>G | p.H297R | Missense | Pathogenic | [18] | Unknown | T (0.56) | B (0.745) | Unaffected 6th relative | VUS |
| ACVRL1 | c.1030T>C | p.C344R | Missense | Pathogenic | [6, 3†] | Unknown | NT (0.00) | Prob D (3.925) | Affected mother, grandfather | Pathogenic |
For SIFT result: NT, not tolerated; T, tolerated.
PSIC: position specific independent count provides a numeric prediction score, with a higher score indicating more confidence in the prediction. B, benign; Prob D, probably damaging; Poss D, possibly damaging.
VUS, variant uncertain significance; N/A, not applicable.
The ESE Finder Program predicts that A311T changes a SF2/ASF binding site in the ACVRL1 gene. The SR protein SF2/ASF has been initially characterized as a splicing factor but has also been shown to mediate post-splicing activities such as mRNA export and translation. Since an unaffected individual carries A311T, this is not likely to be a common splicing mechanism for the ACVRL1 gene.
This is the same family.
Figure 1.
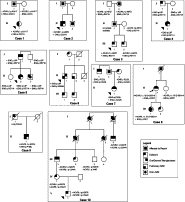
Pedigrees of cases studied. Individuals who were tested for mutations have their test results shown.
Case 1
The proband (II-1) was reported to have epistaxis, pulmonary AVM, and a first degree relative with HHT. The two mutations identified were p.G331S in ENG and p.A311T in ACVRL1. The ENG mutation had been reported twice previously as deleterious, and the ACVRL1 mutation is novel. The ENG mutation was present in the affected father, whereas the ACVRL1 mutation was found in the unaffected mother.
Case 2
This proband (II-1) was reported to have epistaxis, telangiectases, and a pulmonary AVM. Two mutations were detected that had previously been classified as deleterious, p.G331S in ENG and p.A482V in ACVRL1. No family members were available for segregation studies.
Case 3
The proband (III-1) was 11 years of age and reported to have longstanding epistaxis (approximately 3 to 4 times per week), one equivocal telangiectasia and a father affected with HHT who was unavailable for molecular study. A previously reported mutation in ENG (p.R571H) and a previously unreported mutation in ACVRL1 (p.H87D) were detected in the proband. Samples on additional family members were analyzed for co-segregation. Neither mutation was identified in the proband's unaffected mother. The ACVRL1 mutation was identified in the reportedly affected paternal grandmother. The ENG mutation was identified in the asymptomatic paternal grandfather and the proband's 8-year-old brother who, like the proband, was reported to have a history of nosebleeds 3 to 4 times per week since 1 year of age.
Case 4
The proband (II-1). is reported to have epistaxis, multiple oral and dermal telangiectases, pulmonary AVM, and a family history of HHT. Two mutations were identified in ENG; p.L8P, previously published as a deleterious mutation, and p.R571H, previously listed in the HHT international database in an affected individual. Study of parental samples revealed that both mutations were inherited from the proband's symptomatic father and are therefore on the same chromosome (cis).
Case 5
The proband (II-2), reported to have epistaxis, telangiectases, and pulmonary AVM and a family history of HHT, had the same two mutations in ENG as Case 4, p.L8P and p.R571H. A family co-segregation study identified both mutations in the proband's affected father and sibling, and identified neither mutation in the mother or the unaffected sibling. The families are not known to be related; however, haplotype analysis with seven STR markers showed that these two families share at least 4 Mb around the ENG locus (data not shown).
Case 6
The proband (II-1), reported to have characteristic telangiectases, pulmonary AVM, and a family history of HHT, was determined to have two mutations in ENG: p.L8P (also seen in cases 4 and 5) and p.V504M. Both had been previously published as deleterious mutations in affected individuals. Haplotype analysis using the 7 STR markers described above showed a different pattern, suggesting the p.L8P mutation in this patient is an independent event. The proband's symptomatic mother and nephew were shown to have the p.L8P mutation only. The reportedly unaffected brother was shown to have p.V504M.
Case 7
The proband (II-1), a 24-year-old woman, is reported to have epistaxis and telangiectases typical for HHT at her age. She and her affected paternal uncle (I-1) have the nonsense mutation p.C242X in ENG. Her clinically affected father was not available for testing but is presumed to have the ENG p.C242X mutation. In addition, the proband had a novel missense mutation p.P225L in ENG, which was also detected in her mother (I-3). Her mother reports longstanding spontaneous nosebleeds that have required no medical attention but occur often at a frequency of 1 to 3 per week. The mother has not been examined but has no other medical or family history suspicious for HHT.
Case 8
The proband (II-2), with epistaxis, oral and dermal telangiectasia, and a pulmonary AVM, was shown to have ACVRL1 p.P452L, previously reported twice in individuals with HHT6,17 and ENG p. S576G, which was novel. The patient's affected mother and brother were deceased and unavailable for study, but her unaffected father was shown to have ENG p.S576G.
Case 9
The proband (II-1) is reported to have epistaxis, telangiectases, and a pulmonary AVM, as well as a family history of HHT. The following mutations were identified: p.S233L in ACVRL1 (previously published in affected individuals- see Figure 1) and p.P198L in ENG (novel). No family members were available for segregation studies.
Case 10
Targeted sequencing of exon 7 of ACVRL1 was performed on an 11 year-old (V-1) with daily epistaxis since p.C344R had previously been identified in his symptomatic mother, grandfather, aunt and also an affected sixth degree relative. In the process of sequencing this single exon, an additional mutation, p.H297R, was also detected in exon 7 of the 11-year-old. Although p.H297R is predicted to be tolerated or benign by SIFT and Polyphen, it has been previously reported in seven affected members of a family with HHT.18 We have not tested the 11-year-old's father to distinguish paternal vs. de novo origin of p.H297R in this 11-year-old. The father's history with regards to manifestations of HHT is not known.
Discussion
Our clinical laboratory has performed genetics testing for HHT on approximately 400 probands since early 2004. Approximately 35% of affected patients are determined to have at least one missense mutation in either the Endoglin or ACVRL1 gene.2 We report here ten cases in which more than one possibly deleterious mutation was found. Functional studies could help classify these mutations, but the difficulties of these studies prevent them from being routinely used by a clinical laboratory. To help classify mutations as either pathogenic or benign, family co-segregation studies were performed when feasible, in addition to a review of previous case reports and SIFT/Polyphen analyses (Table 1). Our results allow classification of the significance of several novel mutations and, in several cases, change the interpretation of the clinical significance of mutations previously reported as pathogenic.
In Case 1, the presence of ACVRL1 p.A311T in the proband's unaffected mother, and ENG p.G331S in the affected father, as well as a previous published report of p.G331S in an affected individual provide strong evidence that ENG p.G331S is pathogenic and ACVRL1 p.A311T is a benign variant.19 This information helped to clarify the interpretation in Case 2, previously published by our group, in which ENG p.G331S as well as ACVRL1 p.A482V were identified and no family samples were available for study.11 ACVRL1 p.A482V is listed in the HHT mutation database with the following list of citations.19,20,21,22 Review of these papers clarifies that D'Abronzo reports this mutation in one of 64 patients with pituitary gland tumors who were screened for mutations in ACVRL1.20 This patient had no symptoms or family history of HHT. Lesca21 and Schulte22 each report ACVRL1 p.A482V as the sole mutation detected in an affected individual. But it is possible that an additional mutation might have been identified in these cases by full gene sequencing and/or deletion/duplication analysis.21,22 Letteboer19 reports two unrelated affected individuals in which ACVRL1 p.A482V was found, but as in our case a second missense mutation (ACVRL1 p.P424L and ENG p.W261R respectively) was also identified in these individuals. ENG p.W261R was reported in seven additional probands in this paper, but ACVRL1 p.P424L has been reported only in this one case.19 This collective evidence suggests that ACVRL1 p.A482V is a benign variant.
In Case 3, the presence of ACVRL1 p.H87D in the proband's reportedly affected grandmother and of ENG p.R571H in the asymptomatic 60-year-old grandfather, seemed to implicate the novel ACVRL1 mutation as causing HHT in this family. But ENG p.R571H is listed as a mutation in the HHT mutation database, without clinical information. It also has high score from SIFT/Polyphen analysis, which supports it being deleterious. And we also identified ENG p.R571H in the proband's 8-year-old brother who is reported to have 3 to 4 nosebleeds per week since age 1. In this case it was of note that the proband, although suspicious for HHT, did not meet published diagnostic criteria for HHT (Figure 1, case 3). Thus, the reported affected individuals in this family (grandmother and father of proband) have been identified to have ACVRL1 p.H87D alone, and p.H87D combined with p.R571H, respectively. We concluded that based on this evidence we cannot rule out ENG p.R571H as having some deleterious effect on the protein. We currently consider both mutations to be of uncertain clinical significance for the sake of reporting and medical management in the family.
ENG p.R571H was also seen in both Cases 4 and 5, along with ENG p.L8P. In Case 6 ENG p.L8P was seen in combination with ENG p.V504V and the co-segregation study performed in family 6 helped to interpret Cases 4 and 5. First, co-segregation analysis in Cases 4 and 5 allowed confirmation that the mutations were cis-in both families. ENG p.L8P is reported to segregate with disease in one family by Lesca21 but the number and relatedness of the affected family members is not mentioned.
In Case 6, co-segregation analysis suggests that ENG p.L8P is pathogenic and ENG p.V504M is not. This allowed us to resolve that ENG p.L8P is a pathogenic mutation in Cases 4 and 5 as well. But ENG p.V504M is previously reported in two affected individuals by groups doing mutation scanning only,21,23 and the reportedly unaffected brother with ENG p.V504M in family 6 has not been formally evaluated. We thus felt we could not rule out a pathogenic effect for this mutation. Likewise, the evidence does not allow a pathogenic effect to be ruled out for ENG p.R571H, seen in Cases 3, 4, and 5.
Case 7 is the only case in this series in which one of the two detected mutations was a nonsense mutation. What makes this case of interest is that although the clearly affected uncle of the proband (and by inference the untested, affected father) also carried the nonsense mutation, the proband's 51 year-old mother also reported having longstanding nosebleeds, sometimes as often as 3 to 4 times per week. The mother has not been examined, but reports no other manifestations of HHT or suspicious family history. Thus, we could not rule out the possibility of at least a mild effect on the protein by the ENG p.P225L missense mutation detected in both the proband and her mother. There are not published examples of hypomorphic alleles in HHT, but this case and Case 3 suggests the possibility that mutations that cause subtle protein alteration and produce mild manifestations of HHT may exist for these genes. However, this disorder is characterized by such significant variability in clinical expression, even between family members with the same mutation, that evaluating potential genotype/phenotype associations for a particular mutation is difficult.
In Case 8, a mutation (ACVRL1 p.P452L), previously reported twice in an affected individual was seen in combination with a novel missense mutation (ENG p.S576G). Both are predicted by SIFT to be tolerated, and by PolyPhen to be possibly damaging. The co-segregation study in which we proved ENG p.S576G to be inherited from the proband's unaffected father allowed us to conclude that this mutation is benign and adds evidence for the pathogenicity of ACVRL1 p.P452L.
Case 9 is similar to Case 1 in that a novel mutation (ENG p.P198L) was identified in combination with a previously reported missense mutation (ACVRL1 p.S233L). Co-segregation analysis has not been possible to date. There is only one previous report of an individual with HHT and ACVRL1 p.S233L and a large deletion or duplication was not ruled out in this case.24 Although combined SIFT and Polyphen results make it more likely that the ACVRL1 mutation causes HHT in family 9, we have recommended a family co-segregation study to reach sufficient certainty for the purposes of diagnostic testing of at risk family members.
The co-segregation study strongly suggests pathogenicity for p.C344R in ACVRL1 identified in Case 10. We were able to include a quite distant affected relative in the family co-segregation, which increased the power significantly. SIFT and Polyphen suggest that p.C344R, rather than p.H297R is pathogenic. However, it is hard to explain seven affected individuals in one family18 with this mutation (p.H297R) unless the mutation is either pathogenic, or linked to a mutation that is. We were not able to test the proband's father (V-1) to determine whether these mutations are on the same allele.
As a group, these cases demonstrate the limitations of amino acid change and conservation analyses for a clinical laboratory when interpreting the clinical significance of missense mutations. SIFT and Polyphen results can be disparate with each other as was the case for ENG p.G331S and ACVRL1 p.H87D. They can also be in conflict with genetic evidence as in Cases 1, 2, and 6. Thus, clinical laboratories should not rely solely on programs or tools such as SIFT and Polyphen to interpret the clinical significance of missense mutations.
The HHT Mutation Database is a helpful tool for gathering previously reported cases for a particular mutation, but users should review the cited paper(s) for details and confirmation of mutation report. We report here 3 cases (Cases 2, 6, and 10) in which two missense mutations, both previously reported as pathogenic, were detected in an affected individual. These cases argue for caution in assigning a benign interpretation when a novel missense mutation is found in combination with a previously reported missense mutation, or assigning a pathogenic interpretation based on one or two previous reports in affected individuals.
These cases illustrate why molecular analysis should not stop at mutation scanning when a single missense mutation is found, unless evidence for disease causation includes multiple previous reports in affected individuals and/or co-segregation analysis with significant power. This point was also made by a recently published report of an infant with severe symptoms of HHT whose grandmother had previously been shown to have a missense mutation in Endoglin. It was assumed to be the cause of HHT in the family and deletion/duplication analysis was not performed at that time. Later, when an affected newborn was shown not to have this missense mutation previously identified in the family, analysis was done for large gene deletions/duplications and an exon 1 deletion was detected in both infant and grandmother.25
Both of the common HHT genes (ACVRL1 and ENG) have high missense mutation rates and very little information exists from functional studies about these mutations. Currently to classify a novel missense mutation as deleterious in our laboratory, we require a co-segregation study that provides at least an 8 to 1 likelihood ratio that the given mutation is associated with disease in the family, in addition to analysis of amino acid substitution severity. Particularly, diagnostic laboratories performing reflex/sequential HHT testing should be cautious. We believe sequential molecular testing for this disease can easily be misleading. For diagnostic purposes, unless a mutation with a well-known pathogenic effect is identified in a family proband, both genes should be sequenced to completion and deletion/duplication analysis should be also performed.
Acknowledgements
We thank the clinicians and other laboratories who submitted patient samples to our laboratory for testing. We thank Jacquelyn McCowen-Rose for her help in preparation of this manuscript.
Footnotes
Supported by Associated Regional and University Pathologists Institute of Clinical and Experimental Pathology.
References
- 1.Assar A, Friedman C, White R. The natural history of epistaxis in hereditary hemorrhagic telangiectases. Laryngoscope. 1991;101:977–980. doi: 10.1288/00005537-199109000-00008. [DOI] [PubMed] [Google Scholar]
- 2.Bayrak-Toydemir P, Mao R, Lewin S, McDonald J. Hereditary hemorrhagic telangiectasia: an overview of diagnostic and management in the molecular era for clinicians. Genetic in Medicine. 2004;6:175–191. doi: 10.1097/01.gim.0000132689.25644.7c. [DOI] [PubMed] [Google Scholar]
- 3.Bayrak-Toydemir P, McDonald J, Markewitz B, Lewin S, Miller F, Chou LS, Gedge F, Tang W, Coon H, Mao R. Genotype-phenotype correlation in hereditary hemorrhagic telangiectasia: mutations and manifestations. Am J Med Genet A. 2006;140:463–470. doi: 10.1002/ajmg.a.31101. [DOI] [PubMed] [Google Scholar]
- 4.McAllister KA, Grogg KM, Johnson DW, Gallione CJ, Baldwin MA, Jackson CE, Helmbold EA, Markel DS, McKinnon WC, Murrell J, McCormick MK, Pericak-Vance MA, Heutink P, Oostra BA, Haitjema T, Westerman CJJ, Porteous ME, Guttmacher AE, Letarte M, Marchuk DA. Endoglin, a TGF-beta binding protein of endothelial cells, is the gene for hereditary haemorrhagic telangiectasia type 1. Nat Genet. 1994;8:345–351. doi: 10.1038/ng1294-345. [DOI] [PubMed] [Google Scholar]
- 5.Berg JN, Gallione CJ, Stenzel TT, Johnson DW, Allen WP, Schwartz CE, Jackson CE, Porteous MEM, Marchuk DA. The Activin receptor-like kinase 1 gene: genomic structure and mutations in hereditary hemorrhagic telangiectasia type 2. Am J Hum Genet. 1997;61:60–67. doi: 10.1086/513903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bossler AD, Richards J, George C, Godmilow L, Ganguly A. Novel mutations in ENG and ACVRL1 identified in a series of 200 individuals undergoing clinical genetic testing for hereditary hemorrhagic telangiectasia (HHT): correlation of genotype with phenotype. Hum Mutat. 2006;27:667–675. doi: 10.1002/humu.20342. [DOI] [PubMed] [Google Scholar]
- 7.Prigoda N, Savas S, Abdalla S, Piovesan B, Rushlow D, Vandezande K, Zhang E, Ozcelik H, Gallie B, Letarte M. Hereditary hemorrhagic telangiectasia: mutation detection, test sensitivity and novel mutations. J Med Genet. 2006;43:722–728. doi: 10.1136/jmg.2006.042606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cole SG, Begbie ME, Wallace GM, Shovlin CL. A new locus for hereditary haemorrhagic telangiectasia (HHT3) maps to chromosome 5. J Med Genet. 2005;42:577–582. doi: 10.1136/jmg.2004.028712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bayrak-Toydemir P, McDonald J, Akarsu N, Toydemir R, Calderon F, Tuncali T, Tang W, Miller F, Mao R. A fourth locus for hereditary hemorrhagic telangiectasia maps to chromosome 7. Am J Med Genet. 2006;140A:2155–2162. doi: 10.1002/ajmg.a.31450. [DOI] [PubMed] [Google Scholar]
- 10.Gallione CJ, Repetto GM, Legius E, Rustgi AK, Schelley SL, Tejpar S, Mitchell G, Drouin E, Westermann CJ, Marchuk DA. A combined syndrome of juvenile polyposis and hereditary haemorrhagic telangiectasia associated with mutations in MADH4 (SMAD4) Lancet. 2004;363:852–859. doi: 10.1016/S0140-6736(04)15732-2. [DOI] [PubMed] [Google Scholar]
- 11.Gedge F, McDonald J, Phansalkar A, Chou L, Calderon F, Mao R, Lyon E, Bayrak-Toydemir P. Clinical and analytic sensitivities in hereditary hemorrhagic telangiectasia testing and a report of de novo mutations. J Mol Diagn. 2007;9:258–265. doi: 10.2353/jmoldx.2007.060117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shovlin C, Guttmacher A, Buscarini E, Faughnan M, Hyland R, Westermann C, Kjeldsen A, Plauchu H. Diagnostic criteria for hereditary haemorrhagic telangiectasia. Am J Med Genet. 2000;91:66–67. doi: 10.1002/(sici)1096-8628(20000306)91:1<66::aid-ajmg12>3.0.co;2-p. [DOI] [PubMed] [Google Scholar]
- 13.Bayrak-Toydemir P, McDonald J, Mao R, Phansalkar A, Gedge F, Robles J, Goldgar D, Lyon E. Likelihood ratios to assess genetic evidence for clinical significance of uncertain variants: hereditary hemorrhagic telangiectasia as a model. Exp Mol Path. 2008;85:45–49. doi: 10.1016/j.yexmp.2008.03.006. [DOI] [PubMed] [Google Scholar]
- 14.Richards C, Bale S, Bellissimo D, Das S, Grody W, Hegde M, Lyon E, Ward B. ACMG recommendations for standards for interpretation and reporting of sequence variations: revisions 2007. Genet Med. 2008;10:294–300. doi: 10.1097/GIM.0b013e31816b5cae. [DOI] [PubMed] [Google Scholar]
- 15.Ng PC, Henikoff S. Predicting the effects of amino acid substitutions on protein function. Annu Rev Genomics Hum Genet. 2006;7:61–80. doi: 10.1146/annurev.genom.7.080505.115630. [DOI] [PubMed] [Google Scholar]
- 16.Ramensky V, Bork P, Sunyaev S. Human non-synonymous SNPs: server and survey. Nucleic Acids Res. 2002;30:3894–3900. doi: 10.1093/nar/gkf493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Abdalla S, Cymerman U, Rushlow D, Chen N, Stoeber G, Lemire E, Letarte M. Novel mutations and polymorphisms in genes causing hereditary hemorrhagic telangiectasia. Hum Mutat. 2005;25:320–321. doi: 10.1002/humu.9312. [DOI] [PubMed] [Google Scholar]
- 18.Fernandez A, Sanz-Rodriguez F, Zarrabeitia R, Perez-Molino A, Morales C, Restrepo CM, Ramirez JR, Coto E, Lenato GM, Bernabeu C, Botella L. Mutation study of spanish patients with hereditary hemorrhagic telangiectasia and expression analysis of endoglin and ALK1. Human Mutation. 2006;27:295. doi: 10.1002/humu.9413. [DOI] [PubMed] [Google Scholar]
- 19.Letteboer TG, Zewald RA, Kamping EJ, de Haas G, Mager JJ, Snijder RJ, Lindhout D, Hennekam FA, Westermann CJ, Ploos van Amstel JK. Hereditary hemorrhagic telangiectasia: eNG and ALK-1 mutations in Dutch patients. Hum Genet. 2005;116:8–16. doi: 10.1007/s00439-004-1196-5. [DOI] [PubMed] [Google Scholar]
- 20.D'Abronzo FHSB, Klibanski A, Alexander JM. Mutational analysis of activin/transforming growth factor-beta type I and type II receptor kinases in human pituitary tumors. J Clin Endocrinol Metab. 1999;84:1716–1721. doi: 10.1210/jcem.84.5.5704. [DOI] [PubMed] [Google Scholar]
- 21.Lesca G, Plauchu H, Coulet F, Lefebvre S, Plessis G, Odent S, Riviere S, Leheup B, Goizet C, Carette MF, Cordier JF, Pinson S, Soubrier F, Calender A, Giraud S. Molecular screening of ALK1/ACVRL1 and ENG genes in hereditary hemorrhagic telangiectasia in France. Hum Mutat. 2004;23:289–299. doi: 10.1002/humu.20017. [DOI] [PubMed] [Google Scholar]
- 22.Schulte C, Geisthoff U, Lux A, Kupka S, Zenner HP, Blin N, Pfister M. High frequency of ENG and ALK1/ACVRL1 mutations in German HHT patients. Hum Mutat. 2005;25:595. doi: 10.1002/humu.9345. [DOI] [PubMed] [Google Scholar]
- 23.Brusgaard K, Kjeldsen AD, Poulsen L, Moss H, Vase P, Rasmussen K, Kruse TA, Horder M. Mutations in endoglin and in activin receptor-like kinase 1 among Danish patients with hereditary hemorrhagic telangiectasia. Clin Genet. 2004;66:556–561. doi: 10.1111/j.1399-0004.2004.00341.x. [DOI] [PubMed] [Google Scholar]
- 24.Argyriou L, Twelkemeyer S, Panchulidze I, Wehner LE, Teske U, Engel W, Nayernia K. Novel mutations in the ENG and ACVRL1 genes causing hereditary hemorrhagic teleangiectasia. Int J Mol Med. 2006;17:655–659. [PubMed] [Google Scholar]
- 25.Argyriou L, Wirbelauer J, Dev A, Panchulidze I, Shoukier M, Teske U, Nayernia K. A newborn with hereditary haemorrhagic telangiectasia and an unusually severe phenotype. Swiss Med Wkly. 2008;138:432–436. doi: 10.4414/smw.2008.12135. [DOI] [PubMed] [Google Scholar]