

Abstract
We describe the synthesis of activated homopolymers and copolymers of controlled molecular weight based on the controlled radical polymerization of N-acryloyloxysuccinimide (NAS) by reversible addition fragmentation chain transfer (RAFT). We synthesized activated homopolymers in a range of molecular weights with polydispersities between 1 and 1.2. The attachment of an inhibitory peptide to the activated polymer backbone yielded a potent controlled molecular weight polyvalent inhibitor of anthrax toxin. To provide greater control over the placement of the peptides along the polymer backbone, we also used a semi-batch copolymerization method to synthesize copolymers of NAS and acrylamide (AAm). This approach enabled the synthesis of copolymers with control over the placement of peptide-reactive NAS monomers along an inert backbone; subsequent functionalization of NAS with peptide yielded well-defined polyvalent anthrax toxin inhibitors that differed in their potencies. These strategies for controlling molecular weight, ligand density, and ligand placement will be broadly applicable for designing potent polyvalent inhibitors for a variety of pathogens and toxins, and for elucidating structure-activity relationships in these systems.
Keywords: Reversible addition fragmentation chain transfer (RAFT), copolymerization, homopolymers functionalization of polymers, polyvalency
Introduction
Polyvalency – the simultaneous interaction between multiple ligands on one entity and multiple receptors on another – is used extensively in nature to increase the affinity of interactions that may be inherently weak.1–4 We and others have also used this concept to synthesize polyvalent inhibitors for pathogens and toxins that are orders of magnitude more potent than the corresponding monovalent inhibitors.5–16 These polyvalent inhibitors consist of multiple copies of a suitable ligand (e.g., a peptide or a sugar) attached to a scaffold.1–19 Linear polymers are attractive scaffolds for the polyvalent display of ligands1,2,12,13,20–22, and polymeric polyvalent inhibitors are often synthesized by the reaction of ligands with activated polymers – homopolymers or copolymers containing reactive monomers. 5,6,9,11–13,23–26 N-acryloyloxysuccinimide (NAS) is a commonly used reactive monomer and homopolymers of NAS have been widely used to synthesize inhibitors for a variety of pathogens and toxins.5,9,12,27,28 Greater control over the placement of reactive NAS monomers in the polymer scaffolds, and the resulting ability to control ligand density and ligand placement would be important for elucidating structure-activity relationships and for designing well-defined potent polymeric inhibitors. This manuscript describes an approach for achieving such control by using reversible addition-fragmentation chain transfer (RAFT) polymerization.
RAFT polymerization is an attractive controlled radical polymerization technique.29–40 For instance the copolymerization of NAS with N-isopropylacrylamide by RAFT has been used to form core or shell crosslinked thermoresponsive micelles that may find applications in drug delivery, sensors and diagnosis.31,32 Relogio et al. reported the copolymerization of N,N-dimethylacrylamide and NAS by RAFT to obtain random and block copolymer precursors onto which different side-groups (e.g., hydrophobic, ionic, or fluorescent) could be statistically grafted via the reactive NAS units.33 Favier et al. have synthesized copolymers of NAS with N-acryloylmorpholine using RAFT and achieved molecular weights upto 80 kDa, with low polydispersities.34 RAFT has also been used for the synthesis of comb-like branched polymers of poly(n-butyl methacrylate), with controlled backbone and branch chain length, using NAS to generate the branch points.35
Here, we describe the use of RAFT polymerization to synthesize controlled molecular weight homopolymers of NAS (pNAS) and copolymers of acrylamide and NAS (p(AAm-co-NAS)). The technique described here allows for the synthesis of activated homopolymers, as well as copolymers with a well-defined distribution of NAS monomers along the backbone. We also demonstrate the use of these activated scaffolds to design polyvalent inhibitors of anthrax toxin with control over ligand spacing along the polymer backbone (Scheme 1). These controlled molecular weight activated scaffolds should be broadly applicable not only for the design of polyvalent inhibitors for other toxins and pathogens, but also for fundamental studies exploring the recognition of chemically heterogeneous surfaces by both random and well-defined heteropolymeric ligands.41–44
Scheme 1.
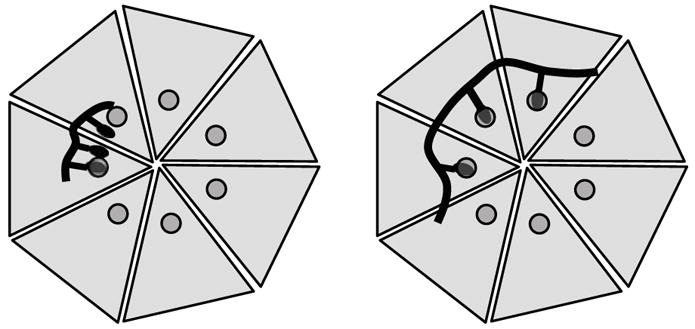
Design of polyvalent inhibitors with control over molecular weight and ligand spacing. The linear polyvalent inhibitors displaying peptides (black ovals) are shown bound to the PA63 heptamer at the peptide-binding sites (circles). The spacing between peptides on the linear scaffold is either too short (left panel) or is sufficient (right panel) to allow a polyvalent interaction.
Experimental
Materials and Methods
Acrylamide (>99%, Aldrich, Allentown, PA) was recrystallized twice from acetone. 1,3,5-Trioxane (99.5%) and anhydrous diethyl ether (99.5%) were obtained from Acros (Somerville, NJ) and used as received. NAS 45 and the chain transfer agent (CTA), 3-benzylsulfanylthiocarbonylsulfanylpropionic acid 46 were synthesized as described in the literature. DMF (99.5%, Acros) and DMSO (99.5%, Acros) were dried over CaH2 and distilled under reduced pressure. Acetone (99.5%, Acros) was dried over K2CO3. 4,4′-Azobis(4-cyanovaleric acid) (AICV), (75%, Aldrich, Allentown, PA) was purified by recrystallization from methanol. Peptide, Ac-HTSTYWWLDGAPK-Am, was purchased from Genemed Synthesis Inc. (San Antonio, TX). Spectra/Por 3 (MWCO 3,500) membrane used for dialysis was obtained from Fisher Scientific (Rancho Domingez, CA). Quantitative 1H NMR spectra of pNAS and p(AAm-co-NAS) were recorded on a Varian Unity 500 MHz spectrometer in DMSO-d6 at room temperature. 13C NMR spectra were also obtained on the same spectrometer in DMSO-d6 at room temperature. No hydrolysis of active ester units was observed during 13C NMR analysis.
Synthesis of Homopolymers of NAS by RAFT
The polymerization reactions were carried out under a dry nitrogen atmosphere using standard Schlenk techniques. A typical polymerization procedure for pNAS with a degree of polymerization (DP) of ~120 (polymer 2, Table 1) is described below. A Schlenk tube (20 mL) was charged with NAS (1.36 g, 8.0 mmol) in anhydrous DMF (4 mL, 2M solution) and 3-benzylsulfanylthiocarbonylsulfanylpropionic acid (10.8 mg, 4×10−5 mol) (CTA). A solution of AICV (50 μL, 1.2 mg, 4×10−6 mol) in anhydrous DMF was transferred to the Schlenk tube. The [M]/[CTA] ratio was maintained at 200, while the [CTA]/[Initiator] ratio was kept at 10. A small fraction (ca. 50 μL) of the sample was taken out from the DMF solution for 1H NMR analysis. The Schlenk tube was flushed with N2 gas for a period of 1h before heating the system at 70 °C for a period of 2 h. Finally, a small fraction of the sample (ca. 50 μL) was taken out and characterized by 1H NMR spectroscopy (in DMSO-d6) to determine monomer conversion (60%). The remaining polymer solution was precipitated with anhydrous acetone (ca. 400 mL) and the polymer was recovered by centrifugation. The polymer was further washed several times with anhydrous acetone and immediately dried under vacuum for a period of 24 h (yield: 0.80 g, ~ 60%). The absence of residual monomers in the polymer was confirmed by 1H NMR (in DMSO-d6). 1H NMR (500 MHz, DMSO-d6, δ ppm): 7.1–7.83 (m), 3–3.2 (br), 2.6–2.9 (br), 1.7–2.3 (br). 13C NMR (500 MHz, DMSO-d6, δ ppm): 25.3, 32.9, 37.8, 16.8, 172.7. Elemental Analysis: Calculated: C = 49.47, H = 4.12, N = 8.24, S = 0.47, Found: C = 49.30, H = 4.12, N = 8.14, S = 0.66.
Table 1.
Polymerization of NAS at various [M]/[CTA] ratiosa
| Run No | [M]/[CTA] | [CTA]/[Initiator] | Polymerization time (h) | Conversionb(mol %) | DP theoreticalc | ASEC resultsd | |||
|---|---|---|---|---|---|---|---|---|---|
| Mn (kDa) | Mw (kDa) | PD | DPe | ||||||
| 1 | 100 | 10 | 1.5 | 77 | 77 | 5.97 | 6.27 | 1.05 | 84 |
| 2 | 200 | 10 | 2.0 | 60 | 120 | 8.96 | 9.85 | 1.10 | 126 |
| 3 | 300 | 5 | 2.5 | 48 | 144 | 1.12 | 1.28 | 1.14 | 158 |
| 4 | 400 | 5 | 3.5 | 48 | 192 | 1.45 | 1.62 | 1.12 | 204 |
Polymerization conditions: NAS (1.34 g) in DMF (4 mL, 2M solution), Temp = 70 °C
NAS conversion determined by 1H NMR
Determined based on conversion
Determined by ASEC after ammonolysis
Calculated based on Mn
Kinetics
The kinetics of the polymerization was followed by withdrawing samples from the polymerization mixture at different intervals of time and analyzing the residual monomer concentrations by 1H NMR in DMSO-d6. Monomer concentrations were calibrated by comparing the integration of the peaks due to the vinyl protons of the monomers with those of an added standard 1,3,5-trioxane.34
Removal of End Groups
The trithiocarbonate end group was removed according to the procedure reported by Perrier et al.47 Typically pNAS (0.2 g, DP 120) and AIBN (100 mg, 25 times higher than pNAS, mol/mol) were dissolved in anhydrous DMF (2 mL). The solution was heated at 80 °C for 90 min. The homopolymer was precipitated in 100 mL of anhydrous acetone, collected by centrifugation, and dried under vacuum for a period of 24 h (yield: 0.19 g, 95%). The absence of the trithiocarbonate end group was confirmed by elemental analysis. A similar procedure was used to remove the terminal trithiocarbonate end group on the copolymers. 1H NMR after end group removal (500 MHz, DMSO-d6, δppm): 7.1–7.83 (m), 3–3.2 (br), 2.6–2.9 (br), 1.7–2.3 (br). 13C NMR (500 MHz, DMSO-d6, δ ppm): 25.3, 32.9, 37.8, 16.8, 172.7. Elemental Analysis after end group removal: Calculated: C = 49.46, H = 4.12, N = 8.24. Found: C = 49.30, H = 4.19, N = 8.34.
Synthesis of copolymers of AAm/NAS by RAFT
Based on knowledge of acrylamide/NAS semi-batch copolymerization kinetics, we synthesized copolymers of acrylamide and NAS with well-defined spacing between NAS units. A representative procedure, for the synthesis of copolymers with an average of 30 acrylamide monomers between two NAS units, is described below. A Schlenk tube (20 mL) (A) was charged with acrylamide (0.84 g, 12.0 mmol), anhydrous DMSO (4.0 mL, 3 M solution), and 3-benzylsulfanylthiocarbonylsulfanylpropionic acid (10.8 mg, 4 × 10−5 mol) (CTA). A solution of AICV (ca. 50 μL, 1.2 mg, 4 × 10−6 mol) in DMSO was transferred to Schlenk tube A. In another Schlenk tube (B), acrylamide (0.28 g, 4 mmol) was dissolved in anhydrous DMSO (4.0 mL, 1.0 M solution). The molar ratio of total monomer to CTA was 400. The contents of both the tubes were flushed with N2 for a period of 1h. The acrylamide solution from Schlenk tube B was placed in a 5 mL airtight syringe and continuously added to acrylamide solution of Schlenk tube A over a period of 6 h at 70 °C using a PHD-2000 model syringe pump (Harvard Apparatus, South Natick, MA). In another Schlenk tube (C) NAS (40 mg, 0.24 mmol) was dissolved in anhydrous DMSO (600 μL), and the contents of this tube were also flushed with N2; 200 μL of this solution was added to Schlenk tube A at three separate times – after 90, 126, and 162 minutes. Samples were withdrawn from Schlenk tube A every hour for analysis of Mn by ASEC.
Biofunctionalization
Biofunctionalization of the polymers (pNAS or poly(AAm-co-NAS)) was carried out as indicated below. A representative procedure for the biofunctionalization of poly(AAm-co-NAS) is described here. The polymer was dissolved in anhydrous dimethyl sulfoxide (DMSO) (100 mg/mL) followed by the addition of peptide (Ac-HTSTYWWLDGAPK-Am; 20mg/mL in DMSO) and triethylamine. The reaction was carried out overnight at room temperature followed by quenching of the unreacted active ester groups with NH4OH. The polymer solution was dialyzed against MilliQ water for 48 h to remove unreacted peptide and other organic reagents. The extent of peptide attached was determined by 1H NMR spectroscopy (yield of peptide attached to NAS side chain ~ 50%). 1H NMR (500 MHz, D2O, δ ppm): 0.55–0.65 (br), 0.8–0.85 (br), 0.9–1.0 (br), 1–1.7 (br), 1.75–1.85 (br), 2–2.2 (br), 2.5–2.95 (br), 3.55–3.85 (br), 4.0–4.5 (br), 4.7–4.8 (br), 4.9–4.95 (br), 6.5–6.8 (br), 7–7.4 (br).
Binding Assay
CHO-K1 cells were grown to confluency in 24-well plates and then incubated on ice with trypsin-activated (2×10−8 M) PA for 2 h. The cells were washed twice with cold PBS and 35S-labeled LFN was added either in the absence or presence of inhibitor for 1 h. The cells were washed three times with cold PBS, lysed, and the cell-associated 35S-LFN was measured by scintillation counting. The level of non-specific binding, determined as the amount of 35S-LFN associated with cells in the absence of PA, was subtracted from values obtained in the presence of PA. The percentage of inhibition was calculated by dividing the radioactivity of the inhibitor-containing samples by that of the sample without inhibitor. The results are the mean ± s.e.m. of three independent experiments.
Cytotoxicity Assay
The efficacy of the polyvalent inhibitor was evaluated by measuring the inhibition of cytotoxicity due to the anthrax lethal toxin in a mouse macrophage cell line, RAW264.7. The cells were grown in RPMI medium supplemented with 5% fetal calf serum (Hyclone) and then plated onto a 96-well plate. A mixture of PA (10−9 M), LF (3 × 10−10 M), and various concentrations of the polyvalent inhibitor was added to the cells. After 4 h of incubation at 37 °C in CO2 (5 %), 20 μL of a mixture of MTS (3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium) and phenazine methosulfate (PMS) was added to each well and incubated for 1 h. The amount of formazan product generated from the reduction of MTS by live cells was determined by measuring the absorbance at 490 nm. The half-maximum inhibitory concentration (IC50) of the polyvalent inhibitor was evaluated by measuring cell viability at different inhibitor concentrations.
Characterization of polymers by aqueous size exclusion chromatography (ASEC)
Polymers (after end group removal) were allowed to react with an aqueous ammonium hydroxide solution followed by dialysis and lyophilization. The molecular weight of the resulting polymers was analyzed by ASEC using a light scattering detector and a phosphate buffer as the eluent (pH 7.0, 100 mM NaCl, 25 mM NaH2PO4, 25 mM Na2HPO4, flow rate = 1 mL/min, dn/dc = 0.165 mL/g). Analysis was carried out at 25 °C. ViscoGEL column (GMPWXL, Mixed Bed, dimensions: 7.8 mm × 30 cm) was used with a Waters 2487 Dual wavelength UV/vis absorbance detector, Waters 2414 refractive index detector, and a Viscotek 270 Trisec Dual Detector (λ = 670 nm) inline with the column. OmniSEC 4.0 software was used to determine the molecular weight.
Results and Discussion
Synthesis of Homopolymers of NAS by RAFT
We first developed a method that uses RAFT to synthesize the activated homopolymer poly (N-acryloyloxysuccinimide) (pNAS) with control over its molecular weight (Fig. 1A). RAFT has emerged as an attractive controlled radical polymerization technique that is compatible with a wide variety of monomers and can even be used to carry out polymerization in aqueous systems. 29,48 RAFT involves a reversible addition-fragmentation chain transfer in the presence of a chain transfer agent (CTA).29 Here we selected 3-benzylsulfanylthiocarbonylsulfanylpropionic acid as the CTA and AICV as the initiator. NAS (2 M solution in DMF) was polymerized using different monomer to CTA ratios ([M]/[CTA]). As shown in Figure 1B, approximately linear consumption of NAS was observed for conversions up to ca. 40 % for [M]/[CTA] ratios between 100 and 400. The rate of polymerization decreased with increase in [M]/[CTA] ratio. However, an induction period was observed for the initial 30 minutes when the polymerization reactions were carried out at [M]/[CTA] ratios ranging from 200–400. Such a “induction period” has been observed in acrylate polymerization by RAFT using trithiocarbonates as chain transfer agent and is attributed to impurities such as oxygen present in the system.49 As seen in Table 1, varying the [M]/[CTA] ratio from 100 – 400 enabled us to synthesize a series of controlled molecular weight pNAS polymers (degree of polymerization, DP, ranging from ca. 80 – 200). Characterization by ASEC revealed that the polymers had a narrow size distribution with polydispersities ranging from 1.1 – 1.2 (Table 1).
Figure 1.

(a) Polymerization of NAS by RAFT. i: 3-benzylsulfanylthiocarbonylsulfanylpropionic acid, DMF; ii: AICV, DMF, 70 °C, 2.5 h; iii: AIBN, DMF, 90 °C, 1.5 h. (b) Plot indicating monomer conversion as a function of time for NAS polymerization at [M]/[CTA] values of 100 (◆), 200 (●), 300 (■), and 400 (▲).
Synthesis of Controlled Molecular Weight Polyvalent Inhibitors of Anthrax Toxin
Next, we demonstrated the use of controlled molecular weight pNAS polymers to synthesize polyvalent inhibitors of anthrax lethal toxin (LeTx). LeTx, which is responsible for symptoms and death in anthrax, consists of two protein components – an enzymatic moiety (LF) and a cell-binding moiety, protective antigen (PA). PA binds to receptors on the surface of mammalian cells, and is then proteolytically cleaved, resulting in the formation of heptamers ([PA63]7) that bind LF.50
To synthesize polyvalent inhibitors, we functionalized the polymers with the peptide Ac-HTSTYWWLDGAPK-Am, which binds to [PA63]7 and inhibits the binding of LF.9 We first allowed the controlled molecular weight pNAS polymers (DP ~ 80 and 120) to react with the peptide Ac-HTSTYWWLDGAPK-Am and then with ammonium hydroxide. The number of peptides per polymer chain was held constant (ca. 3); we reasoned that using pNAS polymers with different degrees of polymerization would enable the average spacing between the peptides on the polymer backbone to be varied (Scheme 1).
We first tested the ability of the inhibitors to prevent toxin assembly – the binding of LF to [PA63]7. The inhibitory potency was quantified in terms of the half-maximal inhibitory concentration (IC50), which we report on a per-peptide basis. As seen in Figure 2A, the IC50 value was significantly lower for the polymeric inhibitor with DP = 120 than for the inhibitor with DP = 80, although the number of peptides per polymer chain was the same in each case. Next, we tested the efficacy of these inhibitors at preventing cell death due to anthrax lethal toxin (Fig. 2B). While the inhibitor with DP = 80 did not significantly inhibit cell death at concentrations as high as 10−6 M in this more stringent cytotoxicity assay, the inhibitor with DP = 120 had an IC50 value of 36 nM on a per-peptide basis. These results suggest that the spacing between peptides for the inhibitor with DP = 120 is more conducive for a polyvalent interaction between the inhibitor and [PA63]7. Control polymers (without pendant peptide) did not significantly inhibit cytotoxicity at all concentrations tested (Fig. 2B). Since the monovalent peptide does not inhibit cytotoxicity at concentrations as high as 2 mM (data not shown), the controlled molecular weight inhibitor shows an enhancement in potency on a per-peptide basis by almost five orders of magnitude.
Figure 2.

Inhibitory activity of polyvalent inhibitors of anthrax toxin derived from controlled molecular weight homopolymer, pNAS. (a) Inhibition of the binding of the anthrax lethal factor, (b) Inhibition of toxin-induced cytotoxicity at various concentrations of polymeric inhibitors with DP = 80 (□) and DP =120 (●). Control polymer (polyacrylamide) did not show any inhibitory activity at concentrations tested (△).
Design of Well-Defined Polyvalent Inhibitors with Control over Ligand Spacing
While the previous results (Fig. 2) suggest that the spacing between peptides influences the potency of polyvalent anthrax toxin inhibitors, it would be desirable to achieve greater control over the placement of peptides on the polymeric scaffold. To that end, we used RAFT for the semi-batch copolymerization of acrylamide (AAm) and NAS. We reasoned that adding the more reactive NAS to the polymerization mixture at specific time intervals would enable the spacing between activated NAS monomers to be controlled, given the greater reactivity of monomers containing active esters13. Subsequent reaction with ligands (e.g., peptides) would generate inhibitors with controlled molecular weights and ligand spacings (Scheme 1).
We first carried out a semi-batch homopolymerization of acrylamide; knowledge of the kinetics of acrylamide polymerization was used to guide the times at which NAS was added to the polymerization mixture to generate copolymers with control over the spacing between NAS groups. For a random heteropolymer containing “i” peptides randomly distributed along a chain composed of DP repeat units, the average spacing between adjacent peptides would be ca. DP/(i+1). For a random heteropolymer with DP = 120 and i = 3 (Fig. 2A), the average spacing between adjacent peptides would therefore be ca. 30 units. We also previously synthesized a series of polyvalent anthrax toxin inhibitors with DP =200 and tested the effect of peptide density on potency.12 We observed an initial sharp increase in potency with increasing peptide density, followed by a plateau where potency was independent of peptide density. The peptide density at the transition corresponded to an average number of peptides per polymer chain of 6±2.12 For a random heteropolymer with DP =200 and i = 6, the average spacing between adjacent peptides would be ca. 28.6 units, which is also close to the spacing of 30 units estimated above. We therefore first synthesized a copolymer, poly(AAm-co-NAS) having three NAS blocks, with ca. 30 acrylamide units between adjacent NAS blocks. NAS was added to the polymerization mixture after 90, 126 and 162 minutes (Fig. 3A). Characterization by NMR spectroscopy indicated that 90% of the added NAS monomer was consumed within 5 min and complete consumption took place within 10 minutes. Characterization by ASEC confirmed that the average spacing between NAS units was 31 ± 1.1 (Fig. 3A). We also synthesized a second copolymer, poly(AAm-co-NAS) having three NAS blocks, in which the average spacing between adjacent blocks was 18 ± 0.8 (Fig. 3B). These well-defined polymers were functionalized with the [PA63]7–binding peptide Ac-HTSTYWWLDGAPK-Am, and the resulting polyvalent inhibitors were tested for their efficacy in neutralizing toxin-induced cytotoxicity. The polyvalent inhibitor with a spacing of 31 units between adjacent peptide-functionalized blocks demonstrated two orders of magnitude greater potency than inhibitors with a spacing of 18 units (Fig. 3C), confirming the ability to tune inhibitor activity by controlling the ligand spacing.
Figure 3.

Characterization of polyvalent inhibitors based on poly(AAm-co-NAS) copolymers. Characterization of molecular weight during semi-batch RAFT copolymerization for polymers with an average spacing of (a) 31 and (b) 18 units between adjacent NAS blocks. (c) The inhibitory activity of the resulting peptide-functionalized polyvalent inhibitors in a cytotoxicity assay.
Conclusion
We have developed a new RAFT-based approach for synthesizing activated homopolymers of NAS of controlled molecular weight and demonstrated the use of this approach to generate potent polyvalent inhibitors of anthrax toxin. We also describe the use of RAFT to synthesize controlled molecular weight copolymers of NAS, in which we can control the placement of NAS groups along the polymer backbone. These methods enable the synthesis of polyvalent inhibitors with control over molecular weight, ligand density, and ligand spacing. Finally, the work also illustrates how results from studies using random heteropolymers (Fig. 2) can be used to guide the design of more well-defined polyvalent inhibitors (Fig. 3). The methods described in this work could be useful for elucidating structure-activity relationships for polyvalent inhibitors for a variety of pathogens and toxins. The ability to synthesize controlled molecular weight random and defined heteropolymers would also be useful for fundamental studies of pattern recognition41–44,51–58, with applications ranging from biosensor design to bioseparations and the design of potent therapeutics.
Acknowledgments
This work was supported by NIH grant U01 AI056546.
References
- 1.Mammen M, Choi SK, Whitesides GM. Angew Chem Intl Ed. 1998;37:2755–2794. doi: 10.1002/(SICI)1521-3773(19981102)37:20<2754::AID-ANIE2754>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 2.Kiessling LL, Strong LE, Gestwicki JE. Annu Rep Med Chem. 2000;35:321–330. [Google Scholar]
- 3.Mulder A, Huskens J, Reinhoudt DN. Org Biomol Chem. 2004;2:3409–3424. doi: 10.1039/b413971b. [DOI] [PubMed] [Google Scholar]
- 4.Kane RS. AIChE J. 2006;52:3638 – 3644. [Google Scholar]
- 5.Mammen M, Dahmann G, Whitesides GMJ. Med Chem. 1995;38:4179–4190. doi: 10.1021/jm00021a007. [DOI] [PubMed] [Google Scholar]
- 6.Sigal GB, Mammen M, Dahmann G, Whitesides GM. J Am Chem Soc. 1996;118:3789–3800. [Google Scholar]
- 7.Kitov PI, Sadowska JM, Mulvey G, Armstrong GD, Ling H, Pannu NS, Read RJ, Bundle DR. Nature. 2000;403:669–672. doi: 10.1038/35001095. [DOI] [PubMed] [Google Scholar]
- 8.Fan EK, Zhang ZS, Minke WE, Hou Z, Verlinde CLMJ, Hol WGJ. J Am Chem Soc. 2000;122:2663–2664. [Google Scholar]
- 9.Mourez M, Kane RS, Mogridge J, Metallo S, Deschatelets P, Sellman BR, Whitesides GM, Collier RJ. Nat Biotechnol. 2001;19:958–961. doi: 10.1038/nbt1001-958. [DOI] [PubMed] [Google Scholar]
- 10.Zhang G, Leibowitz MJ, Sinko PJ, Stein S. Bioconjugate Chem. 2003;14:86–92. doi: 10.1021/bc025526i. [DOI] [PubMed] [Google Scholar]
- 11.Joshi A, Saraph A, Poon V, Mogridge J, Kane RS. Bioconjugate Chem. 2006;17:1265–1269. doi: 10.1021/bc060042y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gujraty KV, Joshi A, Saraph A, Poon V, Mogridge J, Kane RS. Biomacromolecules. 2006;7:2082–2085. doi: 10.1021/bm060210p. [DOI] [PubMed] [Google Scholar]
- 13.Yanjarappa MJ, Gujraty KV, Joshi A, Saraph A, Kane RS. Biomacromolecules. 2006;7:1665–1670. doi: 10.1021/bm060098v. [DOI] [PubMed] [Google Scholar]
- 14.Rai P, Padala C, Poon V, Saraph A, Basha S, Kate S, Tao K, Mogridge J, Kane RS. Nat Biotechnol. 2006;24:582–586. doi: 10.1038/nbt1204. [DOI] [PubMed] [Google Scholar]
- 15.Rai PR, Saraph A, Ashton R, Poon V, Mogridge J, Kane RS. Angew Chem Int Ed Engl. 2007;46:2207–2209. doi: 10.1002/anie.200604317. [DOI] [PubMed] [Google Scholar]
- 16.Basha S, Rai P, Poon V, Saraph A, Gujraty K, Go MY, Sadacharan S, Frost M, Mogridge J, Kane RS. Proc Natl Acad Sci U S A. 2006;103:13509–13513. doi: 10.1073/pnas.0509870103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Joshi A, Punyani S, Bale SS, Yang H, Borca-Tasciuc T, Kane RS. Nat Nanotechnol. 2008;3:41–45. doi: 10.1038/nnano.2007.386. [DOI] [PubMed] [Google Scholar]
- 18.Carrillo A, Yanjarappa MJ, Gujraty KV, Kane RSJ. Poly Sci Part A: Poly Chem. 2005;44:928–939. [Google Scholar]
- 19.Carrillo A, Kane RSJ. Poly Sci Part A: Poly Chem. 2004;42:3352 – 3359. [Google Scholar]
- 20.Bovin NV. Glycoconj J. 1998;15:431–446. doi: 10.1023/a:1006963717646. [DOI] [PubMed] [Google Scholar]
- 21.Godwin A, Hartenstein M, Muller AHE, Brocchini S. Angew Chem Intl Ed. 2001;40:594–597. doi: 10.1002/1521-3773(20010202)40:3<594::AID-ANIE594>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- 22.Polizzotti BD, Maheshwari R, Vinkenborg J, Kiick KL. Macromolecules. 2007;40:7103–7110. doi: 10.1021/ma070725o. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lees WJ, Spaltenstein A, Kingerywood JE, Whitesides GM. J Med Chem. 1994;37:3419–3433. doi: 10.1021/jm00046a027. [DOI] [PubMed] [Google Scholar]
- 24.David A, Kopeckova P, Rubinstein A, Kopecek J. Bioconjugate Chem. 2001;12:890–899. doi: 10.1021/bc010026v. [DOI] [PubMed] [Google Scholar]
- 25.Honda T, Yoshida S, Arai M, Masuda T, Yamashita M. Bioorg Med Chem Lett. 2002;12:1929–1932. doi: 10.1016/s0960-894x(02)00330-x. [DOI] [PubMed] [Google Scholar]
- 26.Carrillo A, Gujraty KV, Rai PR, Kane RS. Nanotechnology. 2005;16:S416–S421. doi: 10.1088/0957-4484/16/7/016. [DOI] [PubMed] [Google Scholar]
- 27.Wang JQ, Chen X, Zhang W, Zacharek S, Chen YS, Wang PG. J Am Chem Soc. 1999;121:8174–8181. [Google Scholar]
- 28.Arranz-Plaza E, Tracy AS, Siriwardena A, Pierce JM, Boons GJ. J Am Chem Soc. 2002;124:13035–13046. doi: 10.1021/ja020536f. [DOI] [PubMed] [Google Scholar]
- 29.Chiefari J, Chong YK, Ercole F, Krstina J, Jeffery J, Le TPT, Mayadunne RTA, Meijs GF, Moad CL, Moad G, Rizzardo E, Thang SH. Macromolecules. 1998;31:5559–5562. [Google Scholar]
- 30.McCormick CL, Lowe AB. Acc Chem Res. 2004;37:312–325. doi: 10.1021/ar0302484. [DOI] [PubMed] [Google Scholar]
- 31.Zhang JY, Jiang X, Zhang YF, Li YT, Liu SY. Macromolecules. 2007;40:9125–9132. [Google Scholar]
- 32.Li YT, Lokitz BS, McCormick CL. Macromolecules. 2006;39:81–89. [Google Scholar]
- 33.Relogio P, Charreyre MT, Farinha JPS, Martinho JMG, Pichot C. Polymer. 2004;45:8639–8649. [Google Scholar]
- 34.Favier A, D’Agosto F, Charreyre MT, Pichot C. Polymer. 2004;45:7821–7830. [Google Scholar]
- 35.Vosloo JJ, Tonge MP, Fellows CM, D’Agosto F, Sanderson RD, Gilbert RG. Macromolecules. 2004;37:2371–2382. [Google Scholar]
- 36.Aamer KA, Tew GN. J Poly Sci Part A: Poly Chem. 2007;45:5618–5625. [Google Scholar]
- 37.Pressly ED, Rossin R, Hagooly A, Fukukawa K, Messmore BW, Welch MJ, Wooley KL, Lamm MS, Hule RA, Pochan DJ, Hawker CJ. Biomacromolecules. 2007;8:3126–3134. doi: 10.1021/bm700541e. [DOI] [PubMed] [Google Scholar]
- 38.Jung IG, Seo J, Chung YK, Shin DM, Chun SH, Son SU. J Poly Sci Part A: Poly Chem. 2007;45:3042–3052. [Google Scholar]
- 39.Barner-Kowollik C, Perrier SJ. Poly Sci Part A: Poly Chem. 2008;46:5715–5723. [Google Scholar]
- 40.Li Y, Yang J, Benicewicz BC. J Poly Sci Part A: Poly Chem. 2007;45:4300–4308. [Google Scholar]
- 41.Srebnik S, Chakraborty AK, Shakhnovich EI. Phys Rev Lett. 1996;77:3157–3160. doi: 10.1103/PhysRevLett.77.3157. [DOI] [PubMed] [Google Scholar]
- 42.Golumbfskie AJ, Pande VS, Chakraborty AK. Proc Natl Acad Sci, USA. 1999;96:11707–11712. doi: 10.1073/pnas.96.21.11707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jayaraman A, Hall CK, Genzer J. Phys Rev Lett. 2005;94:078103. doi: 10.1103/PhysRevLett.94.078103. [DOI] [PubMed] [Google Scholar]
- 44.Mallik S, Plunkett SD, Dhal PK, Johnson RD, Pack D, Shnek D, Arnold FHN. J Chem. 1994;18:299–304. [Google Scholar]
- 45.Pollak A, Blumenfeld H, Wax M, Baughn RL, Whitesides GM. J Am Chem Soc. 1980;102:6324–6336. [Google Scholar]
- 46.Stenzel MH, Davis TP, Fane AG. J Mater Chem. 2003;13:2090–2097. [Google Scholar]
- 47.Perrier S, Takolpuckdee P, Mars CA. Macromolecules. 2005;38:2033–2036. [Google Scholar]
- 48.Thomas DB, Convertine AJ, Myrick LJ, Scales CW, Smith AE, Lowe AB, Vasilieva YA, Ayres N, McCormick CL. Macromolecules. 2004;37:8941–8950. [Google Scholar]
- 49.Wang R, McCormick CL, Lowe AB. Macromolecules. 2005;38:9518–9525. [Google Scholar]
- 50.Collier RJ, Young JAT. Annu Rev Cell Dev Biol. 2003;19:45–70. doi: 10.1146/annurev.cellbio.19.111301.140655. [DOI] [PubMed] [Google Scholar]
- 51.Bratko D, Chakraborty AK, Shakhnovich EI. Chem Phys Lett. 1997;280:46–52. [Google Scholar]
- 52.Bratko D, Chakraborty AK, Shakhnovich EI. Comp Theo Polym Sci. 1998;8:113–126. [Google Scholar]
- 53.Chakraborty AK. Phys Rep. 2001;342:2–61. [Google Scholar]
- 54.Chakraborty AK, Bratko DJ. Chem Phys. 1998;108:1676–1682. [Google Scholar]
- 55.Chakraborty AK, Golumbfskie AJ. Annu Rev Phys Chem. 2001;52:537–573. doi: 10.1146/annurev.physchem.52.1.537. [DOI] [PubMed] [Google Scholar]
- 56.Kriksin YA, Khalatur PG, Khokhlov AR. J Chem Phys. 2005:122. doi: 10.1063/1.1861877. [DOI] [PubMed] [Google Scholar]
- 57.Peppas NA, Huang Y. Pharm Res. 2002;19:578–587. doi: 10.1023/a:1015389609344. [DOI] [PubMed] [Google Scholar]
- 58.Srebnik S, Chakraborty AK, Bratko DJ. Chem Phys. 1998;109:6415–6419. [Google Scholar]