

Abstract
This report describes the synthesis of analogs of 4-[1-(3,5,5,8,8-pentamethyl-5,6,7,8-tetrahydro-2-naphthyl)ethynyl]benzoic acid (1), commonly known as bexarotene, and their analysis in acting as retinoid-X-receptor (RXR)-specific agonists. Compound 1 has FDA approval to treat cutaneous T-cell lymphoma (CTCL); however, its use can cause side effects such as hypothyroidism and increased triglyceride concentrations, presumably by disruption of RXR heterodimerization with other nuclear receptors. The novel analogs in the present study have been evaluated for RXR activation in an RXR mammalian-2-hybrid assay as well as an RXRE-mediated transcriptional assay, and for their ability to induce apoptosis, as well as for their mutagenicity and cytotoxicity. Analysis of 11 novel compounds revealed the discovery of 3 analogs that best induce RXR-mediated transcriptional activity, stimulate apoptosis, have comparable Ki and EC50 values to 1, and are selective RXR agonists. Our experimental approach suggests that rational drug design can develop new rexinoids with improved biological properties.
Introduction
Retinoids are a class of small molecule compounds that play vital roles in the regulation of cellular processes, including transcription of genes, differentiation and proliferation. Two classes of proteins that bind to retinoids, retinoic acid receptors (RARs) and retinoid X receptors (RXRs) have been studied in detail, and three subtypes, α, β and γ, have been identified for both RAR and RXR proteins.1 The receptors for retinoids, as well as for other small lipophilic hormonal ligands, belong to the larger superfamily of receptors for steroids, as well as for thyroid hormone receptor (TR) and vitamin D receptor (VDR), which all function as transcription factors. All of the receptor proteins have an “endogenous ligand” that binds to a specific pocket within the protein, altering the protein’s conformation, and inducing the protein to bind to a specific molecular scaffold on DNA. Most of these proteins, once bound to a signaling lipophilic ligand, interact directly with DNA sequences known as hormone response elements (HREs). Most HREs consist of minimal core hexad sequences that exist as half-sites separated by variable length nucleotide spacers between direct, inverted, or everted repeats2, and are found within the promoter region of target genes. In order to activate transcription, nuclear receptors bind to the HREs as homodimers or heterodimers, with each partner binding to a half-site of the element. The association of the nuclear receptor protein with the DNA results in regulation of target gene expression, ultimately leading to a physiological effect or bio-response.
Although originally proposed to act as homodimers3, TR, RAR and VDR high affinity DNA binding is mediated via a heterodimer of RXR and the appropriate receptor4. RXR also binds to a natural endogenous ligand, 9-cis retinoic acid (9-cis RA; see below), and functions as a homodimer when bound to its cognate ligand, or can function as an un-liganded heterodimeric partner for other nuclear receptors including VDR5. Thus, the RXR “master” partner is central to the function of many nuclear receptors since the RXR protein can form heterodimer complexes with many members of this superfamily that result in specific physiological responses as a result of modulation of gene expression. 6
Interestingly, ligand-induced transcriptional activity for the RXR homodimer is suppressed in most but not all cases when RXR is complexed with a ligand-bound partner such as VDR and TR, and these heterodimers prevent the binding of RXR to its ligand, suggesting that TR and VDR are “nonpermissive” heteropartners for RXR, in which the “primary” receptor (TR or VDR) and its ligand play a dominant role over the “subordinate” RXR coreceptor.5 However, in the case of RAR, which is a primary partner activated by all-trans-retinoic acid (see structure below), the RXR heteropartner is still able to bind 9-cis-RA, and the two retinoids synergistically enhance transactivation from retinoic acid response elements (RAREs). Conversely, when 9-cis-RA or synthetic RXR ligands (rexinoids) are present in excess in the case of VDR-RXR or VDR-TR, these rexinoids divert RXR monomers away from forming heterodimers, instead facilitating RXR homodimers with a resulting attenuation of 1,25(OH)2D3 or thyroid hormone responsiveness. It is now generally recognized that by modifying the structure of nuclear receptor (NR) ligands (and especially the RXR master partner ligand), one can produce specific NR modulators (collectively termed the SNuRMs) with unique new properties that can influence the activity of the NR in novel ways.7
Structure 1.

SNuRMs such as RXR selective molecules (rexinoids) have recently been targets of interest, since the selective activation of RXR proteins versus RAR proteins might confer cancer chemotherapeutic effects 21 without inciting concurrently negative side-effects by interacting with the RAR proteins. 8 The structure of the endogenous 9-cis retinoic acid ligand for RAR and RXR is shown below. Ligand Pharmacueticals, Inc., developed an RXR selective agonist, 4-[1-(3,5,5,8,8-pentamethyl-5,6,7,8-tetrahydro-2-naphthyl)ethynyl]benzoic acid (1)9, commonly known as bexarotene, after lengthy SAR (Structure-Activity-Relationship) studies of several analogous compounds.10 Another analog that was later synthesized that had nearly identical response profiles to 1 was compound 211 (disilabexarotene).
Compound 1 is an FDA approved drug, effective in the treatment of cutaneous T-cell lymphoma (CTCL), and it is being explored for treatment of breast cancer 12, lung cancer13, colon cancer14, and other diseases of uncontrolled cellular proliferation, because activation of RXR and “up-regulation” (or expression) of the genes RXR regulates seems to have a therapeutic effect by slowing or arresting cellular proliferation in these conditions. Analogs of 1 and compound 1 itself have also been explored as possible treatments for non-insulin-dependent diabetes mellitus (NIDDM) in mouse models.15 Despite specific activation of RXR by 1, versus RAR, three drawbacks to the use of 1 include hypothyroidism16, since there may be an unintentional antagonism of the TR receptor with ligand activated RXR17, hyperlipidemia, and cutaneous toxicity as a result of residual RAR agonism at the dose concentration. Thus, there is motivation to pursue novel RXR agonist molecules that avoid these side-effects.
There are several reports of compounds analogous to 1 in the literature. In addition to the disilabexarotene, the trifluoromethyl bexarotene (3)18, the cyclopropyl dienoic acid (4)19, and a host of novel aza-retinoids, including compound 520, as well as amide retinoids21 have been reported. The thiocarbamate analog 622 was shown to induce apoptosis in leukemia HL-60 cells. Several pyridine containing analogs, as in compound 723, and unsaturated analogs, such as compound 824, were synthesized and shown to be potent RXR selective agonists. Boehm and co-workers published a series of papers that developed RXR selective agonists based on aryl-trienoic acids locked by none25, one26, and two rings27, the last of which is demonstrated by compound 927. Notably, addition of fluorine in proximity to the carboxylic acid of the trienoic acids locked by no rings resulted in improved pharmacological profiles28, and we hypothesize that analogs of 1 possessing a fluorine atom close to the carboxylic acid group will possess similarly improved RXR agonist characteristics. Compound 1029 and analogous compounds were identified as potent RXR selective agonists. Finally, Gronemeyer and co-workers used the RXR selective agonist 116 as well as a model compound to design RXR modulating antagonists, several of which were co-crystallized in the ligand binding domain of hRXRα. However, despite the wealth of different RXR agonist compounds that incorporate structural motifs from 1, there are, as yet, no analogs of 1 that contain additional functional moieties such as a nitro group or a halogen atom substituted for hydrogen atoms on the aromatic ring that bears the carboxylic acid.
Therefore, studies in our laboratories have focused on the synthesis, modeling and biological evaluation of novel compounds analogous to 1 such as compounds 12 and 14–21. We have compared these novel analogs to 1 and its ketone analog (22)9.
In the present study, we have synthesized compounds 1 and 12–22 and evaluated these in mammalian 2-hybrid assays and RXR- and RAR-response element (RXRE and RARE) transcriptional activation systems using cultured human cells, as well as in apoptosis, cytotoxicity, and mutagenicity assays.
Results and Discussion
The Chemistry
Synthesis of 1 and isomer 12
In order to prepare a standard sample of 1, as well as the novel isomer (12), 2,5-dimethyl-2,5-hexanediol was converted to 2,5-dichloro-2,5-dimethylhexane (23)9 in 73% recovered yield by treatment with concentrated hydrochloric acid, and the dihalide 23 was reacted with toluene in the presence of aluminum chloride to provide 1,2,3,4-tetrahydro-1,1,4,4,6-pentamethylnaphthalene (24)9 in 94% yield following the procedure in literature (Scheme 1).
Scheme 1.

To make the Friedel-Crafts acylation coupling partners for 24 en route to 1 and its structural isomer (12), commercially available mono methyl terephthalate and mono methyl isophthalate were converted to the corresponding acid chlorides, 25 and 26, respectively, by reaction with thionyl chloride (Scheme 2).
Scheme 2.

Compound 1 was prepared according to the method of Boehm and co-workers. 9 The Friedel-Crafts acylation of 24 with 25 provided ketone 279 in 82% yield. Ketone 27 was converted to alkene-ester 289 in 66% yield following the Wittig reaction with triphenylphosphonium methylide, prepared by the treatment of triphenylphosphonium methyl bromide with sodium amide in THF. Alkene-ester 28 was saponified by treatment with potassium hydroxide in methanol followed by acidification with hydrochloric acid to give 1 in 78% yield (Scheme 3).
Scheme 3.

Analog 12 was prepared by a slightly modified route as reported for 1 (Scheme 4).
Scheme 4.

Despite the report that ketone 1323 is an inactive RXR agonist, we prepared isomer 12.
Synthesis of Analogues 14–21
To prepare analogs 14 – 21, the appropriate acid chloride Friedel-Crafts acylation coupling partners for 26 were synthesized. Thus, trimethyl-1,3,5-benzenetricarboxylate (31) was converted to the mono-acid diester (32)30 according to literature procedures, and compound 32 was refluxed in excess thionyl chloride to give acid chloride 33 (Scheme 5).
Scheme 5.

Dimethyl-nitro-terephthalate (34) was converted to the mono-acid ester (35)31, and compound 35 was refluxed in excess thionyl chloride to give acid chloride 36 (Scheme 6).
Scheme 6.

To prepare the analogs of 1 with fluorine, the method of Kishida and co-workers was used.32 3-Fluoro-4-methylbenzoic acid (37) was dibrominated with NBS and catalytic benzoylperoxide to give compound 3832 which was subsequently treated with silver nitrate in ethanol and water to give aldehyde 3932. Aldehyde 39 was either converted to the methyl-ester 4032 followed by oxidation to the methyl-fluoro-terephthalic acid 4132 and conversion to acid chloride 42, or 3932 was benzylated to give benzyl-ester 4332. Benzyl-ester 43 was oxidized with sodium hypochlorite to acid 4432 which was esterified to methyl-ester 4532, debenzylated to give acid 4632 and converted to acid chloride 4732 (Scheme 7).
Scheme 7.

The analogs 14–21 were synthesized according to a route analogous to the one used to give analogs 13 and 14 (Scheme 8).
Scheme 8.

The X-ray crystal structures of ketones 49, 50, and 51 are shown in Figure 1.
Figure 1.
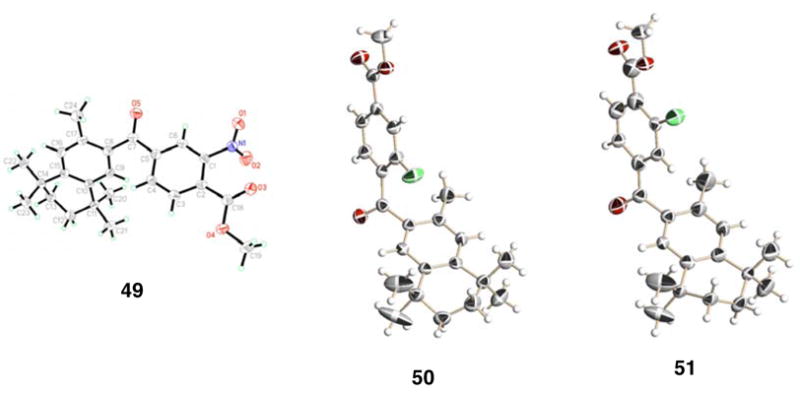
The X-ray crystal structures of ketone-esters 49, 50, and 51. Compound 50 displays twist isomerism in the aliphatic ring, hence, only one of the two isomers in the crystal structure is shown.
The X-ray crystal structures of fluorinated analogs 18 and 20 are shown in Figure 2.
Figure 2.
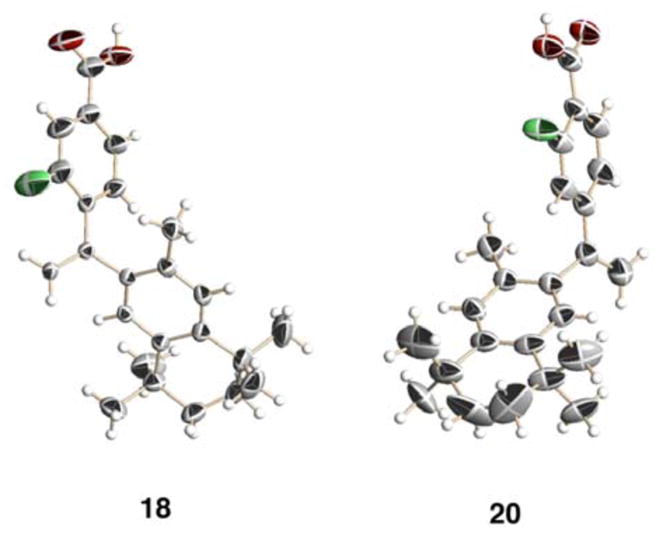
The X-ray crystal structures of fluorinated bexarotene analogs 18 and 20. Compound 18 crystallized in such a way that a channel whose boundaries were formed by the carboxylic acid groups of 18 incorporated a solvent that could not be identified by the model, though likely candidates such as ethyl acetate (the crystallization solvent) were examined. Compound 20 displayed twist-isomerism in the aliphatic ring, as well as partial-filled occupancies of the fluorine atom at both ortho-positions to the carboxylic acid group due to carbon-carbon single bond rotation. Additionally, the carbonyl carbon-oxygen bond of the carboxylic acid group of 20 could not be clearly resolved. Hence, one twist isomer, as well a given rotational isomer for the fluorine and carboxylic acid groups, has been displayed.
Biological Assays and Rationale
A mammalian two hybrid assay reveals that several novel analogs induce RXR homodimerization as well as 1 binds to RXR
Biological assessment of a subset of analogs described above (compounds 12–22) was first carried out employing the mammalian two-hybrid assay in human colon cancer (Caco-2) cells (Figure 3). This assay tests for homodimerization and ligand binding to a recombinant RXR. If the ligand-receptor complex then homodimerizes with an RXR-Gal4 fusion protein, luciferase will be transcribed, as the luciferase gene is downstream of Gal4p DNA binding elements.
Figure 3. Identification of potential RXR agonists via a mammalian two-hybrid screening assay in human colon cancer cells.

Caco-2 human colon cancer cells were cotransfected using both a pCMV-hRXR binding domain vector (BD) as well as an hRXR-activation domain (AD) plasmid along with a pFR-Luc reporter gene containing BD-binding sites, and renilla control plasmid. Cells were transfected for six hours utilizing a liposome-mediated transfection protocol, and then treated with ethanol vehicle or 10−7 M of the indicated compound. After a 24 hour incubation, cells were lysed and a luciferase assay was completed. Analog-mediated RXR binding and homodimerization, as measured by luciferase output, was compared to the RXR agonist parent compound 1 (value set to 1.0).
This initial evaluation revealed that 5 compounds exhibited at least some activity, in the same order of magnitude as 1. More importantly, the initial array of analogs shows a range of receptor binding and RXR homodimerization ability: 16 and 18 bind and mediate homodimerization about half as well as 1, whereas compound 20 binds and activates transcription better than 1 (for compound 20, using a one-tailed heteroscadastic t-test P=0.049, indicating that compound 20 is significantly better than 1). Additionally, ketones 21 and 22 also display a small degree of RXR binding and homodimerization relative to 1. These results imply that compounds modeled after 1 can be synthesized successfully and can possess RXR binding properties, and we are interested in elucidating the factors responsible for eliciting the different response-ranges observed. Moreover, these data suggest that construction of additional analogs of 1 is warranted, especially those compounds that preserve the carboxylic acid position but substitute non-hydrogen atom groups on the aromatic ring that bears the carboxylic acid.
Novel analogs of 1 bind to RXR and mediate transactivation
It is important to point out that the use of the mammalian two hybrid assay as an initial screen for agonist-induced homodimerization of RXR is useful because of the speed, convenience and sensitivity of the assay. However, an additional and vital question in testing RXR agonists is the role of the correct biologically relevant DNA platform, or retinoid X receptor response element (RXRE), that specifically associates with the RXR homodimer in vivo. The RXRE DNA sequence is present in the upstream promoter region of genes controlled by the RXR homodimer in response to the endogenous 9-cis RA ligand, or when RXR is bound to a synthetic rexinoid. It is possible that the RXRE may influence the affinity and/or selectivity of the RXR protein towards potential ligands. Thus, a second screening protocol for our collection of possible RXR agonists included transfection of Caco-2 cells with an expression vector for wild-type human RXRα along with a reporter construct that contains an RXRE driving the expression of the luciferase reporter gene. The results in Figure 4 reveal that of all the compounds tested (12–22), only analog 16, 18 and 20 displayed transcriptional activity significantly above the ethanol control levels (P values of < 0.001 for all, using a one-tailed heteroscedastic t-test).
Figure 4. Identification of potential RXR agonists via an RXRE-luciferase reporter-based screening assay in human colon cancer cells.

Caco-2 cells were transfected with hRXRα, an RXRE-luciferase reporter gene, renilla control plasmid, and carrier DNA (pTZ18U). Cells were transfected for six hours utilizing a liposome-mediated transfection protocol, and then treated with ethanol vehicle or 10−7 M compound 1 or the indicated analog (12–22). After a 24 hour incubation, cells were lysed and a luciferase assay was completed. Analog-stimulated, RXR-mediated transcription, as measured by luciferase output, was compared to the RXR agonist parent compound 1 (value set to 1.0).
These same 3 compounds were also active in the mammalian two-hybrid assay described above (Figure 3).
Determination of RXR binding affinity, EC50 values and quantitation of RAR agonist activity by the most active rexinoids
To better quantitate the affinity and efficacy of the most active novel analogs for RXR binding, we utilized both a ligand binding assay with overexpressed human RXRα as well as the mammalian two-hybrid assay in human colon cancer cells (Figure 3) to evaluate a much larger array of 1 and analog concentrations. The binding affinities (Ki values) of the most active rexinoids were determined by performing competition binding studies via displacement of 10 nM [3H]-9-cis-retinoic acid essentially as described previously.33 The transcriptional efficacy of these same compounds was tested in mammalian two-hybrid dose-response assays carried out with ligand concentrations ranging from 10 × 10−10 M up to 0.5 × 10−5 M. Utilizing this collection of binding affinity and dose-response experiments, we were able to calculate Ki and EC50 values which are listed in Table 1. The Ki value, which is an estimate of ligand affinity for RXR, is similar to that obtained previously for 1 by another group9. Moreover, the data in Figures 3 and 4 suggest that compounds 16 and 18 possess slightly lower RXR binding activity while compound 20 is slightly more active, observations that are entirely consistent with both the Ki and EC50 values in Table 1.
Table 1.
Determination of Binding Affinity, EC50 Values, and Quantitation of RAR Agonist Activity
| Compound | RXRα Binding Affinity (Ki), nM (± S.D.) | EC50 value2 nM (± S.D.) | % RAR Agonist Activity3 at 100 nM (± S.D.) | % RAR Agonist Activity at 1 μM (± S.D.) |
|---|---|---|---|---|
| Compound 1 | 21 (3) | 52 (6) | 23 (5) | 25 (4) |
| Analog 16 | 81 (12) | 200 (28) | 30 (4) | 35 (6) |
| Analog 18 | 161 (28) | 420 (63) | 13 (2) | 14 (1) |
| Analog 20 | 12 (2) | 43 (5) | 25 (6) | 26 (2) |
Binding affinities (Ki values) were determined by competition of 10 nM [3H]-9-cis-retinoic acid (RA) with unlabeled test rexinoids as described in Experimental Section.
EC50 values were determined from full dose-response curves ranging from 10−10 to 10−5 M in transfected Caco-2 cells using an RXR mammalian two-hybrid system.
RAR agonist activity was derived from an RAR/RARE reporter system in transfected Caco-2 cells treated with analog or all-trans RA at 100 nM or 1 μM. The activity with analog (or compound 1) divided by the activity with all-trans RA expressed as a percentage represents the RAR agonist activity.
We also performed an analysis of the “residual” retinoic acid receptor (RAR) agonist activity of the parent compound 1, as well as analogs 16, 18 and 20 versus the authentic RAR ligand (all-trans retinoic acid). The results of this assay, which employed expression of the human RAR and a retinoic acid responsive element (RARE)-luciferase reporter system, revealed that compound 16 possess slightly greater RAR agonist activity, analog 20 is approximately equal to 1 in its activation of RAR, and compound 18 possesses significantly lower RAR binding. Taken together, these results suggest that modification of 1 with a halogen atom on the aromatic ring that bears the carboxylic acid may reduce the activation of RAR (compound 18), or increase its ability to activate RXR (analog 20).
Compound 1 and novel analogs induce apoptosis in a CTCL system
It has been hypothesized that 1 treats CTCL effectively because it induces apoptosis and/or cytotoxicity in the T-lymphocyte.34 Thus, we next tested CTCL cells treated with 1 and promising analogs for their ability to induce classic apoptosis, using an assay for caspases 3 and 7, two executioner caspases in apoptosis.35 We included compounds 16, 18, and 20 because they possess the most potent RXR binding and activation profile, as well as compound 19 which does not bind to RXR and serves as a negative control. Figure 5 illustrates the results of the apoptosis assay, and suggests that some of the analogs that bind and activate RXR (compounds 16 and 20) also posses apoptotic activity that is statistically significantly greater than the ethanol vehicle control (using a one tailed heteroscedastic t-test, P=0.003 for compound 16, and P=0.002 for compound 20), while compound 19, which displays almost no apoptotic activity, also does not bind to RXR. A potent known apoptotic inducer (sodium butyrate, NaBu) serves as a positive control in this system.
Figure 5. Evaluation of Bexarotene (Compound 1) and selected analogs for apoptotic activity utilizing a Caspase 3/7 assay in CTCL cells.

Human T-cell lymphoma cells (CTCL) were plated and immediately dosed with the indicated treatments. Cells were allowed to incubate for 24 hours and the level of apoptosis was measured with a commercial kit (see Experimental Section). Sodium butyrate (Na Bu), a known inducer of apoptosis, was used as a positive control.
Compound 1 and the active analogs display a low level of cytotoxicity
Cytotoxicity results for 1 and active analogs are shown in Figure 6.
Figure 6. Cytotoxocity Analysis of Select Compounds.

Cytotoxicity was assayed by determining lactate dehydrogenase (LDH) release. CTCL cells were left untreated, or were treated for 48 hours with ethanol vehicle, 10−7 M of the indicated compounds, or 10 mM hydroxyurea (HU, positive control). After a 48 hour incubation, the supernatant from cells was removed and assayed for LDH activity (Promega CytoTox 96® Non-Radioactive Cytotoxicity Assay). The total cell activity was determined by lysing untreated cells and assaying the whole cell lysates. Cytotoxcicity was determined as percentage of LDH released, as compared to total activity (total activity set to 1.0) after normalizing for background.
Compound 1 is statistically significantly more cytotoxic in CTCL cells than ethanol vehicle alone (P=0.009, unpaired t-test, one tail, unequal variance); however, the level of cytotoxicity is low (13.2% of total) compared to hydroxyurea (97% of total), a positive control. This is in line with previously reported data for the cytotoxicity of 1.36 All of the analogs are no more cytotoxic than 1 (P<0.01, unpaired t-test, one tail, unequal variance), indicating that these compounds are similar in biological activity to 1.
Compound 1 and the novel analogues are not mutagenic
Mutagenicity of all compounds was tested in a Saccharomyces cerevisiae assay in order to determine if the compounds are potentially suitable to administer in animal models. This assay utilizes a strain of S. cerevisiae in which three phenotypic readouts have been engineered37, 38 in order to determine if the compounds are potentially suitable to administer in animal models. All compounds were tested for mutagenicity compared to DMSO vehicle, and none were mutagenic.
Molecular Modeling
Docking studies of 1 and the novel analogues predict that analogues 16, 18, and 19 will have the best binding affinities
We performed docking studies of 1 and compounds 12, 14, 16, 18, and 20 using the X-ray structure of human RXRα in complex with 5639 (BMS 649) as template. The molecular frameworks of 56, 1 and the compounds are very similar, with 21 carbon atoms at identical positions.
Docking predicted low binding energies for compounds 16, 18, and 20 (Figure 7). The calculated average binding free energies were −14.9 and −14.8 kcal/mol for compound 18 and 20, respectively; this was identical to the calculated binding energy of the docked 1 (−14.9 kcal/mol). Compound 16 was predicted to be the best binder, with an average binding free energy of −16.0 kcal/mol. This lower energy was mostly a result of a more favorable desolvation energy. Overall, the binding of 1 and compounds 16, 18, and 20 were very similar to the binding of 56, with C atom root mean square deviations (rmsds) of 0.35±0.08, 0.50±0.10, 0.43±0.10, and 0.33±0.08Å for the 20 lowest energy structures, respectively. The main deviations stemmed from the benzene ring, which slightly rotated to best accommodate hydrogen bonding between the carboxylate and Arg316. The ligands bound in a large hydrophobic pocket (Figure 8), with the fluoride atom of compounds 18 and 20, and the nitro group of compound 16 pointing towards the C terminus of the H2 helix.
Figure 7.
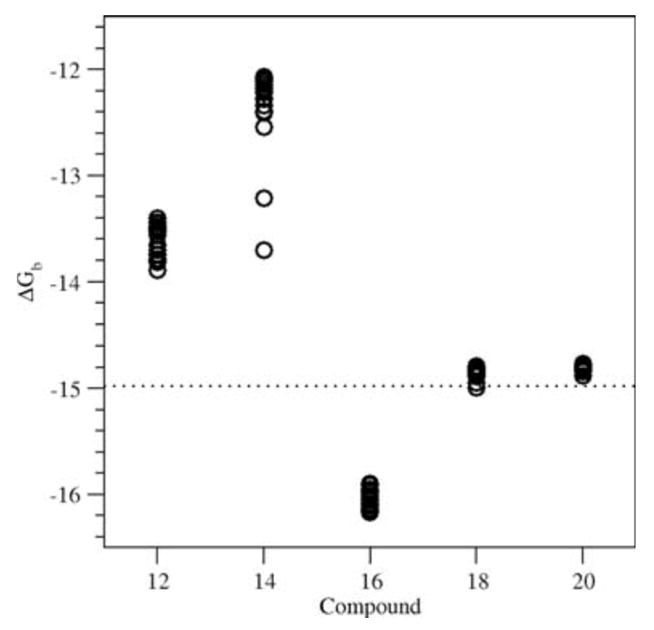
AutoDock binding free energies for compounds 12, 14, 16, 18 and 20. Shown are the 20 lowest energies for each compound. The calculated binding free energy of 1 is shown by the dotted line.
Figure 8.
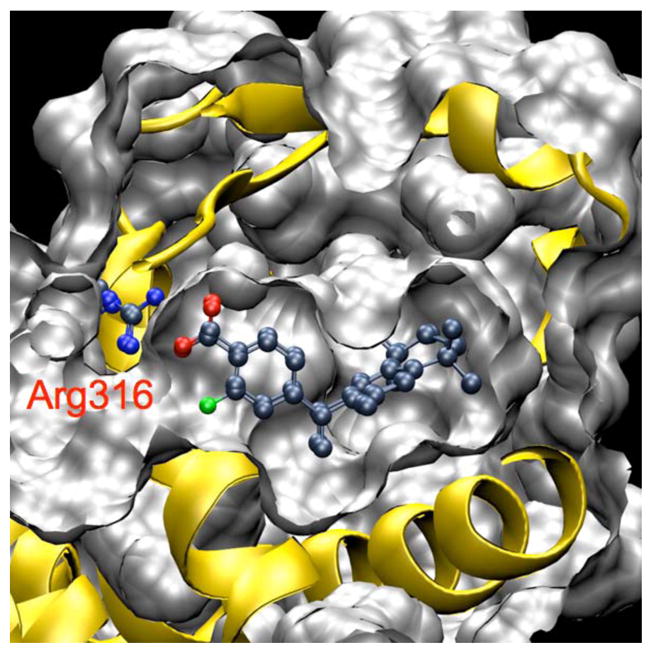
Docked structures of 1 and compound 20 to human RXRα.
Compounds 12 and 14 were predicted to have significantly higher binding energies than 1 (Figure 7). Structural analyses of the docked structures showed that the increase in binding energy was mainly due to the carboxylate at the ortho position. To enable hydrogen bonding between the carboxylate and Arg316, relatively large readjustments of the ligands were needed, resulting in C atom rmsds of 0.95±0.16 and 1.25±0.15Å with 56 for compound 12 and 14, respectively. The readjustments not only involved the benzene ring, but also shifted the rest of the ligand out of the hydrophobic pocket. Such shifts were particularly pronounced for the two lowest energy structures of compound 14 (with binding free energies of −13.7 and −13.2 kcal/mol, see Figure 7). These structures have C atom rmsds of 1.63 and 1.45Å. Although the hydrophobic contacts were less optimal in these structures, overall decreases in binding energy compared to the other docked structures of compound 14 were obtained from better hydrogen bonding with Arg316. Despite these rearrangements, the best docked structures still had much larger binding free energies than 1 or compounds 16, 18 and 20. Therefore, our modeling studies are predictive of the observation that compounds 16, 18, and 20 possessed the greatest RXR/RXRE-mediated transcriptional activation of all novel analogs tested in the RXRE assay, while analogs 12 and 14 did not display significant RXR binding and activation.
Conclusions
We have modeled, synthesized and evaluated several novel analogs of 1 with new functional groups substituting hydrogen atoms on the aromatic ring that bears the carboxylic acid. Biological assays employing these compounds in human Caco-2 and CTCL cells have identified compounds that bind and activate RXR slightly below or near the levels of 1 (16, 18, 21 and 22), and we have also synthesized and identified a compound (20) that possesses an apparent RXR binding affinity that is 75% greater than 1, and that displays a 20% increase in efficacy based on EC50 values (Table 1). Our results suggest that additional novel analogs of 1 that substitute non-hydrogen functional groups on the aromatic ring bearing the carboxylic acid will likely serve as effective, and perhaps more potent, ligands for RXR, that may also have reduced RAR agonist activity (Table 1); thus, these compounds may also possess less detrimental side-effects in cutaneous T-cell lymphoma patients.
Experimental Section
Mammalian two-hybrid assay
Mammalian two hybrid experiments were conducted using Caco-2 human colon cancer cells. Cells were plated at 90,000 cells/well in a 24 well plate and maintained in minimum essential medium (MEM) (Invitrogen, Carlsbad, CA), supplemented with 20% fetal bovine serum (FBS) (Invitrogen), 1 mM sodium pyruvate (Invitrogen), 100 units/mL penicillin and 100 μg/mL streptomycin. The cells were co-transfected utilizing RXR-bait (BD) and RXR-prey (AD) fusion constructs, pFR-Luc reporter gene and renilla control plasmid via liposome-mediated transfection with Lipofectamine LTX and PLUS reagent (Invitrogen). The cells were incubated with the transfection mixture overnight and then treated with ethanol vehicle, 1, or analogs at concentrations ranging from 10 × 10−10 M up to 0.5 × 10−5 M. After incubation with ligands, cells were collected and the amount of reporter gene product (luciferase) produced in the cells was measured using the Dual-Luciferase® Reporter Assay System according to the manufacturer’s protocol (Promega, Madison, WI) in a Sirus FB12 Luminometer (Berthold Detection Systems). Independent experiments were conducted with triplicate samples for each treatment group.
RXRE-mediated transcription assay
Caco-2 human colon cancer cells were plated at 90,000 cells/well in a 24 well plate and maintained as described above. The transfection procedure was adapted from the manufacturer’s protocol (Invitrogen). Briefly, each well received 1 μL of Lipofectamine Reagent, 2 μL of Plus Reagent, 500 ng of pTZ18U carrier DNA plasmid, and 20 ng of pRL-null (constitutively expressing low levels of Renilla reniformis luciferase) to monitor transfection efficiency. Each well also received 250 ng of pLuc-MCS plasmid (Stratagene, La Jolla, CA) containing an oligonucleotide (cloned between the HindIII and BglII sites) with two copies of the retinoid X receptor response element (RXRE) upstream of the firefly (Photinus pyralis) luciferase gene. The RXRE was based on a naturally occurring double repeat responsive element from the rat cellular retinol binding protein II gene. The sequence used was AAAATGAACTGTGACCTGTGACCTGTGACCTGTGAC, with the half elements underlined. The cells were incubated with the transfection mixture overnight and then treated with ethanol vehicle, 10−7 M 1, or analogs for 24 hours. After incubation with ligands, cells were collected and the amount of reporter gene product (luciferase) produced in the cells was measured using a luminometer.
RXR binding assay
[3H]-9-cis-retinoic acid (60 Ci/mmol) was obtained from Perkin-Elmer (Waltham, MA). Assays were carried out essentially as described previously.9,33 Briefly, Caco-2 cells (500,000 cells/60 mm plate) transfected with 50 ng human wild-type RXRα expression plasmid were lysed in KETZD-0.3 buffer (0.3 M KCl, 10 mM Tris-HCl, pH 7.4, 1 mM EDTA, 0.3 mM ZnCl2, 5 mM DTT) containing 0.5% Triton X-100 and supplemented with HALT protease inhibitors (Thermo Scientific, Rockford, IL). Lysates were clarified by centrifugation for 15 min at 16,000 × g at 4 °C and then 40 μg of total protein lysate was incubated with 10 nM [3H]-9-cis-retinoic acid and varying concentrations of competing ligand for 16 h at 2 °C. Bound and free hormone were separated with dextran-coated charcoal for subsequent analysis of ligand binding.
RAR/RARE-agonist activity assay
Caco-2 human colon cancer cells were plated at 90,000 cells/well in a 24 well plate and maintained as described above. The transfection procedure employed 1 μL of Lipofectamine Reagent, 2 μL of Plus Reagent, 50 ng of pTZ18U carrier DNA plasmid, 20 ng of pRL-null (constitutively expressing low levels of Renilla reniformis luciferase) to monitor transfection efficiency, and pCMX-human RARα expression vector. Each well also received 250 ng of the pTK- DR5(X2)-Luc plasmid containing an oligonucleotide with two copies of the retinoic acid response element (RARE) upstream of the firefly (Photinus pyralis) luciferase gene. This RARE is an optimized element that has been described previously40 and is responsive to the RAR ligand, all-trans retinoic acid. The sequence of the double RARE is (5′-AAAGGTCACCGAAAGGTCACCATCCCGGGAAAAGGTCACCGAAAGGTCACC-3′), with the half elements underlined. The cells were incubated with the transfection mixture overnight and then treated with ethanol vehicle, all-trans retinoic acid, or analogs (retinoic acid or analog concentrations ranged from 1 × 10−8 M to 5 × 10−6 M) for 24 hours. After incubation with ligands, cells were collected and the amount of reporter gene product (luciferase) produced in the cells was measured using a luminometer.
Apoptosis assay
Human T-cell lymphoma (CTCL) cells (Hut78) were plated in a 24 well plate and immediately dosed with the indicated treatments, including ethanol, compound 1, analog, water, or sodium butyrate. Cells were allowed to incubate for 24 hours and then lysis was initiated by the addition of a Caspase-Glolysis/substrate mix. Upon programmed cell death, caspases released by the lysed cells cleave the added substrate and generate a measurable luminescent signal, assayed in a luminometer. Analog-induced apoptosis was compared to the parent compound, 1. Sodium butyrate (Na Bu), a known inducer of apoptosis, was used as a positive control.
Cytotoxicity assay
Cytotoxicity was measured in the CTCL cells (Hut78) by performing a lactate dehydrogenase (LDH) assay (Cytotox 96® Non-Radioactive Cytotoxicity Assay, Promega, Madison, WI), whereby induction of LDH leakage is an indication of cytotoxicity. LDH levels are indicated by a change in a tetrazolium salt into a red formazan compound, read by a microtiter plate reader (Bio-Tek Instruments, ELX808.) 15,000 CTCL cells in 50 μL were seeded into 96 well plates in RPMI-1640 media supplemented with 10% FBS. Cells were then treated with 0.5 mL of compound or ethanol vehicle alone or were left untreated. Compound 1 or analog final concentrations were 10−7 M. Hydroxurea’s, a known cytotoxic compound, final concentration was 10 mM. Cells were incubated for 48 hours. Untreated cells were incubated with a final concentration 0.8% Triton-X 100 for one hour at 37C to lyse the cells and release all LDH; this was used as the total cell LDH level. For the remainder of the cells, the supernatant was removed and centrifuged at 500 × g to remove residual cells; these supernatants were then assayed for LDH activity. All assays were carried out in a 96 well plate and included 25 μL of total cell lysate or supernatant, plus 25 μL of water and 50 μL assay reagent. The assay was incubated in the dark for 30 minutes, and then 50 μL stop solution was added. The assay was read in a Biotek microtiter plate reader at 490 nm. Cytotoxicity was graphed in Figure 6 for select analogs. The total cell LDH was set to 100% and the no treatment control was used as 0%. All points treatments were performed six times to generate the data set.
Mutagenicity
Mutagenicity of all compounds was tested in a Saccharomyces cerevisiae assay54 in order to determine if the compounds are potentially suitable to administer in animal models. None were mutagenic in this assay. All compounds were tested for mutagenicity55 with an incubation time of 3 hours; the highest concentration in the dose response curve was 0.15% w/v, compounds were dissolved in DMSO and compared to the nonmutagenic DMSO control.
Instrumentation
All 1H NMR spectra were acquired at 400 MHz or 500 MHz on Bruker or Varian spectrometers. Chemical shifts (δ) are listed in ppm against deuterated solvent peaks as an internal reference. Coupling constants (J) are reported in Hz, and the abbreviations for splitting include: s, single; d, doublet; t, triplet; q, quartet; p, pentet; m, multiplet; br, broad. All 13C NMR spectra were acquired on Bruker instruments at 125.8 MHz or 100.6 MHz. Chemical shifts (δ) are listed in ppm against solvent carbon peaks as an internal reference. Infrared spectra (IR) were assayed on a Perkin Elmer 1600 Series FTIR. High resolution mass spectra were recorded using either a JEOL GCmate(2004), a JEOL LCmate(2002) high resolution mass spectrometer or an ABI Mariner (1999) ESI-TOF mass spectrometer. Melting points were assayed on a Thomas Hoover capillary melting point apparatus.
General Procedures
Tetrahydrofuran, methylene chloride, diethyl ether, and benzene were dried by filtration through alumina according to the procedure described by Grubbs.41 All other solvents were distilled from CaH2 prior to use. Removal of volatile solvents transpired under reduced pressure using a Büchi rotary evaporator and is referred to as removing solvents in vacuo. Thin layer chromatography was conducted on precoated (0.25 mm thickness) silica gel plates with 60F–254 indicator (Merck). Column chromatography was conducted using 230–400 mesh silica gel (E. Merck reagent silica gel 60). All tested compounds were analyzed for purity by combustion analysis through Columbia Analytical Services (formerly Desert Analytics in Tucson, AZ) and were found to be > 95% pure.
2,5-Dichloro-2,5-dimethylhexane (23)
A slightly modified method of Boehm and co-workers9 was followed to make 23. To 2,5-dimethyl-2,5-hexanediol (5.0 g, 34 mmol) in a 100 mL round bottom flask was added concentrated hydrochloric acid (40.0 mL), slowly with gentle swirling. The diol slowly dissolved and a white precipitate formed simultaneously within 10 minutes. After sitting 2 h, the heterogeneous mixture was filtered and washed with copious amounts of water and a small amount of methanol to give crude 23 that was dried under vacuum for 30 minutes to yield a white crystalline solid, m.p. 63–65.8 °C (lit.42 63–66.5 °C): 1H NMR (400 MHz, CDCl3) δ 1.94 (s, 4H), 1.59 (s, 12H); 13C NMR (100.6 MHz, CDCl3) δ 70.3, 41.1 32.5.
1,2,3,4-Tetrahydro-1,1,4,4,6-pentamethylnaphthalene (24)
To a 3-neck, 500 mL round bottom flask charged with 23 (10.0 g, 54.6 mmol) and dry DCM (50.0 mL), fitted with a spiral water condenser, was added aluminum chloride (0.50 grams, 3.7 mmol) in small scoops. The colorless, homogeneous solution of 23 in DCM turned canary yellow concurrent with gas evolution as the aluminum chloride was added. The reaction solution was stirred at room temperature (30 min), and an additional amount of aluminum chloride (100 mg) was added, and the reaction solution was heated to reflux and stirred for 15 minutes. TLC indicated that the reaction was complete. The reaction solution was cooled in an ice bath and quenched with a 20% HCl solution (50 mL). The reaction solution was extracted with hexanes (200 mL, twice), dried over sodium sulfate, concentrated in vacuo and the crude oil was purified by column chromatography (SiO2, Hexanes) to give 24 (10.4 g, 94%) as a white solid, m.p. 34–36 °C: 1H NMR (500 MHz, CDCl3) δ 7.22 (d, J = 8.0, 1H), 7.13 (s, 1H), 6.97 (d, J = 8.4, 1H), 2.31 (s, 3H), 1.68 (s, 4H), 1.29 (s, 6H), 1.28 (s, 6H); 13C NMR (100.6 MHz, CDCl3) δ 144.6, 141.8, 134.7, 126.9, 126.5, 126.4, 35.1, 35.1, 34. 1, 33.8, 31.9, 31.8, 21.1; GC-EI-MS (M+) calcd for C15H22 202.1722, found 202.1751.
Methyl-4-[(3,5,5,8,8-Pentamethyl-5,6,7,8-tetrahydro-2-naphthyl)carbonyl]benzoate (27)
Compound 27 was synthesized according to the methods of Boehm and co-workers.9 Monomethylterephthalic acid chloride (25) was synthesized by refluxing monomethylterephthalic acid (10.1 g, 56.2 mmol) in thionyl chloride (120 mL, 1.65 mol) in a 500 mL one-neck round bottom flask fitted with a water cooled reflux condenser. Excess thionyl chloride was removed in vacuo to give crude 25 as an off-white solid, and this solid was dissolved in dry benzene (ca. 40 mL) and evaporated to dryness three times to remove residual thionyl chloride. The acid chloride 25 was dried on high vacuum to remove residual benzene. To a 3-neck, 500 mL round bottom flask equipped with a reflux condenser and magnetic stir-bar was added 24 (10.0 g, 49.4 mmol) followed by a solution of crude acid chloride 25 (56.2 mmol) in DCM (50 mL). Aluminum chloride (14.0 g, 105 mmol) was added to the reaction solution at room temperature slowly, with stirring, and the reaction solution turned from colorless to red accompanied by the evolution of gas and heat. The reaction was stirred for 30 min and then heated to reflux for 15 min. The reaction was judged to be complete by TLC, and the solution was poured into an ice solution (200 mL) acidified with a 20% HCl solution (50 mL) and ethyl acetate was added (100 mL). The aqueous and organic layers were separated, and the aqueous layer was extracted with ethyl acetate (100 mL, twice). The combined organics were washed with water and brine, dried over sodium sulfate, filtered and rotevapped to give crude 27. Crude 27 was purified by dissolving the crude material in hot ethyl acetated (53 mL) followed by the addition of methanol (100 mL) and slowly cooling the solution to room temperature to yield square plate crystals of 27 (12.86 g, 71%) that were filtered, m.p. 143–144 °C (lit.9 142–143 °C): 1H NMR (400 MHz, CDCl3) δ 8.15 (d, J = 6.8 Hz, 2H), 7.89 (d, J = 6.8, 2H), 7.30 (s, 1H), 7.25 (s, 1H), 4.00 (s, 3H), 2.39 (s, 3H), 1.74 (s, 4H), 1.35 (s, 6H), 1.24 (s, 6H); 13C NMR (100.6 MHz, CDCl3) δ 197.7, 166.4, 148.4, 141.9, 141.8, 134.7, 134.5, 133.4, 129.9, 129.5, 129.4, 128.4, 52.4, 34.9, 34.8, 34.3, 33.8, 31.7, 31.6, 20.0.
Methyl 4-(1-(1,2,3,4-tetrahydro-1,1,4,4,6-pentamethylnaphthalen-7-yl)vinyl)benzoate (28)
The method of Boehm and co-workers9 was followed to prepare compound 28. To a 100 mL round bottom flask charged with methyltriphenylphosphonium bromide (4.72 g, 13.2 mmol) and dry THF (15 mL) under nitrogen was slowly added sodium amide (0.72 g, 18.5 mmol). The heterogeneous solution was allowed to stir for 46 h and it was slowly added to a solution of 27 (3.14 g, 8.62 mmol) in dry THF (20 mL) over 15 min. The reaction solution was stirred for 45 min and then poured into water (200 mL). The aqueous solution was extracted with ethyl acetate (200 mL, twice), and the combined organic layers were washed with water and brine, dried over sodium sulfate, and concentrated in vacuo to give a rouge powder. Crude 28 was dissolved in hot ethyl acetate (10 mL), hot methanol (40 mL) was added, and the solution was allowed to cool to room temperature to give pure 28 as a white powder that was filtered (2.06 g, 66%), m.p. 160–161 °C (lit.9 160–161 °C): 1H NMR (400 MHz, CDCl3) δ 7.96 (d, J = 8.4, 2H), 7.34 (d, J = 8.8, 2H), 7.13 (s, 1H), 7.08 (s, 1H), 5.81 (d, J = 1.2, 1H), 5.33 (d, J = 1.2, 1H), 3.91 (s, 3H), 1.94 (s, 3H), 1.71 (s, 4H), 1.31 (s, 6H), 1.27 (s, 6H); 13C NMR (100.6 MHz, CDCl3) δ 166.9, 149.1, 145.5, 144.3, 142.3, 137.9, 132.7, 129.6, 128.9, 128.1, 128.0, 126.5, 116.8, 52.0, 35.1, 33.9, 33.8, 31.9, 31.8, 19.9.
4-[1-(3,5,5,8,8-pentamethyl-5,6,7,8-tetrahydro-2-naphthyl)ethynyl]benzoic acid (1)
Compound 1 was synthesized according to the method of Boehm and co-workers.9 To a 100 mL round bottom flask charged with 28 (1.48 g, 4.08 mmol) and methanol (20 mL) was added a 5M aqueous solution of potassium hydroxide (2 mL, 10 mmol). A reflux condenser was fitted to the round bottom flask and the reaction solution was refluxed and monitored by TLC. After 70 min at reflux, the reaction solution was cooled to room temperature and quenched with 20% HCl (250 mL). The aqueous solution was extracted with ethyl acetate (200 mL, twice) and the organic extracts were combined, washed with water and brine, dried over sodium sulfate, and concentrated in vacuo to give crude 1. Crude 1 was purified by dissolving the crude material in hot ethyl acetate (19 mL), adding warm hexanes (19 mL), and allowing the solution to cool to room temperature to give crystals of 1 (1.12 g, 78%) m.p. 224–226 °C (lit.9 234 °C): 1H NMR (400 MHz, CDCl3) δ 8.05 (d, J = 8.2, 2H), 7.37 (d, J = 8.2, 2H), 7.14 (s, 1H), 7.09 (s, 1H), 5.84 (s, 1H), 5.36 (s, 1H), 1.94 (s, 3H), 1.70 (s, 4H), 1.32 (s, 6H), 1.29 (s, 6H); 13C NMR (100.6 MHz, CDCl3) δ 172.2, 149.4, 146.7, 144.7, 142.6, 138.1, 132.9, 130.5, 128.3, 128.2, 126.9, 117.4, 105.0, 35.4, 35.4, 34.2, 34.1, 32.1, 32.1, 20.1; LC-APCI-MS (M+) calcd for C24H29O2 349.2168, found 349.2161. Anal. Calcd for C24H29O2: C 82.72; H 8.10. Found: C 82.59; H 8.01.
4-[(3,5,5,8,8-Pentamethyl-5,6,7,8-tetrahydro-2-naphthyl)carbonyl]benzoic acid (22)
Compound 22 was synthesized according to the method of Boehm and co-workers.9 To a 100 mL round bottom flask charged with 27 (0.505 g, 1.39 mmol) and methanol (6.8 mL) was added a 5M aqueous solution of potassium hydroxide (0.71 mL, 3.6 mmol). A reflux condenser was fitted to the round bottom flask and the reaction solution was refluxed and monitored by TLC. After 1 h at reflux, the reaction solution was cooled to room temperature and quenched with 20% HCl (20 mL). The aqueous solution was extracted with ethyl acetate (25 mL, twice) and the organic extracts were combined, washed with water and brine, dried over sodium sulfate, and concentrated in vacuo to give crude 22. Crude 22 was purified by column chromatography (25 mL SiO2, hexanes:ethyl acetate 9:1) to give 22 (0.429 g, 88%) as a white crystalline solid, m.p. 198–199 °C (lit.9 198–199 °C): 1H NMR (400 MHz, CDCl3) δ 8.20 (dd, J = 6.8, 1.8 Hz, 2H), 7.90 (d, J = 8.5, 1.8 Hz, 2H), 7.26 (s, 1H), 7.22 (s, 1H), 2.36 (s, 3H), 1.70 (s, 4H), 1.32 (s, 6H), 1.21 (s, 6H); 13C NMR (100.6 MHz, CDCl3) δ 197.7, 171.3, 148.5, 142.8, 141.9, 134.7, 134.6, 132.4, 130.1, 130.0, 129.5, 128.5, 34.9, 34.8, 34.4, 33.9, 31.7, 31.6, 20.0. Anal. Calcd for C23H26O3: C 78.83; H 7.48. Found: C 78.91; H 7.48.
Methyl 3-[(3,5,5,8,8-Pentamethyl-5,6,7,8-tetrahydronaphthalen-2-yl)carbonyl]benzoate (29)
Compound 2923 was synthesized according to a slightly modified method of Boehm and co-workers.9 Monomethylisophthalic acid chloride (26) was synthesized by refluxing monomethylisophthalic acid (1.30 g, 7.24 mmol) in thionyl chloride (12.0 mL, 165 mmol) in a 100 mL one-neck round bottom flask fitted with a water cooled reflux condenser. Excess thionyl chloride was removed in vacuo to give crude 26 as an off-white solid, and this solid was dissolved in dry benzene (ca. 20 mL) and evaporated to dryness three times to remove residual thionyl chloride. The acid chloride 26 was dried on high vacuum to remove residual benzene. To a 2-neck, 50 mL round bottom flask equipped with a reflux condenser and magnetic stir-bar was added 24 (1.35 g, 6.67 mmol) followed by a solution of crude acid chloride 26 (7.24 mmol) in DCM (15 mL). Aluminum chloride (2.0 g, 15 mmol) was added to the reaction solution at room temperature slowly, with stirring, and the reaction solution turned from colorless to red accompanied by the evolution of gas and heat. The reaction was stirred for 5 min then heated to reflux for 15 min. The reaction was judged to be complete by TLC, and the solution was poured into an ice solution (25 mL) acidified with a 20% HCl solution (8 mL) and ethyl acetate was added (13 mL). The aqueous and organic layers were separated, and the aqueous layer was extracted with ethyl acetate (15 mL, twice). The combined organics were washed with water and brine, dried over sodium sulfate, filtered and rotevapped to give crude 29. Crude 29 was purified by column chromatography (250 mL SiO2, hexanes:ethyl acetate 95:5) to give 29 (2.21 g, 93%) as an oil that crystallized into a solid 110–111 °C (lit.23 110–112 °C): 1H NMR (400 MHz, CDCl3) δ 8.46 (s, 1H), 8.24 (d, J = 8.0, 1H), 8.04 (d, J = 7.6, 1H), 7.55 (t, J = 7.6, 1H), 7.28 (s, 1H), 7.21 (s, 1H); 3.91 (s, 3H), 2.35 (s, 3H), 1.69 (s, 4H), 1.31(s, 6H), 1.21(s, 6H); 13C NMR (100.6 MHz, CDCl3) δ 197.5, 166.5, 148.4, 142.0, 138.8. 134.8, 134.5, 133.7, 131.6, 130.6, 129.7, 128.8, 128.7, 52.5, 35.1, 35.0, 34.5, 34.1, 31.8, 20.2. LC-APCI-MS (M+) calcd for C24H29O3 365.2117, found 365.2133.
Methyl 3-(1-(1,2,3,4-tetrahydro-1,1,4,4,6-pentamethylnaphthalen-7-yl)vinyl)benzoate (30)
To a 100 mL round bottom flask charged with 29 (0.96 g, 2.63 mmol) and dry THF (5 mL) at room temperature was slowly added by equal parts separated by 45 min a triphenylphosphonium methylide solution prepared as follows: methyltriphenylphosphonium bromide (1.9 g, 5.32 mmol) suspended in dry THF (16 mL) in a 100 mL round bottom flask equipped with a stir-bar was stirred for 30 min at room temperature after the addition of a 2.5M solution of n-butyl lithium in hexanes (2.2 mL, 5.5 mmol) which provided a homogeneous dark yellow ylide solution. The reaction was monitored by TLC, and when the reaction was judged to be complete, the reaction solution was poured into water (70 mL) and the aqueous solution was extracted with ethyl acetate (70 mL, twice). The combined organic extracts were washed with water and brine, dried over sodium sulfate, and concentrated in vacuo to give crude 30 which was purified by column chromatography (25 mL SiO2, hexanes:ethyl acetate 97.5:2.5) to give 30 (0.26 g, 27%) as a white solid, m.p. 89–91 °C: 1H NMR (400 MHz, CDCl3) δ 8.10 (s, 1H), 7.92 (d, J = 6.8, 1H), 7.36 (m, 2H), 7.15 (s, 1H), 7.08 (s, 1H), 5.77 (d, J = 1.2, 1H), 5.28 (d, J = 1.2, 1H), 3.91 (s, 3H), 1.96 (s, 3H), 1.71 (s, 4H), 1.31 (s, 6H), 1.28 (s, 6H); 13C NMR (100.6 MHz, CDCl3) δ 167.4, 149.3, 144.4, 142.5, 142.4, 141.8, 138.3, 132.9, 131.6, 131.4, 130.5, 128.7, 128.5, 128.4, 128.3, 127.7, 116.2, 52.3, 35.5, 35.4, 34.2, 34.1, 32.2, 32.1, 30.9, 30.5, 20.2. LC-APCI-MS (M+) calcd for C25H31O2 363.2329, found 363.2324.
3-(1-(1,2,3,4-Tetrahydro-1,1,4,4,6-pentamethylnaphthalen-7-yl)vinyl)benzoic acid (12)
Compound 12 was synthesized from 30 according to the representative procedure for the synthesis of 1 from 28. To a 100 mL round bottom flask charged with 30 (0.3278 g, 0.90 mmol) and methanol (5 mL) was added a 5M aqueous solution of potassium hydroxide (0.50 mL, 2.5 mmol). A reflux condenser was fitted to the round bottom flask and the reaction solution was refluxed and monitored by TLC. After 1 h at reflux, the reaction solution was cooled to room temperature and quenched with 20% HCl (55 mL). The aqueous solution was extracted with ethyl acetate (50 mL, twice) and the organic extracts were combined, washed with water and brine, dried over sodium sulfate, and concentrated in vacuo to give crude 12. Crude 12 was purified by column chromatography (25 mL SiO2, hexanes:ethyl acetate 9:1) to give 12 (0.2623 g, 83%) as a white crystalline solid, m.p. 195–196 °C: 1H NMR (400 MHz, CDCl3) δ 8.16 (s, 1H), 8.02 (m, 1H), 7.42 (m, 2H), 7.15 (s, 1H), 7.09 (s, 1H), 5.79 (s, 1H), 5.30 (s, 1H), 1.96 (s, 3H), 1.71 (s, 4H), 1.31 (s, 6H), 1.29 (s, 6H);13C NMR (100.6 MHz, CDCl3) δ 172.5, 149.2, 144.6, 142.5, 141.9, 138.2, 132.8, 132.5, 129.6, 129.3, 128.7, 128.4, 128.3, 116.4, 35.5, 34.3, 34.2, 32.2, 32.1, 20.2; LC-APCI-MS (M+) calcd for C24H29O2 349.2168, found 349.2149. Anal. Calcd for C24H28O2: C 82.72; H 8.10. Found: C 82.26; H 7.84.
3-[(3,5,5,8,8-Pentamethyl-5,6,7,8-tetrahydronaphthalen-2-yl)carbonyl]benzoic Acid (13)
Compound 1323 was synthesized according to the method of Boehm and co-workers.9 To a 100 mL round bottom flask charged with 29 (0.27 g, 0.74 mmol) and methanol (5 mL) was added a 5M aqueous solution of potassium hydroxide (0.64 mL, 3.2 mmol). A reflux condenser was fitted to the round bottom flask and the reaction solution was refluxed and monitored by TLC. After 1 h at reflux, the reaction solution was cooled to room temperature and quenched with 20% HCl (20 mL). The aqueous solution was extracted with ethyl acetate (25 mL, twice) and the organic extracts were combined, washed with water and brine, dried over sodium sulfate, and concentrated in vacuo to give crude 13. Crude 13 was purified by column chromatography (25 mL SiO2, hexanes:ethyl acetate 9:1) to give 13 (0.2573 g, 99%) as a white crystalline solid, m.p. 192–195 °C (lit.23 192–194 °C): 1H NMR (400 MHz, CDCl3) δ 8.54 (s, 1H), 8.32 (d, J = 7.5, 1H), 8.12 (d, J = 8, 1H), 7.61 (t, J = 8, 1H), 7.30 (s, 1H), 7.24 (s, 1H), 2.37 (s, 3H), 1.71 (s, 4H), 1.34 (s, 6H), 1.22 (s, 6H); 13C NMR (100.6 MHz, CDCl3) δ 197.1, 171.4, 148.4, 141.9, 138.8, 135.1, 134.6, 134.4, 133.9, 132.0, 129.5, 129.4, 128.5, 34.9, 34.8, 34.3, 31.6, 20.0. Anal. Calcd for C23H26O3: C 78.83; H 7.48. Found: C 79.13; H 7.68.
3,5-Di(methoxycarbonyl)benzoicAcid (32)
Compound 32 was synthesized by the method of Dimick and co-workers. 30 To a 250 mL round bottom flask charged with trimethyl 1,3,5-benzenetricarboxylate (31) (1.10 g, 4.36 mmol) and methanol (100 mL) was added a 1M aqueous solution of sodium hydroxide (3.95 mL, 3.95 mmol). The reaction solution was stirred for 18 h, the solvent was removed in vacuo, and the residual solid material was dissolved in saturated NaHCO3 (150 mL) and washed with DCM (75 mL). The aqueous layer was acidified with conc. HCl (18 mL) until the pH ~ 2.0, and the heterogeneous solution was extracted with ethyl acetate (75 mL, twice). The combined extracts were dried over sodium sulfate and the solvents were removed in vacuo to give crude 32. Crude 32 was purified by re-crystallization from boiling water (500 mL/5 g) to give pure 32 (0.817 g, 79%) as a white powder, m.p. 264 °C (decomposition): 1H NMR (400 MHz, CDCl3) δ 8.91 (s, 3H), 3.99 (s, 6H);13C NMR (100.6 MHz, CDCl3) δ 170.0, 165.3, 135.4, 135.1, 131.4, 130.3, 52.7.
4-(Methoxycarbonyl)-3-nitrobenzoic Acid (35)
Compound 35 was synthesized and purified according to a slightly modified method of Keenan and co-workers.31 To a 250 mL round bottom flask charged with dimethylnitroterephthalate (12 g, 50.2 mmol) and dioxane (100 mL) was added 1M sodium hydroxide (50 mL, 50 mmol) dropwise over 30 min at room temperature. The reaction solution was stirred overnight, water was added (100 mL), and the solution was washed with diethyl ether (100 mL, twice). The aqueous layer was acidified with 1M HCl (56 mL) to pH ~ 1–2, then extracted with ethyl acetate (150 mL, thrice). The combined organic extracts were dried over sodium sulfate and removed in vacuo to give crude (35). Crude 35 was re-crystallized in water (600 mL/12 g) to give pure 35 as white crystals (6.0 g, 53%), m.p. 177–179 °C: 1H NMR (400 MHz, CDCl3) δ 8.65 (s, 1H), 8.39 (d, J = 8.0, 1H), 7.84 (d, J = 8.0, 1H), 3.97 (s, 3H).
3-Fluoro-4-formylmethylbenzoate (40)
Compound 40 was synthesized according to the methods of Kishida and co-workers.32 To a 500 mL round bottom flask charged with 3-fluoro-4-methylbenzoic acid (37) (9.0 g, 58.4 mmol) was added NBS (25.0 g, 140 mmol), benzoylperoxide (0.66 g, 2.73 mmol), and carbon tetrachloride (112 mL). The reaction solution was heated to reflux under magnetic stirring for 36 h, cooled to room temperature, and solids were filtered and washed with carbon tetrachloride (~20 mL). The filtrate solvent was removed in vacuo and the crude 4-(dibromomethyl)-3-fluorobenzoic acid (38) was dried on high vacuum and used without further purification. To a 500 mL round bottom flask charged with crude 38 (10.44 g, 58.4 mmol) was added ethanol (148 mL), and a solution of silver nitrate (20.5 g, 120.7 mmol) in warm water (28 mL) was added dropwise while the reaction solution was stirred in an oil bath preheated to 50–55 °C. Upon addition of the silver nitrate solution, a green precipitate formed. After stirring at 50 °C for 45 min, the reaction solution was cooled to room temperature and filtered to remove the green precipitate. The filtrate solvent was concentrated in vacuo, extracted with ethyl acetate (130 mL), and the combined organic extracts were washed with water and brine, dried over sodium sulfate and removed in vacuo to give crude 3-fluoro-4-formylbenzoic acid (39) (8.68 g, 87%) that was used without further purification. To a 500 mL round bottom flask charged with39 (9.87 g, 58.7 mmol) was added dry dimethylformamide (190 mL) and a 60 wt % suspension of NaH in mineral oil (2.75 g, 68.8 mmol) in small aliquots over 20 min. The reaction solution was stirred an additional 20 min, and methyliodide (4.32 mL, 69.4 mmol) was added to the red heterogeneous solution. After stirring 5 h, the reaction solution had become homogeneous, and it was poured into 1N HCl (490 mL), extracted with ethyl acetate (150 mL, twice), and the combined organic extracts were washed with saturated NaHCO3 (75 mL) and brine, dried over sodium sulfate, and removed in vacuo to give crude 40. Crude 40 was purified by column chromatography (150 mL SiO2, hexanes:ethyl acetate 4:1) to give 40 (10.24 g, 95%) as a colorless oil: 1H NMR (400 MHz, CDCl3) δ 10.41 (s, 1H), 7.94 (m, 2H), 7.85 (m, 1H), 3.95 (s, 3H); 13C NMR (100.6 MHz, CDCl3) δ 186.6, 165.2, 164.9, 163.1, 137.2, 132.1, 128.8, 126.8, 125.5, 124.8, 117.9, 117.7, 52.8; LC-APCI-MS (M+) calcd for C9H7O3F 182.0379, found 182.0336.
4-(Methoxycarbonyl)-2-fluorobenzoic acid (41)
Compound 41 was synthesized by the method of Kishida and co-workers.32 To a 250 mL round bottom flask charged with compound 40 (9.22 g, 50.5 mmol) and sulfamic acid (5.40 g, 55.6 mmol) in water (21 mL) and ACN (42 mL) was added a solution of 80% NaClO2 (4.92 g, 53.8 mmol) in water (21 mL) drop-wise at room temperature. After stirring for 1 h, the reaction solution was poured into a saturated, aqueous solution of Na 2SO3 (75 mL) and 1 N HCl (150 mL), and the resulting solution was extracted with ethyl acetate (75 mL, thrice). The combined organic extracts were washed with brine, dried over sodium sulfate, and the solvents were removed in vacuo to give crude 41 (7.56 g, 75%) as a white solid. A small sample was recrystallized from hot ethyl acetate to give pure 41 as white crystals, m.p. 154–155 °C: 1H NMR (400 MHz, CDCl3) δ 10.5 (br s, 1H), 8.10 (t, J = 7.8, 1H), 7.89 (d, J = 8.2, 1H), 7.82 (d, J = 11.0, 1H), 3.97 (s, 3H);13C NMR (100.6 MHz, CDCl3) δ;168.6, 168.5, 165.0, 164.9, 163.4, 160.8, 136.7, 136.6, 132.8, 124.9, 124.8, 121.3, 121.2, 118,4, 118.1, 52.8; LC-APCI-MS (M+) calcd for C9H7O4F 198.0328, found 198.0331.
Benzyl-3-fluoro-4-formylbenzoate (43).32
To a 100 mL round bottom flask charged with a crude sample of 3-fluoro-4-formylbenzoic acid (39) (2.51 g, 14.9 mmol) and dry DMF (45 mL) was slowly added with stirring 60 wt% NaH in mineral oil (0.726 g, 18.2 mmol). The reaction solution was stirred for 45 min, benzyl bromide (2.2 mL, 18.4 mmol) was added dropwise, and the reaction solution was stirred for an additional 5 h. TLC indicated the completion of the reaction, and the reaction solution was poured into 1N HCl (80 mL) and extracted with ethyl acetate (50 mL, twice). The combined organic extracts were washed with saturated NaHCO3 and brine, dried over sodium sulfate, and removed in vacuo to give crude 43 as a yellow oil. Crude 43 was purified by column chromatography (150 mL SiO2, hexanes:ethyl acetate 4:1) to give 43 (3.84 g, 99%) as a colorless oil: 1H NMR (400 MHz, CDCl3) δ 10.31 (s, 1H), 7.83 (m, 2H), 7.76 (m, 1H), 7.31 (m, 5H), 5.29 (s, 2H);13C NMR (100.6 MHz, CDCl3) δ 218.2, 186.6, 186.5, 165.2, 164.3, 164.2, 163.1, 137.3, 137.2, 135.2, 132.1, 128.9, 128.8, 128.7, 128.6, 128.5, 128.4, 128.3, 127.0, 126.9, 125.7, 125.6, 124.8, 118.2, 118.0, 117.8, 67.6, 67.4; LC-APCI-MS (M+) calcd for C15H12O3F 259.0771, found 259.0751.
4-((Benzyloxy)carbonyl)-2-fluorobenzoic Acid (44)
Compound 44 was prepared according to the method of Kishida and co-workers.32 To a 100 mL round bottom flask charged with 43 (3.29 g, 12.7 mmol), sulfamic acid (1.235 g, 12.7 mmol), water (20 mL), and ACN (10 mL) was added a solution of 80% NaClO2 (1.16 g, 12.8 mmol) in water (6.8 mL). After stirring for 1 h, the reaction solution was poured into saturated Na 2SO3 (25 mL) and 1N HCl (50 mL), and the resulting solution was extracted with ethyl acetate (50 mL, thrice). The combined organic extracts were washed with brine, dried over sodium sulfate, and removed in vacuo to give crude 44 (3.14 g, 90%) that was used without further purification. A small sample was purified by column chromatography (25 mL SiO2, hexanes:ethyl acetate 1:1) to give pure 44 as a white powder, m.p. 127–128 °C: 1H NMR (400 MHz, CDCl3) δ 10.91 (br s, 1H), 8.09 (t, J = 7.8, 1H), 7.93 (d, J = 9.2, 1H), 7.85 (d, J = 10.9, 1H), 7.39 (m, 5H), 5.40 (s, 2H); 13C NMR (100.6 MHz, CDCl3) δ 168.8, 168.7, 164.4, 164.3, 163.4, 160.8, 136.8, 136.7, 135.2, 132.8, 128.7, 128.5, 128.3, 125.0, 124.9, 121.3, 121.2, 118.4, 118.2, 67.6.
4-Benzyl 1-methyl 2-fluorobenzene-1,4-dioate (45)
Compound 45 was synthesized according to the method of Kishida and co-workers.32 To a 100 mL round bottom flask charged with compound 44 (2.91 g, 10.6 mmol) was added SOCl2 (9.0 mL, 124 mmol) and the reaction solution was refluxed for 1h. The reaction solution was cooled to room temperature and the excess thionyl chloride was removed in vacuo to give crude 4-chlorocarbonyl-2-fluorobenzoic acid benzyl ester. The crude 4-chlorocarbonyl-2-fluorobenzoic acid in dry toluene (4.5 mL) was added dropwise to a solution of triethylamine (2.9 mL, 20.9 mmol) in methanol (29.8 mL, 736 mmol) over 10 min. The reaction solution was stirred 1h and poured into 1N HCl (80 mL) and extracted with ethyl acetate (80 mL, thrice). The combined organic extracts were washed with brine, dried over sodium sulfate, and removed in vacuo to give crude 45. Crude 45 was purified by column chromatography (150 mL SiO2, hexanes:ethyl acetate 95:5) to give pure 45 as a colorless oil: 1H NMR (400 MHz, CDCl3) δ 7.99 (t, J = 7.8, 1H), 7.88 (d, J = 8.2, 1H), 7.81 (d, J = 10.9, 1H), 7.41 (m, 5H), 5.37 (s, 2H), 3.95 (s, 3H); 13C NMR (100.6 MHz, CDCl3) δ 164.5, 164.4, 164.2, 164.1, 162.7, 160.1, 135.7, 135.6, 135.2, 132.2, 128.7, 128.5, 124.9, 124.8, 122.5, 122.4, 118.3, 118.0, 67.4, 52.6; LC-APCI-MS (M+) calcd for C16H14O4F 289.0876, found 289.0886.
4-(Methoxycarbonyl)-3-fluorobenzoic Acid (46).32
A 3-neck 250 mL round bottom flask charged with 45 (1.87 g, 6.49 mmol), 10% Pd/C (0.191 g), ethanol (11.0 mL), and ethyl acetate (11.0 mL) was evacuated and back-filled with hydrogen gas from a balloon three times, and the reaction solution was allowed to stir under hydrogen at room temperature overnight. The reaction solution was filtered through celite, and the solvents were removed in vacuo to give crude 46 (1.23 g, 95%) as a white crystalline solid that was used without further purification. A small sample of crude 46 was purified by recrystallization from hot ethyl acetate to give pure 46, m.p. 210–211 °C: 1H NMR (400 MHz, DMSO-d6) δ 13.61 (br s, 1H), 7.98 (t, J = 7.8, 1H), 7.83 (d, J = 8.2, 1H), 7.74 (d, J = 11.2, 1H), 3.87 (s, 3H); 13C NMR (100.6 MHz, DMSO-d6) δ 165.9, 165.8, 163.9, 163.8, 162.2, 159.6, 137.4, 137.3, 132.5, 125.6, 125.5, 122.1, 122.0, 117.9, 117.7, 53.0; LC-APCI-MS (M+) calcd for C9H7O4F 198.0328, found 198.0371.
Dimethyl 5-(2-(3,5,5,8,8-pentamethyl-5,6,7,8-tetrahydronaphthalen-2-yl)carbonyl)benzene-1,3- dioate (48)
Compound 48 was synthesized according to the representative procedure for the synthesis of compound 29. Dimethyl 5-(chlorocarbonyl)benzene-1,3-dioate (33) was synthesized by refluxing 3,5-di(methoxycarbonyl)benzoic acid (32) (1.71 g, 7.17 mmol) in thionyl chloride (12.0 mL, 165 mmol) in a 100 mL one-neck round bottom flask fitted with a water cooled reflux condenser. Excess thionyl chloride was removed in vacuo to give crude 33 as an off-white solid, and this solid was dissolved in dry benzene (ca. 20 mL) and evaporated to dryness three times to remove residual thionyl chloride. The acid chloride 33 was dried on high vacuum to remove residual benzene. To a 2-neck, 50 mL round bottom flask equipped with a reflux condenser and magnetic stir-bar was added 24 (1.39 g, 6.86 mmol) followed by a solution of crude acid chloride 33 (7.17 mmol) in DCM (15 mL). Aluminum chloride (2.0 g, 15 mmol) was added to the reaction solution at room temperature slowly, with stirring, and the reaction solution turned from colorless to red accompanied by the evolution of gas and heat. The reaction was stirred for 5 min then heated to reflux for 15 min. The reaction was judged to be complete by TLC, and the solution was poured into an ice solution (25 mL) acidified with a 20% HCl solution (8 mL) and ethyl acetate was added (13 mL). The aqueous and organic layers were separated, and the aqueous layer was extracted with ethyl acetate (15 mL, twice). The combined organics were washed with water and brine, dried over sodium sulfate, filtered and removed in vacuo to give crude 48. Crude 48 was purified by column chromatography (250 mL SiO2, hexanes:ethyl acetate 95:5 to 9:1) to give 48 (1.29 g, 44%) as a white powder, m.p. 143–145 °C: 1H NMR (400 MHz, CDCl3) δ 8.89 (s, 1H), 8.65 (s, 1H), 7.27 (s, 1H), 7.22 (s, 1H), 3.95 (s, 6H), 2.37 (s, 3H), 1.68 (s, 4H), 1.31 (s, 6H), 1.19 (s, 6H); 13C NMR (100.6 MHz, CDCl3) δ 196.2, 165.8, 149.0, 142.2, 139.4, 135.3, 131.2, 130.0, 129.1, 52.8, 35.0, 34.6, 34.1, 31.8, 20.3; LC-APCI-MS (M+H)+ calcd for C26H31O5 423.2171, found 423.2163.
Dimethyl 5-(1-(1,2,3,4-tetrahydro-1,1,4,4,6-pentamethylnaphthalen-7-yl)vinyl)benzene-1,3-dioate (52)
Compound 52 was synthesized following the representative procedure to synthesize compound 30. To a 100 mL round bottom flask charged with 48 (0.92 g, 2.18 mmol) and dry THF (5 mL) at room temperature was slowly added by equal parts separated by 45 min a triphenylphosphonium methylide solution prepared as follows: methyltriphenylphosphonium bromide (1.65 g, 4.62 mmol) suspended in dry THF (10 mL) in a 100 mL round bottom flask equipped with a stir-bar was stirred for 30 min at room temperature after the addition of a 2.5 M solution of n-butyl lithium in hexanes (1.86 mL, 4.65 mmol) which provided a homogeneous dark yellow ylide solution. The reaction was monitored by TLC, and when the reaction was judged to be complete, the reaction solution was poured into water (70 mL) and the aqueous solution was extracted with ethyl acetate (70 mL, twice). The combined organic extracts were washed with water and brine, dried over sodium sulfate, and concentrated in vacuo to give crude 52 which was purified by column chromatography (25 mL SiO2, hexanes:ethyl acetate 97.5:2.5) to give 52 (0.14 g, 16%) as a white solid, m.p. 138 °C: 1H NMR (400 MHz, CDCl3) δ 8.58 (s, 1H), 8.16 (s, 2H), 7.15 (s, 1H), 7.09 (s, 1H), 5.81 (s, 1H), 5.35 (s, 1H), 3.93 (s, 6H), 1.95 (s, 3H), 1.73 (s, 4H), 1.31 (s, 6H), 1.29 (s, 6H); 13C NMR (100.6 MHz, CDCl3) δ 166.5, 148.6, 144.8, 142.6, 137.6, 132.7, 132.2, 130.9, 129.7, 128.5, 128.3, 117.5, 52.6, 35.5, 35.4, 34.2, 34.1, 32.1, 32.0, 20.3; LC-APCI-MS (M+H)+ calcd for C27H33O4 421.2379, found 421.2375.
5-(1-(1,2,3,4-Tetrahydro-1,1,4,4,6-pentamethylnaphthalen-7-yl)vinyl)benzene-1,3-dioic Acid (14)
Compound 14 was synthesized following the representative procedure for the synthesis of compound 12. To a 100 mL round bottom flask charged with 52 (0.15 g, 0.36 mmol) and methanol (5 mL) was added a 5M aqueous solution of potassium hydroxide (0.43 mL, 2.15 mmol). A reflux condenser was fitted to the round bottom flask and the reaction solution was refluxed and monitored by TLC. After 1 h at reflux, the reaction solution was cooled to room temperature and quenched with 20% HCl (49 mL). The precipitate product was filtered to give crude 14 that appeared to be pure by TLC. A small sample of crude 14 was purified by recrystallization from hot ethyl acetate to give pure 14 as a white crystalline solid, m.p. 258–260 °C: 1H NMR (400 MHz, MeOH-d4) δ 8.55 (s, 1H), 8.12 (s, 2H), 7.15 (s, 2H), 5.87 (s, 1H), 5.28 (s, 1H), 1.95 (s, 3H), 1.72 (s, 4H), 1.30 (s, 6H), 1.28 (s, 6H); 13C NMR (100.6 MHz, MeOH-d4) δ 167.5, 148.9, 144.5, 142.4, 142.1, 137.9, 132.5, 131.6, 131.5, 129.6, 128.2, 127.8, 116.0, 35.2, 35.1, 33.7, 33.6, 31.1, 31.0, 18.9; LC-APCI-MS (M-H)- calcd for C25H27O4 391.1909, found 391.1942. Anal. Calcd for C25H28O4: C 76.50; H 7.19. Found: C 74.10; H 6.96.
5-(1-(3,5,5,8,8-pentamethylnaphthalen-2-yl)carbonyl)benzene-1,3-dioic Acid (15)
Compound 15 was synthesized following the representative procedure for the synthesis of compound 13. To a 100 mL round bottom flask charged with 48 (0.31 g, 0.74 mmol) and methanol (5 mL) was added a 5M aqueous solution of potassium hydroxide (0.87 mL, 4.4 mmol). A reflux condenser was fitted to the round bottom flask and the reaction solution was refluxed and monitored by TLC. After 1 h at reflux, the reaction solution was cooled to room temperature and quenched with 20% HCl (20 mL). The precipitate was filtered to give crude 15 (0.28 g, 97%) that appeared to be pure by TLC. A sample of 15 was purified by recrystallization in hot ethyl acetate to give pure 15 as a white crystalline solid, m.p. 301–303 °C: 1H NMR (400 MHz, MeOH-d4) δ 8.85 (s, 1H), 8.57 (s, 2H), 7.33 (s, 1H), 7.31 (s, 1H), 2.34 (s, 3H), 1.73 (s, 4H), 1.34 (s, 6H), 1.22 (s, 6H); 13C NMR (100.6 MHz, MeOH-d4) δ 196.8, 166.6, 148.7, 142.1, 139.1, 134.7, 134.6, 134.2, 132.0, 129.6, 128.5, 106.4, 105.0, 34.8, 34.7, 34.2, 33.7, 30.8, 18.8. Anal. Calcd for C24H26O5: C 73.08; H 6.64. Found: C 72.95; H 6.75.
Methyl 4-(1-(3,5,5,8,8-pentamethyl-5,6,7,8-tetrahydronaphthalen-2-yl)carbonyl)-2-nitrobenzoate (49)
Compound 49 was synthesized following the representative procedure for the synthesis of compound 29. Methyl 4-(chlorocarbonyl)-2-nitrobenzoate (36) was synthesized by refluxing 4-(methoxycarbonyl)-3-nitrobenzoic acid (35) (2.46 g, 10.9 mmol) in thionyl chloride (22.0 mL, 302 mmol) in a 100 mL one-neck round bottom flask fitted with a water-cooled reflux condenser. Excess thionyl chloride was removed in vacuo to give crude 36 as an off-white solid, and this solid was dissolved in dry benzene (ca. 20 mL) and evaporated to dryness three times to remove residual thionyl chloride. The acid chloride 36 was dried on high vacuum to remove residual benzene. To a 2-neck, 50 mL round bottom flask equipped with a reflux condenser and magnetic stir-bar was added 24 (2.55 g, 12.6 mmol) followed by a solution of crude acid chloride 36 (10.9 mmol) in DCM (15 mL). Aluminum chloride (4.0 g, 30 mmol) was added to the reaction solution at room temperature slowly, with stirring, and the reaction solution turned from colorless to red accompanied by the evolution of gas and heat. The reaction was stirred for 5 min then heated to reflux for 15 min. The reaction was judged to be complete by TLC, and the solution was poured into an ice solution (50 mL) acidified with a 20% HCl solution (16 mL) and ethyl acetate was added (13 mL). The aqueous and organic layers were separated, and the aqueous layer was extracted with ethyl acetate (25 mL, twice). The combined organics were washed with water and brine, dried over sodium sulfate, filtered and removed in vacuo to give crude 49. Crude 49 was purified by column chromatography (250 mL SiO2, hexanes:ethyl acetate 95:5 to 9:1) to give 49 (4.08 g, 91%) as a yellow, crystalline solid, m.p. 115–116 °C: 1H NMR (400 MHz, CDCl3) δ 8.32 (s, 1H), 8.09 (d, J = 8, 1H), 7.82 (d, J = 8, 1H), 7.25 (m, 1H), 3.95 (s, 3H), 2.36 (s, 3H), 1.69 (s, 4H), 1.31 (s, 6H), 1.20 (s, 3H); 13C NMR (100.6 MHz, CDCl3) δ 194.8, 165.5, 149.6, 148.2, 142.5, 141.7, 135.4, 134.2, 133.3, 130.7, 130.2, 128.9, 125.6, 53.7, 35.0, 34.9, 34.7, 34.1, 31.9, 31.7, 20.3; LC-APCI-MS (M+H)+ calcd for C24H28NO5 410.1967, found 410.1959.
Methyl 4-(1-(1,2,3,4-tetrahydro-1,1,4,4,6-pentamethylnaphthalen-7-yl)vinyl)-2-nitrobenzoate (53)
Compound 53 was synthesized following the representative procedure for the synthesis of compound 30. To a 100 mL round bottom flask charged with 52 (0.895 g, 2.19 mmol) and dry THF (5 mL) at room temperature was slowly added by equal parts separated by 45 min a triphenylphosphonium methylide solution prepared as follows: methyltriphenylphosphonium bromide (1.7 g, 4.76 mmol) suspended in dry THF (16 mL) in a 100 mL round bottom flask equipped with a stir-bar was stirred for 30 min at room temperature after the addition of a 2.5M solution of n-butyl lithium in hexanes (1.86 mL, 4.65 mmol) which provided a homogeneous dark yellow ylide solution. The reaction was monitored by TLC, and when the reaction was judged to be complete, the reaction solution was poured into water (70 mL) and the aqueous solution was extracted with ethyl acetate (70 mL, twice). The combined organic extracts were washed with water and brine, dried over sodium sulfate, and concentrated in vacuo to give crude 53 which was purified by column chromatography (25 mL SiO2, hexanes:ethyl acetate 97.5:2.5) to give 53 (0.10 g, 11%) as an off-white solid: 1H NMR (400 MHz, CDCl3) δ 7.77 (s, 1H), 7.68 (d, J = 8, 1H), 7.50 (d, J = 7.6, 1H), 7.10 (m, 2H), 5.87 (s, 1H), 5.43 (s, 1H), 3.90 (s, 3H), 1.95 (s, 3H), 1.70 (s, 4H), 1.30 (s, 6H), 1.27 (s, 6H); 13C NMR (100.6 MHz, CDCl3) δ 165.5, 149.0, 147.1, 145.4, 145.0, 142.7, 136.5, 132.4, 130.3, 130.1, 128.4, 128.0, 125.2, 121.5, 118.8, 104.7, 53.1, 35.1, 35.0, 34.0, 33.9, 31.9, 31.8, 19.9; LC-APCI-MS (M+H)+ calcd for C25H30NO4 408.2175, found 408.2169.
4-(1-(1,2,3,4-Tetrahydro-1,1,4,4,6-pentamethylnaphthalen-7-yl)vinyl)-2-nitrobenzoic Acid (16)
Compound 16 was synthesized following the representative procedure for the synthesis of compound 12. To a 100 mL round bottom flask charged with 53 (0.099 g, 0.24 mmol) and methanol (5 mL) was added a 5M aqueous solution of potassium hydroxide (0.12 mL, 0.61 mmol). A reflux condenser was fitted to the round bottom flask and the reaction solution was refluxed and monitored by TLC. After 1 h at reflux, the reaction solution was cooled to room temperature and quenched with 20% HCl (15 mL). The aqueous solution was extracted with ethyl acetate (50 mL, twice) and the organic extracts were combined, washed with water and brine, dried over sodium sulfate, and concentrated in vacuo to give crude 16. Crude 16 was purified by column chromatography (25 mL SiO2, ethyl acetate) to give 16 (0.077 g, 80%) as an off-white crystalline solid, m.p. 212–214 °C: 1H NMR (400 MHz, CDCl3) δ 8.60 (br s, 1H), 7.86 (d, J = 8, 1H), 7.71 (s, 1H), 7.52 (d, J = 8, 1H), 7.12 (s, 1H), 7.11 (s, 1H), 5.92 (s, 1H), 5.47 (s, 1H), 1.98 (s, 3H), 1.72 (s, 4H), 1.32 (s, 6H), 1.29 (s, 6H); 13C NMR (100.6 MHz, CDCl3) δ 169.9, 150.1, 147.3, 146.8, 145.4, 143.0, 136.7, 132.7, 131.2, 130.1, 128.7, 128.3, 123.6, 121.7, 119.4, 35.4, 35.3, 34.3, 34.2, 32.1, 32.0, 20.2; LC-APCI-MS (M-H)- calcd for C24H26NO4 392.1862, found 392.1872. Anal. Calcd for C24H27O4N: C 73.26; H 6.92; N 3.56. Found: C 72.92; H 6.71; N 3.76.
4-(1-(3,5,5,8,8-pentamethyl-5,6,7,8-tetrahydronaphthalen-2-yl)carbonyl)-2-nitrobenzoic Acid (17)
Compound 17 was synthesized following the representative procedure for the synthesis of compound 13. To a 100 mL round bottom flask charged with 52 (0.30 g, 0.73 mmol) and methanol (5 mL) was added a 5M aqueous solution of potassium hydroxide (0.37 mL, 1.85 mmol). A reflux condenser was fitted to the round bottom flask and the reaction solution was refluxed and monitored by TLC. After 1 h at reflux, the reaction solution was cooled to room temperature and quenched with 20% HCl (20 mL). The aqueous solution was extracted with ethyl acetate (25 mL, twice) and the organic extracts were combined, washed with water and brine, dried over sodium sulfate, and concentrated in vacuo to give crude 17. Crude 17 was purified by column chromatography (25 mL SiO2, ethyl acetate) to give 17 (0.28 g, 97%) as a yellow crystalline solid, m.p. 167–169 °C: 1H NMR (400 MHz, CDCl3) δ 9.00 (br s, 1H), 8.29 (s, 1H), 8.11 (d, J = 7.5, 1H), 7.97 (d, J = 7.5, 1H), 7.26 (m, 2H), 2.39 (s, 3H), 1.71 (s, 4H), 1.33 (s, 6H), 1.22 (s, 6H); 13C NMR (100.6 MHz, CDCl3) δ 194.7, 169.2, 149.5, 148.5, 142.4, 142.0, 135.2, 133.7, 133.0, 130.4, 130.0, 129.2, 128.7, 125.2, 34.8, 34.7, 34.4, 33.9, 31.7, 31.5, 31.4, 20.1. Anal. Calcd for C23H25O5N: C 69.86; H 6.37; N 3.54. Found: C 68.35; H 6.75; N 3.34.
Methyl 3-fluoro-4-(1-(3,5,5,8,8-pentamethyl-5,6,7,8-tetrahydronaphthalen-2-yl)carbonyl)benzoate (50)
Compound 50 was synthesized following the representative procedure for the synthesis of compound 29. Methyl 4-(chlorocarbonyl)-3-fluorobenzoate (42) was synthesized by refluxing 4-(methoxycarbonyl)-2-fluorobenzoic acid (41) (1.34 g, 6.76 mmol) in thionyl chloride (12.0 mL, 165 mmol) in a 100 mL one-neck round bottom flask fitted with a water cooled reflux condenser. Excess thionyl chloride was removed in vacuo to give crude 42 as an off-white solid, and this solid was dissolved in dry benzene (ca. 20 mL) and evaporated to dryness three times to remove residual thionyl chloride. The acid chloride 42 was dried on high vacuum to remove residual benzene. To a 2-neck, 50 mL round bottom flask equipped with a reflux condenser and magnetic stir-bar was added 24 (1.35 g, 6.67 mmol) followed by a solution of crude acid chloride 42 (6.25 mmol) in DCM (15 mL). Aluminum chloride (2.0 g, 15 mmol) was added to the reaction solution at room temperature slowly, with stirring, and the reaction solution turned from colorless to red accompanied by the evolution of gas and heat. The reaction was stirred for 5 min then heated to reflux for 15 min. The reaction was judged to be complete by TLC, and the solution was poured into an ice solution (25 mL) acidified with a 20% HCl solution (8 mL) and ethyl acetate was added (13 mL). The aqueous and organic layers were separated, and the aqueous layer was extracted with ethyl acetate (15 mL, twice). The combined organics were washed with water and brine, dried over sodium sulfate, filtered and rotevapped to give crude 50. Crude 50 was purified by column chromatography (250 mL SiO2, hexanes:ethyl acetate 95:5 to 92.5:7.5) to give 50 (2.50 g, 97%) as a colorless crystalline solid, m.p. 114–117 °C: 1H NMR (400 MHz, CDCl3) δ 7.90 (d, J = 8.0, 1H), 7.80 (d, J = 10.1, 1H), 7.60 (t, J = 7.4, 1H), 7.32 (s, 1H), 7.20 (s, 1H), 3.96 (s, 3H), 2.51 (s, 3H), 1.67 (s, 4H), 1.29 (s, 6H), 1.14 (s, 6H); 13C NMR (100.6 MHz, CDCl3) δ 193.9, 165.4, 165.3, 161.1, 158.6, 149.9, 142.2, 136.0, 134.4, 134.3, 134.0, 132.4, 132.3, 131.0, 130.9, 130.3, 129.9, 125.2, 125.1, 117.5, 117.3, 52.6, 34.8, 34.7, 34.4, 33.8, 31.5, 31.4, 20.9; LC-APCI-MS (M+H)+ calcd for C24H28O3F 383.2023, found 383.2021.
Methyl 3-fluoro-4-(1-(1,2,3,4-tetrahydro-1,1,4,4,6-pentamethylnaphthalen-7-yl)vinyl)benzoate (54)
Compound 54 was synthesized following a slightly modified procedure for the synthesis of compound 30. To a 20-dram vial containing 50 (0.79 g, 2.07 mmol) and dry THF (3 mL) at room temperature was slowly added a triphenylphosphonium methylide solution prepared as follows: to a 20-dram vial equipped with a Teflon magnetic stir-bar and contining dry THF (2.0 mL) was added iPr2NH (0.66 mL, 4.67 mmol) and a 2.5M solution of n-butyl lithium in hexanes (1.7 mL, 4.25 mmol), and the solution was stirred for 30 min at room temperature at which point, methyl triphenylphosphonium bromide (1.13 g, 3.19 mmol) was added and the solution was stirred an additional 20 min to provide a homogeneous dark yellow ylide solution. The reaction was monitored by TLC, and when the reaction was judged to be complete, the reaction solution was poured into water (50 mL) and the aqueous solution was extracted with ethyl acetate (50 mL, twice). The combined organic extracts were washed with water and brine, dried over sodium sulfate, and concentrated in vacuo to give crude 54 which was purified by column chromatography (25 mL SiO2, hexanes:ethyl acetate 97.5:2.5) to give 54 (0.254 g, 32%) as a white solid, m.p. 107–109 °C: 1H NMR (400 MHz, CDCl3) δ 7.73 (d, J = 10.8, 1H), 7.70 (d, J = 7.8, 1H), 7.15 (s, 1H), 7.10 (t, J = 8.0, 1H), 7.05 (s, 1H), 5.86 (s, 1H), 5.56 (s, 1H), 3.91 (s, 3H), 1.98 (s, 3H), 1.69 (s, 4H), 1.29 (s, 6H), 1.28 (s, 6H); 13C NMR (100.6 MHz, CDCl3) δ 165.9, 165.8, 161.0, 158.5, 144.3, 143.5, 142.3, 138.3, 133.8, 133.7, 132.3, 130.7, 130.6, 130.5, 130.4, 128.0, 127.8, 125.0, 124.9, 121.3, 121.2, 117.2, 117.0, 52.2, 35.2, 35.1, 33.9, 33.8, 31.9, 31.8, 19.7; LC-APCI-MS (M+H)+ calcd for C25H30O2F 381.2230, found 381.2220.
3-Fluoro-4-(1-(1,2,3,4-tetrahydro-1,1,4,4,6-pentamethylnaphthalen-7-yl)vinyl)benzoic acid (18)
Compound 18 was synthesized following the representative procedure for the synthesis of compound 12. To a 100 mL round bottom flask charged with 54 (0.2541 g, 0.90 mmol) and methanol (5 mL) was added a 5M aqueous solution of potassium hydroxide (0.35 mL, 1.74 mmol). A reflux condenser was fitted to the round bottom flask and the reaction solution was refluxed and monitored by TLC. After 1 h at reflux, the reaction solution was cooled to room temperature and quenched with 20% HCl (42 mL). The aqueous solution was extracted with ethyl acetate (50 mL, twice) and the organic extracts were combined, washed with water and brine, dried over sodium sulfate, and concentrated in vacuo to give crude 18. Crude 18 was purified by column chromatography (25 mL SiO2, hexanes:ethyl acetate 9:1) to give 18 (0.24 g, 98%) as a white crystalline solid, m.p. 188–189 °C: 1H NMR (400 MHz, CDCl3) δ 10.80 (br s, 1H), 7.80 (d, J = 9.9, 1H), 7.78 (d, J = 7.0, 1H), 7.15 (s, 1H), 7.13 (t, J = 8.1, 1H), 7.07 (s, 1H), 5.89 (s, 1H), 5.59 (s, 1H), 1.99 (s, 3H), 1.70 (s, 4H), 1.30 (s, 6H), 1.29 (s, 6H); 13C NMR (100.6 MHz, CDCl3) δ 171.0, 170.9, 161.0, 158.5, 144.4, 143.4, 142.4, 138.2, 134.8, 134.7, 132.3, 130.8, 130.7, 129.5, 129.4, 128.1, 127.8, 125.7, 125.6, 121.7, 121.6, 117.8, 117.6, 35.1, 33.9, 33.8, 31.9, 31.8, 19.7; LC-APCI-MS (M+H)+ calcd for C24H28O2F 367.2073, found 367.2032. Anal. Calcd for C24H27O2F: C 78.66; H 7.43; F 5.18. Found: C 78.34; H 7.52; F 4.8.
3-Fluoro-4-(1-(3,5,5,8,8-pentamethyl-5,6,7,8-tetrahydronaphthalen-2-yl)carbonyl)benzoic acid (19)
Compound 19 was synthesized following the representative procedure used to synthesize compound 13. To a 100 mL round bottom flask charged with 50 (0.50 g, 1.31 mmol) and methanol (5 mL) was added a 5M aqueous solution of potassium hydroxide (0.74 mL, 3.71 mmol). A reflux condenser was fitted to the round bottom flask and the reaction solution was refluxed and monitored by TLC. After 1 h at reflux, the reaction solution was cooled to room temperature and quenched with 20% HCl (20 mL). The aqueous solution was extracted with ethyl acetate (25 mL, twice) and the organic extracts were combined, washed with water and brine, dried over sodium sulfate, and concentrated in vacuo to give crude 19. Crude 19 was purified by column chromatography (25 mL SiO2, hexanes:ethyl acetate 4:1 to 3:2) to give 19 (0.409 g, 84%) as a white crystalline solid, m.p. 228–229 °C: 1H NMR (400 MHz, DMSO-d6) δ 13.58 (br s, 1H), 7.88 (d, J = 8.0, 1H), 7.76 (d, J = 10.4, 1H), 7.64 (t, J = 7.4, 1H), 7.32 (s, 1H), 7.28 (s, 1H), 2.40 (s, 3H), 1.61 (s, 4H), 1.26 (s, 6H), 1.09 (s, 6H); 13C NMR (100.6 MHz, DMSO-d6) δ 193.5, 165.6, 160.4, 157.9, 149.4, 141.9, 135.6, 135.5, 135.1, 133.9, 131.4, 131.3, 131.0, 129.7, 129.5, 125.3, 116.9, 116.6, 34.2, 34.1, 33.4, 31.2, 31.1, 20.3; LC-APCI-MS (M+H)+ calcd for C23H26O3F 369.1866, found 369.1877. Anal. Calcd for C23H25O3F: C 74.98; H 6.84; F 5.16. Found: C 74.92; H 7.21; F 4.9.
methyl 2-fluoro-4-(1-(3,5,5,8,8-pentamethyl-5,6,7,8-tetrahydronaphthalen-2-yl)carbonyl)benzoate (51)
Compound 51 was synthesized following the representative procedure for the synthesis of compound 29. Methyl 4-(chlorocarbonyl)-2-fluorobenzoate (47) was synthesized by refluxing 4-(methoxycarbonyl)-3-fluorobenzoic acid (46) (1.21 g, 6.11 mmol) in thionyl chloride (12.0 mL, 165 mmol) in a 100 mL one-neck round bottom flask fitted with a water-cooled reflux condenser. Excess thionyl chloride was removed in vacuo to give crude 47 as an off-white solid, and this solid was dissolved in dry benzene (ca. 20 mL) and evaporated to dryness three times to remove residual thionyl chloride. The acid chloride 47 was dried on high vacuum to remove residual benzene. To a 2-neck, 50 mL round bottom flask equipped with a reflux condenser and magnetic stir-bar was added 24 (1.34 g, 6.61 mmol) followed by a solution of crude acid chloride 47 (6.11 mmol) in DCM (15 mL). Aluminum chloride (2.0 g, 15 mmol) was added to the reaction solution at room temperature slowly, with stirring, and the reaction solution turned from colorless to red accompanied by the evolution of gas and heat. The reaction was stirred for 5 min then heated to reflux for 15 min. The reaction was judged to be complete by TLC, and the solution was poured into an ice solution (25 mL) acidified with a 20% HCl solution (8 mL) and ethyl acetate was added (13 mL). The aqueous and organic layers were separated, and the aqueous layer was extracted with ethyl acetate (15 mL, twice). The combined organics were washed with water and brine, dried over sodium sulfate, filtered and concentrated to give crude 51. Crude 51 was purified by column chromatography (250 mL SiO2, hexanes:ethyl acetate 95:5 to 92.5:7.5) to give 51 (1.76 g, 75%) as a white, crystalline solid, m.p. 105–106 °C: 1H NMR (400 MHz, CDCl3) δ 8.00 (t, J = 7.4, 1H), 7.59 (d, J = 8.4, 1H), 7.56 (d, J = 11.6, 1H), 7.24 (s, 1H), 7.21 (s, 1H), 3.96 (s, 3H), 2.34 (s, 3H), 1.69 (s, 4H), 1.31 (s, 6H), 1.20 (s, 6H); 13C NMR (100.6 MHz, CDCl3) δ 196.1, 164.3, 164.2, 162.8, 160.2, 148.8, 143.9, 143.8, 142.0, 134.7, 134.0, 132.1, 129.5, 128.4, 125.3, 125.2, 121.9, 121.8, 118.3, 118.1, 52.6, 34.8, 34.7, 34.3, 33.8, 31.6, 31.5, 20.0; LC-APCI-MS (M+H)+ calcd for C24H28O3F 383.2023, found 383.2036.
methyl 2-fluoro-4-(1-(1,2,3,4-tetrahydro-1,1,4,4,6-pentamethylnaphthalen-7-yl)vinyl)benzoate (55)
Compound 55 was synthesized following a representative procedure for the synthesis of compound 54. To a 20-dram vial containing 51 (0.79 g, 2.07 mmol) and dry THF (3 mL) at room temperature was slowly added a triphenylphosphonium methylide solution prepared as follows: to a 20-dram vial equipped with a Teflon magnetic stir-bar and contining dry THF (2.0 mL) was added iPr2NH (0.66 mL, 4.67 mmol) and a 2.5M solution of n-butyl lithium in hexanes (1.7 mL, 4.25 mmol), and the solution was stirred for 30 min at room temperature at which point, methyl triphenylphosphonium bromide (1.13 g, 3.19 mmol) was added and the solution was stirred an additional 20 min to provide a homogeneous dark yellow ylide solution. The reaction was monitored by TLC, and when the reaction was judged to be complete, the reaction solution was poured into water (50 mL) and the aqueous solution was extracted with ethyl acetate (50 mL, twice). The combined organic extracts were washed with water and brine, dried over sodium sulfate, and concentrated in vacuo to give crude 55 which was purified by column chromatography (25 mL SiO2, hexanes:ethyl acetate 97.5:2.5) to give 54 (0.301 g, 38%) as a white solid, m.p. 130–132 °C: 1H NMR (400 MHz, CDCl3) δ 7.86 (t, J = 8.0, 1H), 7.14 (d, J = 8.2, 1H), 7.12 (d, J = 6.0, 1H), 7.02 (d, J = 12.4, 1H), 5.82 (s, 1H), 5.36 (s, 1H), 3.93 (s, 3H), 1.95 (s, 3H), 1.70 (s, 4H), 1.31 (s, 6H), 1.27 (s, 6H); 13C NMR (100.6 MHz, CDCl3) δ 164.8, 164.7, 163.3, 160.7, 148.1, 147.8, 147.7, 144.6, 142.4, 137.2, 132.6, 132.0, 128.1, 128.0, 122.0, 117.7, 117.1, 117.0, 115.0, 114.8, 52.2, 35.2, 35.1, 33.9, 33.8, 31.9, 31.8, 19.8; LC-APCI-MS (M+H)+ calcd for C25H30O2F 381.2230, found 381.2254.
2-fluoro-4-(1-(1,2,3,4-tetrahydro-1,1,4,4,6-pentamethylnaphthalen-7-yl)vinyl)benzoic acid (20)
Compound 20 was synthesized following the representative procedure for the synthesis of compound 12. To a 100 mL round bottom flask charged with 55 (0.25 g, 0.66 mmol) and methanol (5 mL) was added a 5M aqueous solution of potassium hydroxide (0.35 mL, 1.76 mmol). A reflux condenser was fitted to the round bottom flask and the reaction solution was refluxed and monitored by TLC. After 1 h at reflux, the reaction solution was cooled to room temperature and quenched with 20% HCl (42 mL). The aqueous solution was extracted with ethyl acetate (50 mL, twice) and the organic extracts were combined, washed with water and brine, dried over sodium sulfate, and concentrated in vacuo to give crude 20. Crude 20 was purified by column chromatography (25 mL SiO2, hexanes:ethyl acetate 9:1) to give 12 (0.24 g, 99%) as a white crystalline solid, m.p. 212–214 °C: 1H NMR (400 MHz, CDCl3) δ 11.12 (br s, 1H), 7.97 (t, J = 7.9, 1H), 7.18 (d, J = 8.3, 1H), 7.11 (s, 1H), 7.10 (s, 1H), 7.06 (d, J = 12.3, 1H), 5.86 (s, 1H), 5.39 (s, 1H), 1.96 (s, 3H), 1.71 (s, 4H), 1.31 (s, 6H), 1.28 (s, 6H); 13C NMR (100.6 MHz, CDCl3) δ 169.5, 169.4, 164.0, 161.4, 149.0, 148.9, 148.0, 144.7, 142.5, 137.1, 132.7, 132.6, 128.1, 128.0, 122.2, 122.1, 118.1, 115.9, 115.8, 115.1, 114.9, 35.2, 35.1, 34.0, 33.8, 31.9, 31.8, 19.8; LC-APCI-MS (M+H)+ calcd for C24H28O2F 367.2073, found 367.2097. Anal. Calcd for C24H27O2F: C 78.66; H 7.43; F 5.18. Found: C 78.45; H 7.45; F 5.0.
2-fluoro-4-(1-(3,5,5,8,8-pentamethyl-5,6,7,8-tetrahydronaphthalen-2-yl)carbonyl)benzoic acid (21)
Compound 21 was synthesized following the representative procedure for the synthesis of compound 13. To a 100 mL round bottom flask charged with 51 (0.5065 g, 1.34 mmol) and methanol (5 mL) was added a 5M aqueous solution of potassium hydroxide (0.73 mL, 3.66 mmol). A reflux condenser was fitted to the round bottom flask and the reaction solution was refluxed and monitored by TLC. After 1 h at reflux, the reaction solution was cooled to room temperature and quenched with 20% HCl (20 mL). The aqueous solution was extracted with ethyl acetate (25 mL, twice) and the organic extracts were combined, washed with water and brine, dried over sodium sulfate, and concentrated in vacuo to give crude 21. Crude 21 was purified by column chromatography (25 mL SiO2, hexanes:ethyl acetate 9:1) to give 21 (0.477 g, 97%) as a white crystalline solid, m.p. 174–175 °C: 1H NMR (400 MHz, CDCl3) δ 11.00 (br s, 1H), 8.14 (t, J = 7.5, 1H), 7.63 (d, J = 9.1, 1H), 7.60 (d, J = 12.3, 1H), 7.27 (s, 1H), 7.22 (s, 1H), 2.36 (s, 3H), 1.70 (s, 4H), 1.32 (s, 6H), 1.22 (s, 6H); 13C NMR (100.6 MHz, CDCl3) δ 196.1, 168.3, 168.7, 163.5, 160.9, 148.9, 144.9, 144.8, 142.1, 134.8, 133.8, 132.7, 129.6, 128.5, 125.3, 125.2, 120.7, 120.6, 118.5, 118.3, 34.8, 34.7, 34.4, 33.9, 31.6, 31.5, 20.0; LC-APCI-MS (M+H)+ calcd for C23H26O3F 369.1866, found 369.1867. Anal. Calcd for C23H25O3F: C 74.98; H 6.84; F 5.16. Found: C 74.95; H 6.98; F 5.0.
Supplementary Material
Structure 2.

Structure 3.

Structure 4.
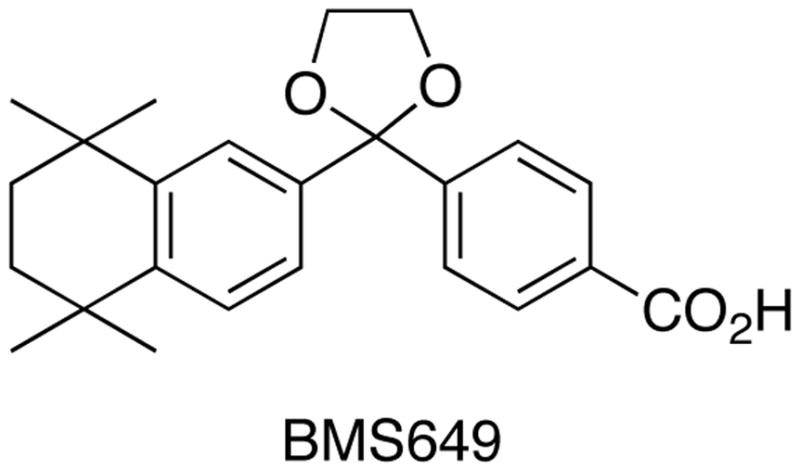
Acknowledgments
We thank the Chemistry Division of the National Science Foundation for financial support of this work (Grant CHE-0741978). Thanks is also given to Dr. Natalya Zolotova of the High-Resolution Mass Spectrometry Lab at ASU Tempe. Belinda V. Miguel was supported in part by the NIH, in the form of a MARC grant, Tempe, AZ. Julie K. Furmick wishes to thank ASU Tempe for a School of Life Sciences Undergraduate Research (SOLUR) fellowship. We also thank Z. Nevin Gerek and J. Spiriti for technical assistance, and the Fulton High Performance Computing Initiative at Arizona State University for computer time. pCMX-hRARα and pTK-RARE(2)-Luc were generous gifts from Dr. Paul Thompson, School of Biomedical Sciences, University of Ulster, Coleraine, UK.
Abbreviations
- RXR
retinoid-X-receptor
- RAR
retinoic-acid-receptor
- CTCL
cutaneous T-cell lymphoma
- RXRE
retinoid-X-receptor element
- HRE
hormone response element
- TR
thyroid receptor
- VDR
vitamin D receptor
- SNuRMs
specific nuclear receptor modulators.
Footnotes
Supporting Information Available: 1H-NMR and 13C-NMR spectra of all compounds reported in the the experimentals as well as X-ray data for compounds 12, 18, 20, 49, 50, and 51. The X-ray data can be found at the Cambridge Crystallographic Data Centre under the identification numbers: 738925-73830. The spectra is available free of charge via the Internet at http://pubs.acs.org.
Contributor Information
Carl E. Wagner, Division of Mathematical and Natural Sciences, New College of Interdisciplinary Arts and Sciences, Arizona State University, 4701 W. Thunderbird Road, Glendale, AZ 85306.
Peter W. Jurutka, Division of Mathematical and Natural Sciences, New College of Interdisciplinary Arts and Sciences, Arizona State University, 4701 W. Thunderbird Road, Glendale, AZ 85306
Pamela A. Marshall, Division of Mathematical and Natural Sciences, New College of Interdisciplinary Arts and Sciences, Arizona State University, 4701 W. Thunderbird Road, Glendale, AZ 85306
Thomas L. Groy, Department of Chemistry and Biochemistry, College of Liberal Arts and Sciences, Arizona State University, Tempe, AZ, 85287
Arjan van der Vaart, Department of Chemistry, University of South Florida, 4202 E Fowler Ave, CHE 205, Tampa, FL, 33620.
Joseph W. Ziller, Department of Chemistry, University of California, Irvine, 576 Rowland Hall, Irvine, CA 92697
Julie K. Furmick, Division of Mathematical and Natural Sciences, New College of Interdisciplinary Arts and Sciences, Arizona State University, 4701 W. Thunderbird Road, Glendale, AZ 85306
Mark E. Graeber, Division of Mathematical and Natural Sciences, New College of Interdisciplinary Arts and Sciences, Arizona State University, 4701 W. Thunderbird Road, Glendale, AZ 85306
Erik Matro, Division of Mathematical and Natural Sciences, New College of Interdisciplinary Arts and Sciences, Arizona State University, 4701 W. Thunderbird Road, Glendale, AZ 85306.
Belinda V. Miguel, Division of Mathematical and Natural Sciences, New College of Interdisciplinary Arts and Sciences, Arizona State University, 4701 W. Thunderbird Road, Glendale, AZ 85306
Ivy T. Tran, Division of Mathematical and Natural Sciences, New College of Interdisciplinary Arts and Sciences, Arizona State University, 4701 W. Thunderbird Road, Glendale, AZ 85306
Jeng Eun S. Kwon, Division of Mathematical and Natural Sciences, New College of Interdisciplinary Arts and Sciences, Arizona State University, 4701 W. Thunderbird Road, Glendale, AZ 85306
Jamie N. Tedeschi, Division of Mathematical and Natural Sciences, New College of Interdisciplinary Arts and Sciences, Arizona State University, 4701 W. Thunderbird Road, Glendale, AZ 85306
Shahram Moosavi, Division of Mathematical and Natural Sciences, New College of Interdisciplinary Arts and Sciences, Arizona State University, 4701 W. Thunderbird Road, Glendale, AZ 85306.
Amina Danishyar, Division of Mathematical and Natural Sciences, New College of Interdisciplinary Arts and Sciences, Arizona State University, 4701 W. Thunderbird Road, Glendale, AZ 85306.
Joshua S. Philp, Division of Mathematical and Natural Sciences, New College of Interdisciplinary Arts and Sciences, Arizona State University, 4701 W. Thunderbird Road, Glendale, AZ 85306
Reina O. Khamees, Division of Mathematical and Natural Sciences, New College of Interdisciplinary Arts and Sciences, Arizona State University, 4701 W. Thunderbird Road, Glendale, AZ 85306
Jevon N. Jackson, Division of Mathematical and Natural Sciences, New College of Interdisciplinary Arts and Sciences, Arizona State University, 4701 W. Thunderbird Road, Glendale, AZ 85306
Darci K. Grupe, Division of Mathematical and Natural Sciences, New College of Interdisciplinary Arts and Sciences, Arizona State University, 4701 W. Thunderbird Road, Glendale, AZ 85306
Syed L. Badshah, Department of Chemistry and Biochemistry, College of Liberal Arts and Sciences, Arizona State University, Tempe, AZ, 85287
Justin W. Hart, Division of Mathematical and Natural Sciences, New College of Interdisciplinary Arts and Sciences, Arizona State University, 4701 W. Thunderbird Road, Glendale, AZ 85306
References
- 1.(a) Mangelsdorf DJ, Umesono K, Evans RM. The Retinoids. Academic Press; Orlando, FL: 1994. The Retinoid Recpetors; pp. 319–349. [Google Scholar]; (b) Leid M, Kastner P, Chambon P. Multiplicity Generates Diversity in the Retinoic Acid Signaling Pathways. Trends Biochem Sci. 1992;17:427–433. doi: 10.1016/0968-0004(92)90014-z. [DOI] [PubMed] [Google Scholar]
- 2.Olefsky JM. Nuclear Receptor Minireview Series. J Biol Chem. 2001;276(40):36863–36864. doi: 10.1074/jbc.R100047200. [DOI] [PubMed] [Google Scholar]
- 3.Forman BM, Yang CR, Au M, Casanova J, Ghysdael J, Samuels HH. A domain containing leucin-zipper-like motifs mediates novel in vitro interactions between the thyroid hormone and retinoic acid receptors. Mol Endocrinol. 1989;3:1610–1626. doi: 10.1210/mend-3-10-1610. [DOI] [PubMed] [Google Scholar]
- 4.Mangelsdorf DJ, Evans RM. The RXR heterodimers and orphan receptors. Cell. 1995;83:841–850. doi: 10.1016/0092-8674(95)90200-7. [DOI] [PubMed] [Google Scholar]
- 5.Thompson PD, Remus LS, Hsieh J-C, Jurutka PW, Whitfield GK, Galligan MA, Encinas Dominguez C, Haussler CA, Haussler MR. Distinct retinoid X receptor activation dunction-2 residues mediate transactivation in homodimeric and vitamin D receptor heterodimeric contexts. J Mol Endocrinol. 2001;27(2):211–227. doi: 10.1677/jme.0.0270211. [DOI] [PubMed] [Google Scholar]
- 6.Nahoum V, Perez E, Germain P, Rodriquez-Barrios F, Manzo F, Kammerer S, Lemaire G, Hirsch O, Royer CA, Gronemeyer H, de Lera AR, Bourguet W. Modulators of the structural dynamics of the retinoid X receptor to reveal receptor function. PNAS. 2007;104:17323–17328. doi: 10.1073/pnas.0705356104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Altucci L, Leibowitz MD, Ogilvie KM, de Lera AR, Gronemeyer H. RAR and RXR modulation in cancer and metabolic disease. Nature Rev Drug Discov. 2007;6:793–810. doi: 10.1038/nrd2397. [DOI] [PubMed] [Google Scholar]
- 8.Lehmann JM, Jong L, Fanjul A, Cameron JF, Lu XP, Haefner P, Dawson MI, Pfahl M. Retinoids selective for retinoid X receptor pathways. Science. 1992;258:1944–1946. doi: 10.1126/science.1335166. [DOI] [PubMed] [Google Scholar]
- 9.Boehm MF, Zhang L, Badea BA, White SK, Mais DE, Berger E, Suto CM, Goldman ME, Heyman RA. Synthesis and Structure-Activity Relationships of Novel Retinoid X Receptor-Selective Retinoids. J Med Chem. 1994;37:2930–2941. doi: 10.1021/jm00044a014. [DOI] [PubMed] [Google Scholar]
- 10.(a) Jong L, Lehmann JM, Hobbs PD, Harlev E, Huffman JC, Pfahl M, Dawson MI. Conformational effects on retinoid receptor selectivity. 1. Effect of 9-double bond geometry on retinoid X receptor activity. J Med Chem. 1993;36:2605–2613. doi: 10.1021/jm00070a003. [DOI] [PubMed] [Google Scholar]; (b) Dawson MI, Jong L, Hobbs PD, Cameron JF, Chao W-R, Pfahl M, Lee M-O, Shroot B, Pfahl M. Conformational Effects on Retinoid Receptor Selectivity 2 Effects of Retinoid Bridging Group on Retinoid X Receptor Activity and Selectivity. J Med Chem. 1995;38:3368–3383. doi: 10.1021/jm00017a021. [DOI] [PubMed] [Google Scholar]
- 11.Daiss JO, Burschka C, Mills JS, Montana JG, Showell GA, Fleming I, Gaudon C, Ivanova D, Gronemeyer H, Tacke R. Synthesis, Crystal Structure Analysis, and Pharmacological Characterization of Disila-bexarotene, a Disila-Analogue of the RXR-Selective Retinoid Agonist Bexarotene. Organometallics. 2005;24:3192–3199. [Google Scholar]
- 12.Yen W-C, Prudente RY, Lamph WW. Synergistic effect of a retinoid X receptor-selective ligand bexarotene (LGD1069, Targretin) and paclitaxel (Taxol) in mammary carcinoma. Breast Cancer Res Treat. 2004;88:141–148. doi: 10.1007/s10549-004-1426-5. [DOI] [PubMed] [Google Scholar]
- 13.Yen WC, Corpuz MR, Prudente RY, Cooke TA, Bissonnette RP, Negro-Vilar A, Lamph WW. A Selective Retinoid X Receptor Agonist Bexarotene (Targretin) Prevents and Overcomes Acquired Paclitaxel (Taxol) Resistance in Human Non-Small Cell Lung Cancer. Clin Cancer Res. 2004;10:8656–8664. doi: 10.1158/1078-0432.CCR-04-0979. [DOI] [PubMed] [Google Scholar]
- 14.Cesario RM, Stone J, Yen WC, Bissonnette RP, Lamph WW. Differentiation and growth inhibition mediated via the RXR:PPARγ heterodimer in colon cancer. Cancer Letters. 2006;240:225–233. doi: 10.1016/j.canlet.2005.09.010. [DOI] [PubMed] [Google Scholar]
- 15.Mukherjee R, Davies PJA, Crombie DL, Bischoff ED, Cesario RM, Jow L, Hamanns LG, Boehm MF, Mondon CE, Nadzan AM, Paterniti JR, Heyman RA. Sensitization of diabetic and obese mice to insulin by retinoid X receptor agonists. Nature. 1997;386:407–410. doi: 10.1038/386407a0. [DOI] [PubMed] [Google Scholar]
- 16.Sherman SI, Gopal J, Haugen BR, Chiu AC, Whaley K, Nowlakha P, Duvic M. Central hypothyroidism associated with retinoid X receptor-selective ligands. N Engl J Med. 1999;340:1075–1079. doi: 10.1056/NEJM199904083401404. [DOI] [PubMed] [Google Scholar]
- 17.Li D, Li T, Wang F, Tian H, Samuels HH. Functional Evidence for Retinoid X Receptor (RXR) as a Nonsilent Partner in the Thyroid Hormone Receptor/RXR Heterodimer. Mol Cell Biol. 2002;22:5782–5792. doi: 10.1128/MCB.22.16.5782-5792.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Qing FL, Fan J. A Suzuki coupling approach to the trifluoromethyl derivative of Targretin (LGD 1069) Bioorg Med Chem Lett. 1997;7:2117–2120. [Google Scholar]
- 19.Vuligonda V, Thacher SM, Chandraratna RAS. Enantioselective Synthesis of Potent Retinoid X Receptor Ligands: Differential Biological Activities of Individual Antipodes. J Med Chem. 2001;44:2298–2303. doi: 10.1021/jm0100584. [DOI] [PubMed] [Google Scholar]
- 20.Farmer LJ, Marron KS, Canan Koch SS, Hwang CK, Kallel EA, Zhi L, Nadzan AM, Robertson DW, Bennani YL. Aza-retinoids as novel retinoid X receptor-specific agonists. Bioorg Med Chem Lett. 2006;16:2352–2356. doi: 10.1016/j.bmcl.2005.12.003. [DOI] [PubMed] [Google Scholar]
- 21.Kagechika H, Kawachi E, Hashimoto Y, Himi T, Shudo K. Retinobenzoic Acids. 1. Structure-Activitiy Relationships of Aromatic Amides with Retinoidal Activity. J Med Chem. 1988;31:2182–2192. doi: 10.1021/jm00119a021. [DOI] [PubMed] [Google Scholar]
- 22.Winum JY, Baghdiguian S, Commes T, Leydet A, Montero JL. Synthesis of New Targretin® Analogues that Induce Apoptosis in Leukemia HL-60 Cells. Bioorg Med Chem Lett. 2002;12:3529–3532. doi: 10.1016/s0960-894x(02)00803-x. [DOI] [PubMed] [Google Scholar]
- 23.Boehm MF, Zhang L, Zhi L, McClurg MR, Berger E, Wagoner M, Mais DE, Suto CM, Davies PJA, Heyman RA, Nadzan AM. Design and Synthesis of Potent Retinoid X Recptor Selective Ligands That Induce Apoptosis in Leukemia Cells. J Med Chem. 1995;38:3146–3155. doi: 10.1021/jm00016a018. [DOI] [PubMed] [Google Scholar]
- 24.Faul MM, Ratz AM, Sullivan KA, Trankle WG, Winneroski LL. Synthesis of Novel Retinoid X Receptor-Selective Retinoids. J Org Chem. 2001;66:5772–5782. doi: 10.1021/jo0103064. [DOI] [PubMed] [Google Scholar]
- 25.Michellys PY, Ardecky RJ, Chen JH, Crombie DL, Etgen GJ, Faul MM, Faulkner AL, Grese TA, Heyman RA, Karanewsky DS, Klausing K, Leibowitz MD, Liu S, Mais DA, Mapes CM, Marschke KB, Reifel-Miller A, Ogilvie KM, Rungta D, Thompson AW, Tyhonas JS, Boehm MF. Novel (2E,4E,6Z)-7-(2-Alkoxy-3,5-dialkylbenzene)-3-methylocta-2,4,6-trienoic Acid Retinoid X Receptor Modulators are Active in Models of Type 2 Diabetes. J Med Chem. 2003;46:2683–2696. doi: 10.1021/jm020340q. [DOI] [PubMed] [Google Scholar]
- 26.Michellys P-Y, Ardecky RJ, Chen J-H, D’Arrigo J, Grese TA, Karanewsky DS, Leibowitz MD, Liu S, Mais DA, Mapes CM, Montrose-Rafizadeh C, Ogilvie KM, Reifel-Miller A, Rungta D, Thompson AW, Tyhonas JS, Boehm MF. Design, Synthesis, and Structure-Activity Relationship Studies of Novel 6,7-Locked-[7-(2-alkoxy-3,5-dialkylbenzene)-3-methylocta]-2,4,6-trienoic Acids. J Med Chem. 2003;46:4087–4103. doi: 10.1021/jm020401k. [DOI] [PubMed] [Google Scholar]
- 27.Michellys PY, Darrigo J, Grese TA, Karanewsky DS, Leibowitz MD, Mais DA, Mapes CM, Reifel-Miller A, Rungta D, Boehm MF. Design, synthesis and structure-activity relationship of novel RXR-selective modulators. Bioorg Med Chem Lett. 2004;14:1593–1598. doi: 10.1016/j.bmcl.2003.12.089. [DOI] [PubMed] [Google Scholar]
- 28.(a) Gernert DL, Ajamie R, Ardecky RA, Bell MG, Leibowitz MD, Mais DA, Mapes CM, Michellys PY, Rungta D, Reifel-Miller A, Tyhonas JS, Yumibe N, Grese TA. Design and Synthesis of Fluorinated RXR Modulators. Bioog Med Chem Lett. 2003;13:3191–3195. doi: 10.1016/s0960-894x(03)00703-0. [DOI] [PubMed] [Google Scholar]; (b) Michellys P-Y, Boehm MF, Chen J-H, Grese TA, Karanewsky DS, Leibowitz MD, Liu S, Mais DA, Mapes CM, Reifel-Miller A, Ogilvie KM, Rungta D, Thompson AW, Tyhonas JS, Yumbie N, Ardecky RJ. Design and Synthesis of Novel RXR-Selective Modulators with Improved Pharmacological Profile. Biorg Med Chem Lett. 2003;13:4070–4075. doi: 10.1016/j.bmcl.2003.08.048. [DOI] [PubMed] [Google Scholar]
- 29.Lehmann JM, Jong L, Fanjul A, Cameron JF, Lu XP, Haefner P, Dawson MI, Pfahl M. Retinoids Selective for Retinoid X Receptor Response Pathways. Science. 1992;258:1944–1946. doi: 10.1126/science.1335166. [DOI] [PubMed] [Google Scholar]
- 30.Dimick SM, Powell SC, McMahon SA, Moothoo DN, Naismith JH, Toone EJ. On the Meaning of Affinity: Cluster Glycoside Effects and Concanavalin A. J Am Chem Soc. 1999;121:10286–10296. [Google Scholar]
- 31.Keenan RM, Callahan JF, Samanen JM, Bondinell WE, Calvo RR, Chen L, DeBrosse C, Egglesont DS, Haltiwanger RC, Hwang SM, Jakas DR, Ku TW, Miller WH, Newlander KA, Nichols A, Parker MF, Southhall LS, Uzinskas I, Vasko-Moser JA, Venslavsky JW, Wong AS, Huffman WF. Conformational Preferences in a Benzodiazepine Series of Potent Nonpeptide Fibrogen Receptor Antagonists. J Med Chem. 1999;42:545–559. doi: 10.1021/jm980166z. [DOI] [PubMed] [Google Scholar]
- 32.Sakaki J, Kishida M, Konishi K, Gunji H, Toyao A, Matsumoto Y, Kanazawa T, Uchiyama H, Fukaya H, Mitani H, Arai Y, Kimura M. Synthesis and structure-activity relationship of novel RXR antagonists: Orally active anti-diabetic and anti-obesity agents. Bioorg & Med Chem Lett. 2007;17:4804–4807. doi: 10.1016/j.bmcl.2007.06.080. [DOI] [PubMed] [Google Scholar]
- 33.Koch SSC, Dardashti LJ, Hebert JJ, White SK, Croston GE, Flatten KS, Heyman RA, Nadzan AM. Identification of the First Retinoid X Receptor Homodimer Antagonist. J Med Chem. 1996;39:3229–3234. doi: 10.1021/jm960311d. [DOI] [PubMed] [Google Scholar]
- 34.Zhang C, Hazarika P, Ni X, Weidner DA, Duvic M. Induction of Apoptosis by Bexarotene in Cutaneous T-Cell Lymphoma Cells. Clin Cancer Res. 2002;8:1234–1240. [PubMed] [Google Scholar]
- 35.Budihardjo I, Holt O, Lutter M, Luo X, Wang X. Biochemical Pathways of Caspase Activation During Apoptosis. Ann Rev Cell Develop Biol. 1999;15:269–290. doi: 10.1146/annurev.cellbio.15.1.269. [DOI] [PubMed] [Google Scholar]
- 36.Yen WC, Prudente RY, Corpuz MR, Negro-Villar A, Lamph WW. A selective retinoid X receptor agonist bexarotene (LGD1069, targretin) inhibits angiogenesis and metastasis in solid tumours. Br J Cancer. 2006;94:654–660. doi: 10.1038/sj.bjc.6602995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.(a) Zimmerman FK. Procedures used in the induction of mitotic recombination and mutation in the yeast Saccharomyces cerevisiae. Mutat Res. 1975;31:71–86. doi: 10.1016/0165-1161(75)90069-2. [DOI] [PubMed] [Google Scholar]; (b) Zimmermann FK, Kern R, Rasenberger H. A yeast strain for simultaneous detection of mitotic crossing over, mitotic gene conversion, and reverse mutation. Mutat Res. 1975;28:381–388. [Google Scholar]
- 38.Marshall PA. Using Saccharomyces cerevisiae to Test the Mutagenicity of Household Compounds: An Open Ended Hyothesis-Driven Teaching Lab. CBE-LSE. 2007;6:307–315. doi: 10.1187/cbe.06-12-0204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Egea PF, Mitschler A, Moras D. Molecular Recognition of Agonist Ligands by RXRs. Mol Endocrin. 2002;16:987–997. doi: 10.1210/mend.16.5.0823. [DOI] [PubMed] [Google Scholar]
- 40.Forman BM, Umesono K, Chen J, Evans RM. Unique response pathways are established by allosteric interactions among nuclear hormone receptors. Cell. 1995;81:541–550. doi: 10.1016/0092-8674(95)90075-6. [DOI] [PubMed] [Google Scholar]
- 41.Pangborn AB, Giardello MA, Grubbs RH, Rosen RK, Timmers FJ. Safe and Convenient Procedure for Solvent Purification. Organometallics. 1996;15:1518–1520. [Google Scholar]
- 42.Condon FE. Aluminum Chloride-Catalyzed Formation of 2,5-Dichloro-2,5-dimethylhexane from tert-Butyl Chloride. J Org Chem. 1956;21:761–763. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.