

Abstract
Mutations in the androgen receptor (AR) that enable activation by antiandrogens occur in hormone-refractory prostate cancer, suggesting mutant ARs are selected by treatment. To validate this hypothesis, we compared AR variants in metastases obtained by rapid autopsy of patients treated with flutamide or bicalutamide, or by excision of lymph node metastases from hormone-naïve patients. AR mutations occurred at low levels in all specimens, reflecting genetic heterogeneity of prostate cancer. Base changes recurring in multiple samples or multiple times per sample were considered putative selected mutations. Of 26 recurring missense mutations, most in the N-terminal domain (NTD) occurred in multiple tumors, while those in the ligand binding domain (LBD) were case-specific. Hormone-naïve tumors had few recurring mutations and none in the LBD. Several AR variants were assessed for mechanisms that might underlie treatment resistance. Selection was evident for the promiscuous receptor AR-V716M, which dominated three metastases from one flutamide-treated patient. For the inactive cytoplasmically restricted splice variant AR23, co-expression with AR enhanced ligand response, supporting a decoy function. A novel NTD mutation, W435L, in a motif involved in intramolecular interaction influenced promoter-selective, cell-dependent transactivation. AR-E255K, mutated in a domain that interacts with an E3 ubiquitin ligase, led to increased protein stability and nuclear localization in the absence of ligand. Thus treatment with antiandrogens selects for gain-of-function AR mutations with altered stability, promoter preference, or ligand specificity. These processes reveal multiple targets for effective therapies regardless of AR mutation.
Keywords: androgen receptor, prostate cancer, antiandrogen resistance, flutamide, bicalutamide
INTRODUCTION
Tumors arise through the accumulation of somatic mutations that allow uncontrolled growth and lead to general genomic instability and acquisition of random mutations (1). This creates a heterogeneous tumor population that is able to adapt to changes in environment (2). In the case of prostate cancer, this “mutator phenotype” may contribute to the relatively rapid development of treatment resistance.
Because prostate cancer is initially androgen responsive, standard treatment uses combined androgen blockade: reduction of androgen synthesis and direct antagonism of the androgen receptor (AR) with antiandrogens (3). Therapy ultimately fails, indicated by increasing prostate specific antigen (PSA) levels and recurrent tumor growth (4). Despite castrate androgen levels, AR is still highly expressed and active in hormone-refractory tumors implying a switch to alternative mechanisms of activation (5). Among mechanisms proposed for AR activity at no or low hormone levels are AR gene amplification, increased coactivator expression, activation by growth factors and selection of somatic AR mutations (6). Therapy-specific selection of AR mutants may underlie antiandrogen withdrawal syndrome where tumors regress upon treatment cessation (7, 8), and may explain why tumors resistant to one antagonist may respond favorably to another (9, 10).
Many AR mutations have been reported in prostate cancer, but their prevalence and influence on disease progression are unclear due to few comprehensive sequencing studies, variable treatment regimens, and limited access to high-quality samples. Many previous studies focused on the ligand binding domain (LBD), although recent examinations of the entire AR coding region have identified N-terminal domain (NTD) mutations as well (11-13). Apart from the T878A mutation that is reported in about one-third of hormone-refractory tumors (10, 14), most mutations appear to be rare (15).
Studies in mouse prostate cancer models, where treatment is experimentally controlled, add compelling evidence for treatment selection. In the transgenic adenocarcinoma of mouse prostate (TRAMP) model, intact vs. castrate hormonal status selects for AR mutations in different domains (16). Our lab recently identified mutations in tumors from TRAMP mice expressing a “humanized” AR (17). Mutations in AR were frequent but at low levels, generally comprising 10% or less of the tumor RNA. Examination of recurring alterations identified ones distinct between flutamide- and bicalutamide-treated mice, as well as clustered mutations shared among groups. Characterization of select mutants revealed altered AR function, including differential activation of androgen-responsive promoters.
Here we extend this analysis to a set of high quality patient samples with detailed treatment records from the University of Michigan Specialized Program of Research Excellence (SPORE) in Prostate Cancer. To determine whether antiandrogens impose treatment-specific selection pressure, AR mutations were compared from flutamide-treated, bicalutamide-treated, and hormone-naïve patients. Functional analysis of known as well as novel variants provides insight into alternative mechanisms of antiandrogen resistance.
MATERIALS AND METHODS
Patient Samples
RNA from metastases of patients treated with bicalutamide or flutamide were obtained from the University of Michigan SPORE in Prostate Cancer rapid autopsy program; tissue was procured as described (18). Biopsies of treatment-naïve lymph node metastases were obtained from the University Hospital in Ulm, Germany as part of the UM SPORE-Ulm Cooperative Collaborative Clinical Case Procurement Program (19).
Mutation Identification
1 μg of RNA was reverse transcribed using SuperscriptII reverse transcriptase (Invitrogen, Carlsbad, CA) with 0.5 μg oligo (dT) in a 20 μl reaction. Two reverse transcription (RT) reactions were performed per sample to control for error. The entire AR coding region was PCR-amplified in 5 fragments, using primers listed below. 25 μl reactions contained 2.5 units Platinum Pfx DNA polymerase (Invitrogen), 2X Buffer and 1X GC enhancer (supplied by the manufacturer), 1.5 mM MgSO4, 0.3 mM dNTPs , 0.5 μM each primer, and 1−3 μl of RT reaction. Primer Pairs:
AR1 Forward, position 1074: 5’ CGGGGTAAGGGAAGTAGGTG 3’
AR1 Reverse, position 1732: 5’ CTGCCTTCGGATACTGCTTC 3’
AR2 Forward, position 1689: 5’ CAACTCCTTCAGCAACAG 3’
AR2 Reverse, position 2448: 5’ CAGTTGTATGGACCGTGT 3’
AR3 Forward, position 2412: 5’ TCATCCTGGCACACTCTCTTCACA 3’
AR3 Reverse, position 2693: 5’ GGGGCCCATTTCGCTTTTGACACA 3’
AR4 Forward, posotion 2639: 5’ GGTGAGCAGAGTGCCCTATC 3’
AR4 Reverse, position 3399: 5’ TCCTGGAGTTGACATTGGTG 3’
AR5 Forward, position 3312: 5’ GACCAGATGGCTGTCATTCA 3’
AR5 Reverse, position 3982: 5’ GAAATTCCCCAAGGCACTG 3’
Products were processed as described (17). Briefly, products were visualized on 1% agarose gels; bands were excised and purified with the QiaexII gel extraction kit (Qiagen, Valencia CA). 3’-A overhangs were added by incubation with Taq polymerase (Invitrogen) at 70°C for 30 min. Products were ligated into pGEM-T easy (Promega, Madison, WI) and transfected into DH5α chemically competent bacteria (Invitrogen). DNA from 20 clones/sample (10 clones/RT reaction) was purified with QIAprep Spin Miniprep columns (Qiagen) and sequenced by the University of Michigan DNA Sequencing Core.
Sequence was compared to the human AR (Genbank Accession# NM_000044) using Sequencher software (version 4.1, Gene Codes, Ann Arbor, MI), and mutations checked against the Androgen Receptor Gene Mutations Database (http://www.androgendb.mcgill.ca/) (15).
Mutant AR Plasmids
Mutations E255K and W435L were introduced into the pCMV5 hAR expression vector using the Quickchange Site Directed Mutagenesis kit (Stratagene, La Jolla CA) and the primers below. DMSO was added to the mutant strand synthesis to prevent Q and G tract contraction. Plasmids were sequenced to verify the mutation and the original number of Q and G codons. Mutation primers:
E255K Sense: 5’ GTGTGGAGGCGTTGAAGCATCTGAGTCCAGGG 3’
E255K Antisense: 5’ CCCTGGACTCAGATGCTTCAACGCCTCCACAC 3’
W435L Sense: 5’ CGCTTCCTCATCCTTGCACACTCTCTTCACAGC 3’
W435L Antisense: 5’ GCTGTGAAGAGAGTGTGCAAGGATGAGGAAGCG 3’
A mutant with the 69 bp DBD insertion was constructed by ligating a HindIII/Tth111I fragment into pCMV5-hAR; insert and junctions were verified by sequencing. To introduce W435L into the mammalian two-hybrid plasmid VP16-ARTAD (20), a BstEII/HindIII fragment of wtAR NTD was substituted with the analogous pCMV5-AR-W435L fragment.
Transfection Assays
CV-1 cells were cultured in DMEM and PC-3 cells in RPMI, supplemented with 10% fetal bovine serum, 1% Glutamax, and 1% penicillin/streptomycin. RWPE cells were grown in complete keratinocyte-serum-free medium (KSFM). Cells were seeded at 5 × 104 (CV-1) or 1 × 105 (PC-3, RWPE) in 12-well plates. Four hours before transfection, media was replaced with standard DMEM or RPMI + 2.5% charcoal-stripped NuSerum + 1% Glutamax. Cells were transfected with Fugene 6 reagent (Roche, Nutley, NJ) at 3 volumes of Fugene/μg DNA with 4 ng pCMV5-AR (wt or mutant), 400 ng luciferase (luc) reporter plasmid and 100 ng promoterless renilla (Promega) for normalization. PSA-luc includes the distal PSA enhancer (−5323 to −4023) and promoter (−542 to +12) (21). C’Δ9, HRE3 and HRE2 reporters have been described (22). NF-κB expression plasmid and pBVIx-luc (6XNF-κB) reporter were from G. Nunez (23). 24 hours post-transfection, cells were rinsed in 1X PBS and fed phenol red-free media + 10% charcoal-stripped Nuserum +/− hormone. Cells were harvested 48 hours post-transfection into 1X Passive Lysis Buffer (Promega). Luciferase activity was measured using the Dual Luciferase Reporter Assay System (Promega) on a Veritas Microplate Luminometer (Turner Biosystems Inc., Sunnyvale, CA).
Immunoblotting
CV-1 cells were seeded at 4 × 105 cells/ 60 mm dish, fed phenol red-free media +/− 1 nM R1881 four hours prior to transfecting as above with 100 ng receptor (wtAR or mutant) and 1.9 μg vector (pCMV5). 24 hours later, cells were rinsed in cold 1X PBS, harvested in 100 μl RIPA buffer + protease inhibitors, lysed at 4°C for 10 minutes and centrifuged at 4°C for 10 minutes. Protein was quantified by the Dc Protein Assay (Bio-Rad, Hercules CA). 20 μg protein was run on 5% stacking/8% separating SDS-polyacrylamide gels and transferred to nitrocellulose. The blot was probed with antibody to the AR N-terminus (N20, Santa Cruz Biotechnology, Santa Cruz, CA) (1:500) and HRP-conjugated ECL anti-rabbit IgG (GE Healthcare, Piscataway, NJ) (1:5,000) for 45 minutes. Bands were detected with ECL western blotting reagents (Pierce Biotechnology, Rockford IL).
Cycloheximide and Lactacystin Treatments
CV-1 cells were transfected with wtAR or AR-E255K as above. After 24 hours, cells were rinsed with PBS and incubated in media containing 30 μM cycloheximide (Sigma, St. Louis MO) +/− 1 nM R1881 for times indicated, or treated with 10 μM lactacystin (Cayman Chemical, Ann Arbor, MI) for 18 hours. At indicated times, cells were lysed in RIPA buffer plus inhibitors as above. Immunoblot bands were quantitated by densitometry using ImageJ (NCBI). AR levels were normalized to ß-tubulin and % protein determined relative to amount at time 0 (100%).
Immunocytochemistry
4 × 104 PC-3 cells were seeded onto 4-chamber slides and transfected with 100 ng receptor in phenol red-free RPMI + 10% charcoal-stripped Nuserum. 24 hours later, cells were fed fresh media +/− 10 nM R1881and incubated 24 hours. Cells were rinsed in ice-cold PBS, fixed on ice in 4% paraformaldehyde for 5 minutes, permeabilized in 0.1% triton-X PBS for 10 minutes, blocked in 5% heat-inactivated goat serum (Invitrogen) in 0.1% triton-X PBS for 1 hour, incubated in AR N20 (Santa Cruz Biotechnology) (1:500) overnight and FITC-conjugated goat anti-rabbit antibody (1:1000) for 1 hour. Slides were mounted with Prolong Gold plus DAPI (Invitrogen). Images were captured using an Olympus BX-51 microscope with an Olympus DP-70 high-resolution digital camera.
RESULTS
Identification of AR Mutations in Prostate Cancer Metastases
To examine directly whether AR mutations differ between treated and untreated tumors, whether mutation frequency increases following antiandrogen treatment, and whether different antiandrogens select for distinct mutations, the AR coding region was sequenced from prostate cancer metastases collected in the University of Michigan Rapid Autopsy Program (18). Because secondary hormone therapy is often used after relapse, only eight of thirty patients met the criterion of treatment with only one antiandrogen - 4 were treated with flutamide and 4 with bicalutamide (Table 1). AR from 3 hormone naïve lymph node metastases from patients at the University of Ulm Hospital (Ulm, Germany) was sequenced for comparison (19). RNA from all samples was reverse transcribed and the entire AR coding region amplified, subcloned, sequenced and mutations compared within and between groups.
Table 1.
Summary of sample information.
| Patient #* | Source of metastasis | Hormone therapy | Months on therapy | TMPRSS 2-ETS gene fusions† | Base pair changes | |
|---|---|---|---|---|---|---|
| multiple clones/patient | multiple patients | |||||
| 5 | Liver | Flu** | 22 | No | ΔQ86, AR23 | Q58L, ΔQ86 T440P, G456S, 69 bp ins |
| 12 | Liver | Flu | 36 | Yes | E255K, L446S, K610E, L798P, L874P | R485C, 69 bp ins, R787X |
| 28 | Lung | Flu | 12 | Yes | V716M | ΔQ86, G457D, 69 bp ins |
| 18 | Liver | Flu | 48 | Yes | none | Q58L, T229C A253V, W435L |
| 23 | Soft Tissue | Bic†† | 48 | No | none | V509L, 69 bp ins |
| 24 | Liver | Bic | 10 | Yes | Q58L, L595M, Q262X | Q58L, ΔQ86, 69 bp ins, E666D |
| 26 | Kidney | Bic | 60 | Yes | ΔQ86, G456S | Q58L, ΔQ86, A253V, G456S |
| 30 | Soft Tissue | Bic | 18 | No | Q868X | ΔQ86, T229C, W435L,T440P, T498I, V509L, |
| LK8 | Lymph node | Naïve | 0 | N/A*** | T440I | Q58L, ΔQ86 G457D |
| 2C | Lymph node | Naïve | 0 | N/A | none | R485C, T498I |
| 12A | Lymph node | Naïve | 0 | N/A | ΔQ86 | ΔQ86, R787X Q868X |
Sequencing the equivalent of 20 full-length AR mRNAs per metastasis (10 from two independent RT reactions) identified 280 single nucleotide changes in 191 codons. The average alteration rate within the population was 4.1 base changes/10,000 bps, comparable to the rate observed in h/mAR-TRAMP tumors using the same method (17). For that study, baseline error due to sequence peculiarities (e.g., variability in CAG (Q) and GGN (G) tracts and high GC content overall) and methodology (e.g., RT and subcloning error) was established by sequencing Ar RNA from mouse testis. Testis samples carried 2.2 base changes/10,000 bp indicating that about half the differences in tumor samples by this approach are likely somatic mutations. A similar error level was reported in comparable studies using RT and PCR amplification (24).
Of the total base alterations, 160 were missense, with 10% in the polyQ and G-tracts, and 69 were silent mutations, 30% of which were in the polymorphic G-tract. A breakdown of mutation types per treatment group is available (Supplemental Table). There were no significant differences between treatment groups in total number or types of mutations. Mutations in the NTD (amino acids 1−535) were over-represented relative to AR length, accounting for 73% of mutations from all groups (excluding the polyamino acid tracts). Most mutations were present in one or two clones per sample, or 5−10% of the RNA population, similar to mutation frequencies in mouse (16, 17). Since it is difficult to distinguish between true mutations that occur in a single clone and methodological errors, analysis was restricted to mutations that occurred in multiple clones.
Treatment-specific Patterns of Recurring Mutations
Mutations that provide a growth advantage are likely to be more common within the tumor. Mutations occurred more than once in 36 codons, either in multiple cases (24 codons) (Fig. 1A) or in multiple clones within a tumor (17 codons) (Fig. 1B). Recurring missense mutations include those that alter a codon to different residues (away from wild type, e.g. L194F/R) or to the same new residue (e.g. Q58L). Both types could be functionally significant. All but two missense mutations identified in multiple cases were located in the NTD, with few specific to a single group - half occurred only with antiandrogen treatment and half were shared by treated and untreated patients (Fig. 1A). In contrast, the 13 missense and 2 nonsense mutations present in multiple clones per tumor were case-specific and not restricted by domain (Fig. 1B). 10 silent mutations recurred, six of which were in the G-tract. A silent change at E213 is a known polymorphism (25), occurring in all 20 clones of 3 samples, but also in four and seven clones from two other samples.
Figure 1.

Recurring AR mutations from prostate cancer metastases. A. Mutations found in multiple cases. For codons carrying mutations to different amino acids, both changes are shown. B. Mutations in multiple clones per sample. Only ΔQ86 was shared among groups. AR domains and repeats are boxed. Mutations above the map are silent or nonsense; mutations below are missense. Codon color indicates treatment group. Q: Polyglutamine tract, NTD: N-terminal domain, G: Polyglycine tract, DBD: DNA binding domain, H: Hinge region, LBD: Ligand binding domain. C. V716M was the only AR sequence in 3 metastases from patient 28, but did not occur in normal kidney. Electropherograms, left to right: amplified cDNA clone from metastasis 1 with G3261A (numbering from Genbank NM_000044) resulting in V716M; wild type sequence from normal kidney genomic DNA; cDNA and genomic DNA of metastasis 2. Green arrow: mutation; black arrow: wild type base.
Differences between treatments were most apparent for mutations that occurred multiple times per tumor (Fig. 1B). Only two of these mutations occurred in hormone-naïve samples, while flutamide- and bicalutamide-treated tumors carried eight and seven mutations, respectively. Three antiandrogen-treated tumors carried most of the recurring mutations (Table 1). Length of treatment did not affect frequency of mutations, although power was limited by sample size.
All recurring mutations in the LBD were from antiandrogen-treated tumors, suggesting selection for altered conformation of, or ligand contacts within, the binding pocket. Half of the antiandrogen-treated tumors carried at least one recurring LBD mutation, but none overlapped between the flutamide and bicalutamide groups, indicating distinct selection conferred by each drug. The flutamide-treated group had three missense mutations in the LBD - V716M, L798P, and L874P (Fig. 1B). V716M, discussed below, creates a promiscuous receptor (26). L798P is a novel mutation within an E3 ubiquitin ligase interacting area (27). L874P, also novel, lies near codons H875 and T878 that when mutated allow flutamide to activate AR (28, 29).
Multiple Metastases from One Patient Express Only AR-V716M
The mutation, V716M, was present in all 20 clones sequenced from the lung metastasis of flutamide-treated patient 28 (Table 1). To rule out a germline mutation, 281 bps around V716M were amplified and sequenced from the patient's normal kidney genomic DNA. Only the wild type G occurred at position 3261 indicating that the mutation was somatic (Fig. 1C). Additional examination of cDNA and/or genomic DNA from two other metastases from this patient yielded only mutant sequence with no detectable wild type (Fig. 1C), indicating that a clonal population carrying AR-V716M accounted for all three metastases. No other mutations recurred in this sample. Given that AR-V716M is activated by a wide array of ligands (26), its predominance in this patient's cancer supports its role in treatment resistance.
The Splice Variant AR23 was Only in Antiandrogen-treated Cases
A variant generated by the use of a cryptic splice site in intron 2 was identified in one or more clones in 5 of 8 tumors from treated patients, but in none of the hormone-naïve tumors. Alternative splicing inserted 69 bps of intron 2 in frame to add 23 amino acids between the zinc fingers of the DNA binding domain (DBD). This variant, AR23, was previously found in Androgen Insensitivity Syndrome due to a mutation upstream of exon 3 that altered splicing (30). Recently, AR23 was identified in a prostate metastasis from a bicalutamide-treated patient (31). AR23 was engineered into an expression plasmid and its activity assayed after transfection. As also shown by Jagla et al. (31), AR23 was incapable of nuclear localization upon hormone addition but rather formed cytoplasmic speckles (Fig. 2A) and failed to activate androgen-responsive reporters (Fig. 2B). Previously AR23 was shown to increase endogenous AR-T878A activity when over expressed in LNCaP cells (31). In Fig. 2B, AR23 also increased wtAR activation (2-fold greater PSA-luc activity) following co-expression in PC-3 cells. Moreover, in the presence of AR23, wtAR was less inhibited by hydroxyflutamide or bicalutamide. This effect was not specific to AR since transactivation by NF-κB and AP-1 also increased with AR23 (31), and AR23 reduced glucocorticoid receptor inhibition by RU-486, which antagonizes both receptors (Supplemental Data). AR23 could not transrepress activated NF-κB-induced transcription, unlike wtAR (Fig. 2C) (32). Thus cytoplasmic activity of AR23 broadly, but not universally, influences nuclear activities.
Figure 2.

Splice variant AR23 has altered subcellular localization and enhances wild type AR (wtAR) activity. A. Punctate cytoplasmic localization of AR23. AR23 transfected into PC-3 cells shows diffuse cytoplasmic localization without ligand (top) like wtAR (not shown), but forms cytoplasmic puncta with 10 nM R1881 (middle) unlike wtAR nuclear localization (bottom). AR detection used AR N20 and FITC-conjugated secondary antibody. B. Transactivation of wtAR, AR23, or 1:1 wtAR:AR23 (4 ng each) with 200 ng PSA-luc and 100 ng promoterless renilla in PC-3 cells. Cells were harvested 24 hours after agonist or antagonist treatment (HOF, hydroxyflutamide; Bic, bicalutamide) and luciferase activity assayed. Average normalized values of three independent trials are presented as percent wtAR transactivation at 1 nM R1881. C. Transrepression of NF-κB activity in CV-1 cells. WtAR or AR23 was transfected with the NF-κB reporter pBVIx-luc and NF-kB was activated with TPA. WtAR reduced activation to 20% of vector alone with 10 nM R1881 + 1 nM TPA; with AR23, NF-κB activity remained 80% of control. Error bars are standard error of the mean (SEM). * Significant difference based on p<0.05 by Student's t-test.
Novel Mutations in the AR NTD in Conserved Functional Motifs
The largely unstructured NTD contains two activation functions (AF1, AF5) that bind coactivators and are critical for AR activity (33). The NTD directs intramolecular amino-carboxy (N-C) interactions, via FxxLF and WxxLF motifs, that stabilize ligand-bound AR. In this study, 14 of 19 mutations in the NTD fell into four regions: the polymorphic Q-tract, the COOH-terminus of Hsp70-Interacting Protein (CHIP) interaction domain, the WxxLF motif, and the end of AF5 involved in coactivator interactions (34) (Fig. 1). Mutations in the CHIP interacting domain were previously discovered in TRAMP: AR-E231G causes cancer as a prostate-specific transgene, highlighting AR's oncogenic potential (16, 35). The novel mutations W435L and E255K were engineered into expression vectors for functional characterization.
W435L Alters an AR N-C Interaction Motif
The mutation, W435L, was identified in one clone each of two antiandrogen-treated patients. Its position within the WxxLF motif suggests this mutation may influence N-C interactions. To determine the effect on transactivation, AR-W435L was co-transfected into CV-1 fibroblasts, immortalized prostate RWPE cells, and prostate cancer PC-3 cells along with varied reporters. Androgen responsive elements are generally either canonical inverted repeats of a TGTTCT half-site that bind multiple steroid receptors (e.g., HRE3), or direct repeats that are weaker but AR-selective (e.g., HRE2) (36). Natural promoters often contain both element types as well as binding sites for other factors. AR-W435L increased transactivation preferentially for MMTV in CV-1 cells and PSA in RWPE, likely due to greater efficacy on AR-selective elements (Fig 3). This promoter-specific effect was also cell-type dependent since there was minimal effect in PC-3 cells. To probe W435L action farther, we used a mammalian two-hybrid system in which the ability of the AR NTD (fused to the VP16 activation domain) to bind the LBD (fused to the Gal4DBD) is assessed by luciferase activity driven by Gal4 DNA elements (20). When W435L was introduced into the NTD-VP16 fusion, activity was more than 50% greater than for wtAR, confirming that this mutation enhances N-C interaction (Fig 3D).
Figure 3.
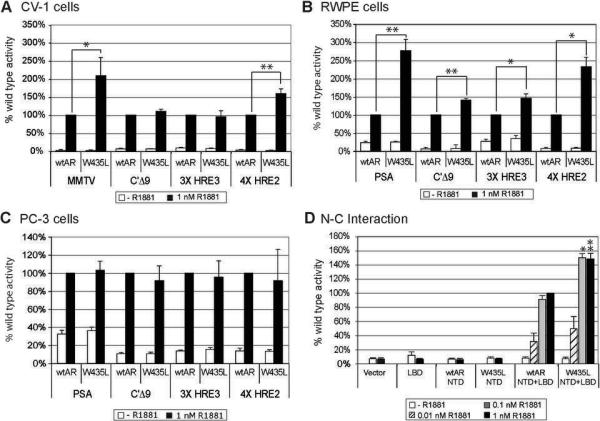
Promoter- and cell-context dependence of AR-W435L. Transactivation of ARW435L was assessed in CV-1 (A), RWPE (B), and PC-3 (C) cells, revealing promoter-specific increases most pronounced in RWPE and absent in PC-3 cells. Cells were transfected with 4 ng wtAR or AR-W435L and 400 ng of the indicated reporters. PSA activated poorly in CV-1 cells so MMTV was tested instead. After 24 hours cells were fed with phenol red free medium +/− 1 nM R1881. Average values normalized to renilla for three trials are represented as percent induced wtAR activity. D. AR-W435L increased N-C interaction in mammalian 2-hybrid assays compared to wtAR. PC-3 cells were transfected with 100 ng VP16-wtAR-NTD or W435LNTD, 100 ng Gal4-AR LBD, 200 ng Gal4-luc and 100 ng renilla. 24 hours after transfection cells were fed +/− R1881 for 24 hours. Activity was compared to induced wtAR NTD + LBD (100%). Values are the average of 5 trials +/− standard error of the mean (SEM). * p<0.05, ** p<0.005, significant differences between wtAR and AR-W435L based on Student's t-test.
Mutations in the Conserved CHIP Interacting Domain
Two mutations from treated patients, A253V and E255K, lie adjacent to the most highly conserved region of the NTD where interaction with CHIP, an E3-ubiquitin ligase, promotes AR degradation (37). To determine whether E255K enhances AR stability, CV-1 cells transfected with wtAR or AR-E255K were treated with cycloheximide to inhibit protein synthesis; cells were harvested at times thereafter to detect protein degradation. R1881 greatly stabilized both mutant and wild type AR protein as expected (Fig. 4A). However, AR-E255K half-life was extended compared to wtAR, particularly in the absence of ligand (12.5 vs. 5.2 hours, respectively). E255K migrated slower than wtAR, which may be due to differential protein modification.
Figure 4.
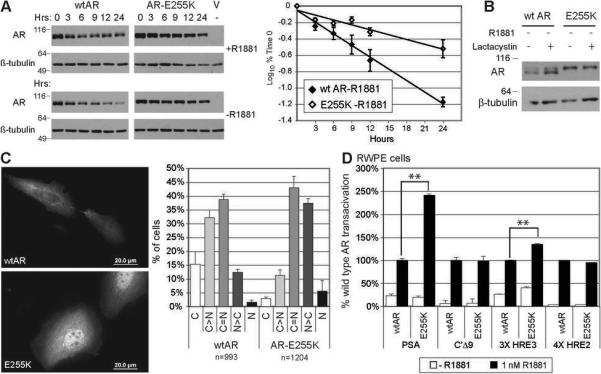
AR-E255K has increased stability and ligand-independent nuclear localization. A. AR degradation following cycloheximide treatment. 100 ng wtAR or AR-E255K plasmid was transfected into CV-1 cells, which were treated after 24 hours with 30 μM cycloheximide. Cells were harvested at times indicated and 20 μg of total protein electrophoresed (left). AR bands from scanned immunoblots were quantified using ImageJ, values normalized to the amount of protein at time 0 (100%). The log10 of the percentage was plotted versus time for wtAR and ARE255K without hormone (right). Half-life was calculated as log10 of 50% based on the linear regression. AR-E255K shows a longer half-life (t1/2= 12.5 hrs) than wtAR (t1/2=5.2 hrs). Full gels are in Supplementary Data. B. Proteasome inhibition with lactacystin increased unliganded wtAR but not AR-E255K levels. CV-1 cells were transfected as above, treated after 24 hours with 10μM lactacystin, harvested 18 hours later, and immunoblotted. C. Following transfection into PC-3 cells, wtAR was largely cytoplasmic without hormone (top) while most cells with ARE255K showed more nuclear staining (bottom). Color images and composite are in Supplementary Data. AR was detected as for Fig. 2. Percent of cells with cytoplasmic to nuclear AR fluorescence are graphed as: C, exclusively cytoplasmic; C>N, cytoplasmic greater than nuclear; C=N, equal cytoplasmic and nuclear; N>C, nuclear greater; N, exclusively nuclear. n= number of cells counted for all three trials. Mean percentages +/− SEM are shown. D. AR-E255K showed increased transactivation of PSA-luc in RWPE cells, compared to wtAR. Bars are average % wtAR activation of three trials +/− SEM. ** p<0.005 based on Student's t-test.
To explore whether E255K stabilization was influenced by the 20S proteasome, cells were treated with the proteasome inhibitor lactacystin. Without ligand, proteasome inhibition increased wtAR steady state levels as expected (38). However, AR-E255K levels were unaffected, indicating that proteasome activity has little impact on this mutant (Fig 4B).
Because both the proteasome and chaperones are implicated in nuclear transit, ARE255K localization was examined by immunocytochemistry. Without R1881, wtAR was mostly cytoplasmic as expected, but AR-E255K showed significant nuclear localization (Fig. 4C). Tallying the localization in cells showed skewing of AR-E255K to the nucleus without ligand compared to wtAR (Fig. 4C).
AR-E255K induced reporter gene expression similarly to wtAR in CV-1 and PC-3 cells with no increased activity without androgen or with added coactivators ARA70 and SRC-1 (not shown). However, in RWPE cells, AR-E255K increased transactivation of PSA-luc 2.5-fold relative to wtAR (Fig. 4D). This may be due to host cofactor differences, as well as somewhat greater activity on canonical elements like HRE3. Thus AR-E255K exhibited increased stability, substantial nuclear localization without ligand, and cell-type dependent differential promoter activation.
DISCUSSION
This study reveals a low level of mutation throughout the AR coding region in metastases from antiandrogen-treated as well as hormone-naïve patients, providing evidence for genetic heterogeneity and a “mutator phenotype” in prostate cancer (1). Very few mutations in the hormone naïve samples occur in multiple clones per case suggesting that most provide little growth advantage and may be random “passenger” mutations. However, antiandrogen treatment leads to more mutations in greater abundance, suggesting that treatment selects for a subset of AR mutations within this diverse population.
Examination of recurring mutations within and between samples indicates specific codons that may provide a selective advantage during cancer progression. Remarkably, mutations recurring in multiple samples are mostly in the NTD and are shared across treatment groups. This emphasizes the broad function of the NTD in growth factor and coactivator interactions and in receptor stability, and suggests some AR variants provide general growth advantages regardless of treatment. In contrast, all missense mutations in the LBD are case-specific and are only found in antiandrogen-treated patients, evidencing their likely selection by treatment. Further, the lack of overlap in mutations between bicalutamide and flutamide treatment suggests these antagonists select for distinct variants.
Although the patient samples are fewer than the 40 tumors we examined from h/mAR-TRAMP mice (17), and are metastases rather than primary tumors, similarities emerge. Overall mutation frequency is comparable, although there are more mutations present in multiple clones per human sample, likely reflecting the clonal nature of metastases and extended time with disease. Q58L and ΔQ86 are common in both mice and men regardless of treatment. In both species, there are fewer recurring mutations in hormone-naïve tumors, substantiating selection pressure of therapy.
Mutations also occur in similar domains in human and mouse ARs, particularly following flutamide treatment. Mutations in flutamide-treated tumors occur in two regions important for ligand specificity: the highly conserved signature sequence (i.e., mAR-W719C, hAR-V716M) and the distal region where some mutations allow promiscuous ligand recognition (i.e., mARP893S, hAR-L874P) (17, 39). While this study did not find the common T878A variant, L874P may act similarly (40), perhaps displacing the T878 residue that extends into the ligand pocket thus accommodating the larger hydroxyflutamide.
The capacity of LBD mutations to affect disease progression is highlighted by the dominance of AR-V716M in three metastases examined from one flutamide-treated patient. We infer that V716M arose either within the primary tumor or early in metastatic invasion. This sample had no other recurring mutations suggesting that an effective variant reduces the selective value of other mutations. Interestingly, this patient survived much longer than the other cases. In eight patients this was the only case of fixation of an AR mutation, indicating that this is a relatively rare event; most cancers may instead have subsets of cells with different mutations, each providing a similar growth advantage.
Only one LBD mutation recurred following bicalutamide treatment, perhaps because a single residue change is unlikely sufficient to accommodate this bulkier antagonist in a manner compatible with agonism. Only mutation of W742 has been shown to allow bicalutamide to activate AR (41). The single recurring LBD mutation in a bicalutamide-treated patient, R761K, is at a residue commonly mutated in castrated h/mAR-TRAMP mice (17), implying the mechanism is not partial agonism.
Not only mutants but also splice variants may be subject to treatment selection, as shown recently for variant ARs that lack LBDs in hormone-refractory prostate cancer (42). The AR23 splice variant found in antiandrogen-treated patients here may be present in cells along with wtAR, but has effects on other nuclear factors as well. Although itself inactive, AR23 increases wtAR transactivation when co-expressed. Cytoplasmic aggregates of liganded AR23 may sequester antiandrogens or interacting partners, or participate in intracellular signaling, via intact NTD and LBD domains, allowing wtAR to function (31). Alternatively, AR23 in an unfolded state, suggested by aggregation, may compromise the cell chaperone system, allowing AR and other proteins to evade degradation for generally enhanced activity. This decoy function may be valuable against treatment since AR23 is absent in untreated patients.
The W435L mutation increases transactivation of AR-selective promoters in some cells. This contrasts with an h/mAR-TRAMP mutant, AR-R753Q, that functions on canonical but not selective elements (17). Selection for differential promoter usage may change over the course of disease and incorporate multiple mechanisms. The effect of W435L might also vary with disease stage or cell type. Recently the WxxLF motif has been implicated in ligand-independent AR activation (43). Mutation to LxxLF could weaken normal competition with the FxxLF motif thus increasing ligand-dependent activity, while increasing ligand-independent function via greater mimicry of coactivator interactions. Alternatively, W435L may affect AR stability via altered exposure of FQNLF, which helps target AR to the proteasome (44). Since steady-state levels of AR-W435L appear unaffected, it is more likely that W435L impacts transcription and coactivator interactions, either directly or via influence on FxxLF function, as supported by greater N-C interaction demonstrated in the mammalian two-hybrid assay.
Mutation of the highly conserved CHIP interaction domain in both murine and human tumors underscores the importance of this region, and illustrates the utility of mouse models for obtaining clinically relevant insights. E255K stabilizes AR and increases nuclear localization in the absence of hormone. This may have a similar effect to AR amplification, seen often in metastatic prostate cancer (45). Increased AR levels may enhance response to low ligand concentrations, increase ligand-independent activation, or promote agonism of antiandrogens (46). Although transactivation by AR-E255K is similar to wild type in transfection, overexpression may mask relevant differences. The analogous mAR-E231G shows modest differences in transfection but is oncogenic as a prostate-specific transgene (35).
In summary, this study identified a greater number of recurring mutations in metastases from treated versus untreated patients. Furthermore, the variety of mutations identified indicates that antagonist treatment does not select for a few common mutations, but instead selects for numerous rare mutations many of which may affect AR function and might be overlooked using bulk sequencing methods. Combining the novel mutations identified here with those from previous studies highlights AR domains within which mutations share a similar phenotype (47). These mutations affect diverse AR processes beyond transcriptional potency, including cell localization, stability, and promoter-selectivity. Better understanding of these processes may present new targets for therapies that obviate AR's ability to evade antiandrogen treatment.
Supplementary Material
ACKNOWLEDGEMENTS
We thank E.L. Yong (Singapore) for the AR mammalian 2-hybrid plasmids, Jorge Iniguez for RU486, and Elizabeth Starnes for assistance with transfections.
Grant support: DOD17-02-1-0099, NCI-P50 CA69568 and NIDDK-RO1-56356 (DMR), W81XWH-05-1-0105 (OAM), NIH-T32-HD075005 (MPS). Additional support came from the University of Michigan Cancer Center Support Grant (5 P30 CA46592) and the Michigan Diabetes Research and Training Center (NIH5P60 DK20572).
REFERENCES
- 1.Bielas JH, Loeb KR, Rubin BP, True LD, Loeb LA. Human cancers express a mutator phenotype. Proc Natl Acad Sci U S A. 2006;103:18238–42. doi: 10.1073/pnas.0607057103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Loeb LA, Bielas JH, Beckman RA. Cancers exhibit a mutator phenotype: clinical implications. Cancer Res. 2008;68:3551–7. doi: 10.1158/0008-5472.CAN-07-5835. discussion 7. [DOI] [PubMed] [Google Scholar]
- 3.Labrie F, Dupont A, Belanger A, et al. New approach in the treatment of prostate cancer: complete instead of partial withdrawal of androgens. Prostate. 1983;4:579–94. doi: 10.1002/pros.2990040605. [DOI] [PubMed] [Google Scholar]
- 4.Pienta KJ, Bradley D. Mechanisms underlying the development of androgen-independent prostate cancer. Clin Cancer Res. 2006;12:1665–71. doi: 10.1158/1078-0432.CCR-06-0067. [DOI] [PubMed] [Google Scholar]
- 5.Scher HI, Buchanan G, Gerald W, Butler LM, Tilley WD. Targeting the androgen receptor: improving outcomes for castration-resistant prostate cancer. Endocr Relat Cancer. 2004;11:459–76. doi: 10.1677/erc.1.00525. [DOI] [PubMed] [Google Scholar]
- 6.Feldman BJ, Feldman D. The development of androgen-independent prostate cancer. Nat Rev Cancer. 2001;1:34–45. doi: 10.1038/35094009. [DOI] [PubMed] [Google Scholar]
- 7.Hara T, Miyazaki J, Araki H, et al. Novel mutations of androgen receptor: a possible mechanism of bicalutamide withdrawal syndrome. Cancer Res. 2003;63:149–53. [PubMed] [Google Scholar]
- 8.Scher HI, Kelly WK. Flutamide withdrawal syndrome: its impact on clinical trials in hormone-refractory prostate cancer. J Clin Oncol. 1993;11:1566–72. doi: 10.1200/JCO.1993.11.8.1566. [DOI] [PubMed] [Google Scholar]
- 9.Suzuki H, Okihara K, Miyake H, et al. Alternative nonsteroidal antiandrogen therapy for advanced prostate cancer that relapsed after initial maximum androgen blockade. J Urol. 2008;180:921–7. doi: 10.1016/j.juro.2008.05.045. [DOI] [PubMed] [Google Scholar]
- 10.Taplin ME, Bubley GJ, Ko YJ, et al. Selection for androgen receptor mutations in prostate cancers treated with androgen antagonist. Cancer Res. 1999;59:2511–5. [PubMed] [Google Scholar]
- 11.Chen G, Wang X, Zhang S, et al. Androgen receptor mutants detected in recurrent prostate cancer exhibit diverse functional characteristics. Prostate. 2005;63:395–406. doi: 10.1002/pros.20191. [DOI] [PubMed] [Google Scholar]
- 12.Hyytinen ER, Haapala K, Thompson J, et al. Pattern of somatic androgen receptor gene mutations in patients with hormone-refractory prostate cancer. Lab Invest. 2002;82:1591–8. doi: 10.1097/01.lab.0000038924.67707.75. [DOI] [PubMed] [Google Scholar]
- 13.Tilley WD, Buchanan G, Hickey TE, Bentel JM. Mutations in the androgen receptor gene are associated with progression of human prostate cancer to androgen independence. Clin Cancer Res. 1996;2:277–85. [PubMed] [Google Scholar]
- 14.Gaddipati JP, McLeod DG, Heidenberg HB, et al. Frequent detection of codon 877 mutation in the androgen receptor gene in advanced prostate cancers. Cancer Res. 1994;54:2861–4. [PubMed] [Google Scholar]
- 15.Gottlieb B, Beitel LK, Wu JH, Trifiro M. The androgen receptor gene mutations database (ARDB): 2004 update. Hum Mutat. 2004;23:527–33. doi: 10.1002/humu.20044. [DOI] [PubMed] [Google Scholar]
- 16.Han G, Foster BA, Mistry S, et al. Hormone status selects for spontaneous somatic androgen receptor variants that demonstrate specific ligand and cofactor dependent activities in autochthonous prostate cancer. J Biol Chem. 2001;276:11204–13. doi: 10.1074/jbc.M008207200. [DOI] [PubMed] [Google Scholar]
- 17.O'Mahony OA, Steinkamp MP, Albertelli MA, Brogley M, Rehman M, Robins D. Profiling human androgen receptor mutations reveals treatment effects in a mouse model of prostate cancer. Molecular Cancer Research in press. doi: 10.1158/1541-7786.MCR-08-0273. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shah RB, Mehra R, Chinnaiyan AM, et al. Androgen-independent prostate cancer is a heterogeneous group of diseases: lessons from a rapid autopsy program. Cancer Res. 2004;64:9209–16. doi: 10.1158/0008-5472.CAN-04-2442. [DOI] [PubMed] [Google Scholar]
- 19.Hofer MD, Kuefer R, Huang W, et al. Prognostic factors in lymph node-positive prostate cancer. Urology. 2006;67:1016–21. doi: 10.1016/j.urology.2005.10.055. [DOI] [PubMed] [Google Scholar]
- 20.Lim J, Ghadessy FJ, Abdullah AA, Pinsky L, Trifiro M, Yong EL. Human androgen receptor mutation disrupts ternary interactions between ligand, receptor domains, and the coactivator TIF2 (transcription intermediary factor 2). Mol Endocrinol. 2000;14:1187–97. doi: 10.1210/mend.14.8.0499. [DOI] [PubMed] [Google Scholar]
- 21.Perez-Stable CM, Pozas A, Roos BA. A role for GATA transcription factors in the androgen regulation of the prostate-specific antigen gene enhancer. Mol Cell Endocrinol. 2000;167:43–53. doi: 10.1016/s0303-7207(00)00300-2. [DOI] [PubMed] [Google Scholar]
- 22.Robins DM. Androgen receptor and molecular mechanisms of male-specific gene expression. Novartis Found Symp. 2005;268:42–52. discussion 3−6, 96−9. [PubMed] [Google Scholar]
- 23.Inohara N, Koseki T, Lin J, et al. An induced proximity model for NF-kappa B activation in the Nod1/RICK and RIP signaling pathways. J Biol Chem. 2000;275:27823–31. doi: 10.1074/jbc.M003415200. [DOI] [PubMed] [Google Scholar]
- 24.Arezi B, Hogrefe HH. Escherichia coli DNA polymerase III epsilon subunit increases Moloney murine leukemia virus reverse transcriptase fidelity and accuracy of RT-PCR procedures. Anal Biochem. 2007;360:84–91. doi: 10.1016/j.ab.2006.10.009. [DOI] [PubMed] [Google Scholar]
- 25.Riva A, Kohane IS. A SNP-centric database for the investigation of the human genome. BMC Bioinformatics. 2004;5:33. doi: 10.1186/1471-2105-5-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Culig Z, Hobisch A, Cronauer MV, et al. Mutant androgen receptor detected in an advanced-stage prostatic carcinoma is activated by adrenal androgens and progesterone. Mol Endocrinol. 1993;7:1541–50. doi: 10.1210/mend.7.12.8145761. [DOI] [PubMed] [Google Scholar]
- 27.Rees I, Lee S, Kim H, Tsai FT. The E3 ubiquitin ligase CHIP binds the androgen receptor in a phosphorylation-dependent manner. Biochim Biophys Acta. 2006;1764:1073–9. doi: 10.1016/j.bbapap.2006.03.013. [DOI] [PubMed] [Google Scholar]
- 28.Veldscholte J, Ris-Stalpers C, Kuiper GG, et al. A mutation in the ligand binding domain of the androgen receptor of human LNCaP cells affects steroid binding characteristics and response to anti-androgens. Biochem Biophys Res Commun. 1990;173:534–40. doi: 10.1016/s0006-291x(05)80067-1. [DOI] [PubMed] [Google Scholar]
- 29.Fenton MA, Shuster TD, Fertig AM, et al. Functional characterization of mutant androgen receptors from androgen-independent prostate cancer. Clin Cancer Res. 1997;3:1383–8. [PubMed] [Google Scholar]
- 30.Bruggenwirth HT, Boehmer AL, Ramnarain S, et al. Molecular analysis of the androgen-receptor gene in a family with receptor-positive partial androgen insensitivity: an unusual type of intronic mutation. Am J Hum Genet. 1997;61:1067–77. doi: 10.1086/301605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jagla M, Feve M, Kessler P, et al. A splicing variant of the androgen receptor detected in a metastatic prostate cancer exhibits exclusively cytoplasmic actions. Endocrinology. 2007;148:4334–43. doi: 10.1210/en.2007-0446. [DOI] [PubMed] [Google Scholar]
- 32.Palvimo JJ, Reinikainen P, Ikonen T, Kallio PJ, Moilanen A, Janne OA. Mutual transcriptional interference between RelA and androgen receptor. J Biol Chem. 1996;271:24151–6. doi: 10.1074/jbc.271.39.24151. [DOI] [PubMed] [Google Scholar]
- 33.Shen HC, Coetzee GA. The androgen receptor: unlocking the secrets of its unique transactivation domain. Vitam Horm. 2005;71:301–19. doi: 10.1016/S0083-6729(05)71010-4. [DOI] [PubMed] [Google Scholar]
- 34.Irvine RA, Ma H, Yu MC, Ross RK, Stallcup MR, Coetzee GA. Inhibition of p160-mediated coactivation with increasing androgen receptor polyglutamine length. Hum Mol Genet. 2000;9:267–74. doi: 10.1093/hmg/9.2.267. [DOI] [PubMed] [Google Scholar]
- 35.Han G, Buchanan G, Ittmann M, et al. Mutation of the androgen receptor causes oncogenic transformation of the prostate. Proc Natl Acad Sci U S A. 2005;102:1151–6. doi: 10.1073/pnas.0408925102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Robins DM. Multiple mechanisms of male-specific gene expression: lessons from the mouse sex-limited protein (Slp) gene. Prog Nucleic Acid Res Mol Biol. 2004;78:1–36. doi: 10.1016/S0079-6603(04)78001-0. [DOI] [PubMed] [Google Scholar]
- 37.He B, Bai S, Hnat AT, et al. An androgen receptor NH2-terminal conserved motif interacts with the COOH terminus of the Hsp70-interacting protein (CHIP). J Biol Chem. 2004;279:30643–53. doi: 10.1074/jbc.M403117200. [DOI] [PubMed] [Google Scholar]
- 38.Lin HK, Altuwaijri S, Lin WJ, Kan PY, Collins LL, Chang C. Proteasome activity is required for androgen receptor transcriptional activity via regulation of androgen receptor nuclear translocation and interaction with coregulators in prostate cancer cells. J Biol Chem. 2002;277:36570–6. doi: 10.1074/jbc.M204751200. [DOI] [PubMed] [Google Scholar]
- 39.Culig Z, Stober J, Gast A, et al. Activation of two mutant androgen receptors from human prostatic carcinoma by adrenal androgens and metabolic derivatives of testosterone. Cancer Detect Prev. 1996;20:68–75. [PubMed] [Google Scholar]
- 40.Bohl CE, Miller DD, Chen J, Bell CE, Dalton JT. Structural basis for accommodation of nonsteroidal ligands in the androgen receptor. J Biol Chem. 2005;280:37747–54. doi: 10.1074/jbc.M507464200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yoshida T, Kinoshita H, Segawa T, et al. Antiandrogen bicalutamide promotes tumor growth in a novel androgen-dependent prostate cancer xenograft model derived from a bicalutamide-treated patient. Cancer Res. 2005;65:9611–6. doi: 10.1158/0008-5472.CAN-05-0817. [DOI] [PubMed] [Google Scholar]
- 42.Hu R, Dunn TA, Wei S, et al. Ligand-independent androgen receptor variants derived from splicing of cryptic exons signify hormone-refractory prostate cancer. Cancer Res. 2009;69:16–22. doi: 10.1158/0008-5472.CAN-08-2764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dehm SM, Regan KM, Schmidt LJ, Tindall DJ. Selective role of an NH2-terminal WxxLF motif for aberrant androgen receptor activation in androgen depletion independent prostate cancer cells. Cancer Res. 2007;67:10067–77. doi: 10.1158/0008-5472.CAN-07-1267. [DOI] [PubMed] [Google Scholar]
- 44.Chandra S, Shao J, Li JX, Li M, Longo FM, Diamond MI. A common motif targets huntingtin and the androgen receptor to the proteasome. J Biol Chem. 2008;283:23950–5. doi: 10.1074/jbc.M800467200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Haapala K, Kuukasjarvi T, Hyytinen E, Rantala I, Helin HJ, Koivisto PA. Androgen receptor amplification is associated with increased cell proliferation in prostate cancer. Hum Pathol. 2007;38:474–8. doi: 10.1016/j.humpath.2006.09.008. [DOI] [PubMed] [Google Scholar]
- 46.Chen CD, Welsbie DS, Tran C, et al. Molecular determinants of resistance to antiandrogen therapy. Nat Med. 2004;10:33–9. doi: 10.1038/nm972. [DOI] [PubMed] [Google Scholar]
- 47.Buchanan G, Greenberg NM, Scher HI, Harris JM, Marshall VR, Tilley WD. Collocation of androgen receptor gene mutations in prostate cancer. Clin Cancer Res. 2001;7:1273–81. [PubMed] [Google Scholar]
- 48.Mehra R, Tomlins SA, Yu J, et al. Characterization of TMPRSS2-ETS gene aberrations in androgen-independent metastatic prostate cancer. Cancer Res. 2008;68:3584–90. doi: 10.1158/0008-5472.CAN-07-6154. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.