

Abstract
In a previous study we showed that pharmacological blockade of the neurokinin-1 receptors attenuated the methamphetamine-induced toxicity of the striatal dopamine terminals. In the present study we examined the role of the neurokinin-1 receptors on the methamphetamine-induced apoptosis of some striatal neurons. To that end, we administered a single injection of METH (30 mg/kg, i.p.) to male mice. METH induced the apoptosis (TUNEL) of approximately 20% of striatal neurons. This percentage of METH-induced apoptosis was significantly attenuated by either a single injection of the neurokinin-1 receptor antagonist WIN-51,708 (5 mg/kg, i.p.) or the ablation of the striatal interneurons expressing the neurokinin-1 receptors (cholinergic and somatostatin) with the selective neurotoxin [Sar9,Met(O2)11] substance P-saporin. Next we assessed the levels of striatal 3-nitrotyrosine (3-NT) by HPLC and immunohistochemistry. METH increased the levels of striatal 3-NT and this increase was attenuated by pretreatment with WIN-51,708. Our data support the hypothesis that METH-induced striatal apoptosis occurs via a mechanism involving the neurokinin-1 receptors and the activation of nitric oxide synthesis. Our findings are relevant for the treatment of METH abuse and may be relevant to certain neurological disorders involving the dopaminergic circuitry of the basal ganglia.
Keywords: methamphetamine, neurotoxicity, substance P, apoptosis, nitric oxide, striatum
Methamphetamine (METH) is a widely abused drug in the United States. A recent study shows that 7.3% of individuals between the ages of 19–30 reported the lifetime use of METH (Johnston et al., 2007). Several studies demonstrate that METH is damaging to the brain. For example, METH has been known to induce the depletion of dopamine in the striatum (Seiden et al., 1975), depletion of the dopamine terminal markers tyrosine hydroxylase and dopamine transporters (Ricaurte et al., 1980; Schmidt et al., 1985), inactivation and oligomerization of dopamine transporters (Fleckenstein et al., 1997; Baucum et al., 2004), activation of striatal microglia (Thomas et al., 2004, 2008), accumulation of intracellular inclusions in some striatal neurons (Fornai et al., 2004), degeneration of dopamine terminals (Ricaurte et al., 1982, 1984; O’Callaghan and Miller, 1994) and the loss of some striatal and cortical neurons (Pu et al., 1996; Eisch and Marshall, 1998; Deng et al., 2001; Bowyer et al., 2008). Moreover, some of these neural deficits have also been observed in human METH users. For example, after three years abstaining from METH use, PET scans revealed a loss of dopamine transporters among former METH users (Wilson et al., 1996; McCann et al., 1998; Volkow et al., 2001a,b), and MRI studies suggest METH also induced cell death (Ernst et al., 2000). These findings suggest that METH use poses a serious problem (Freese et al., 2002). Despite a wealth of information on the damaging effects of METH both in laboratory animal species and humans, the mechanism by which METH induces neural damage in the brain remains poorly understood.
Treatment with METH augments the release of glutamate in the striatum (Nash and Yamamoto, 1992) inducing neurotoxicity in this brain region (Sonsalla et al., 1991). For example, the N-methyl-D-aspartic acid (NMDA) receptor antagonist MK-801 decreases METH-induced striatal dopamine overflow, attenuating damage to the dopamine terminals (O’Dell et al., 1992). Moreover, agents that abrogate the increase of extracellular glutamate also prevent METH-induced decreases in dopamine content (Stephans and Yamamoto, 1994). Compelling evidence suggests oxidative stress to be attributable to METH-induced neurodegeneration (De Vito and Wagner, 1989). METH-induced dopaminergic neurotoxicity in the striatum depends on the extracellular accumulation of dopamine (O’Dell et al., 1993) and its auto-oxidation (Fridovich, 1986). Pharmacological inhibition of nitric oxide synthase (NOS) and the deletion of the gene for this enzyme in mice protects the striatum from METH suggesting a role for nitric oxide (Itzhak and Ali, 1996; Imam et al., 2001a,b).
The neuropeptide substance P participates in neurodegeneration. For example, the release of substance P induces glutamate release in the hippocampus of the rodent brain (Liu et al., 1999a). Exposure to the excitotoxin kainate in wild-type mice results in the death of neurons in the hippocampus, but mice lacking the preprotachykinin-A gene (encoding substance P) treated with kainate do not display hippocampal cell death (Liu et al., 1999b). Our laboratory has demonstrated that neurokinin-1 receptor (substance P receptor) antagonists prevent METH-induced loss of neurochemical markers of toxicity such as tyrosine hydroxylase, dopamine transporters, and tissue dopamine content in the striatum (Yu et al., 2002, 2004). The neurokinin-1 receptor has been associated with the formation of nitric oxide. For example, in cultured synoviocytes substance P acting via the neurokinin-1 receptor increases nitric oxide production (O’Shaughnessy et al., 2006). Moreover, in rat skin microvasculature substance P mediates inflammation by increasing nitric oxide (Ralevic et al., 1995). Activation of the NMDA receptor has been linked to production of nitric oxide (Dawson and Dawson, 1996) and substance P modulates the synthesis of nitric oxide. In this study we demonstrate the involvement of the striatal neurokinin-1 receptor in the METH-induced apoptosis of some striatal neurons. We combined pharmacological and lesion studies to demonstrate the role of the neurokinin-1 receptor in this phenomenon. Moreover, we provide biochemical evidence supporting the hypothesis that the striatal neurokinin-1 receptors affect the METH-induced production of nitric oxide in this brain region (we measured 3-NT, an indirect index of nitric oxide synthesis).
MATERIALS & METHODS
Animals
Male ICR mice (Taconic, Germantown, NY) between 10 to 13 weeks of age were housed individually on a 12-h light/dark cycle with food and water available ad libitum. The mice were habituated for two weeks prior to commencement of intraperitoneal (i.p.) drug administration or intrastriatal microinjection. All procedures regarding animal use were performed in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and were approved by the Institutional Animal Care Committee at Hunter College of the City University of New York.
Drug Preparation and Administration
Methamphetamine (METH) (30 mg/kg of body weight) (Sigma, St. Louis, MI) was dissolved in phosphate-buffered saline (PBS) and administered as a single i.p. injection. METH was administered to either 10-week-old ICR mice or 13-week-old ICR mice three weeks after their instriatal microinjections of saporin (SAP) (Advanced Targeting Systems, San Diego, CA) and [Sar9,Met(O2)11] substance P-saporin (SSP-SAP) (Advanced Targeting Systems, San Diego, CA). The nonpeptide neurokinin-1 receptor antagonist, WIN 51,708 (Sigma/RBI, St. Louis, MI), was dissolved in 45% 2-hydroxylpropyl-β-cyclodextrin (Sigma/RBI, St. Louis, MI) and PBS (1:4) and administered i.p. 30 minutes prior to METH treatment. SAP and SSP-SAP were dissolved in PBS and administered by intrastriatal microinjections. One side received SSP-SAP and the contralateral side SAP serving as an internal control.
After one, two, or three weeks post-intrastriatal microinjections of SAP and SSP-SAP, animals were perfused transcardially with 30 ml of PBS with heparin followed by 30 ml of 4% paraformaldehyde in PBS. The mice were anesthetized with i.p. injections of ketamine/acepromazine (100mg/kg, 3mg/kg of body weight) prior to transcardial perfusion. The brains were post-fixed in 4% paraformaldehyde in PBS overnight and cryoprotected in 30% sucrose in PBS at 4 °C. All tissues were then frozen and stored at −80 °C until use.
Intrastriatal Microinjections
Mice were anesthetized with inhaled isoflurane (2.5% for induction, 2% for maintenance) and their heads were placed in a stererotaxic frame (Model 5000, David Kopf Instruments, Tujunga, CA). A hole was drilled on the skull and a 25 gauge 2 μl Hamilton microinjection syringe was lowered into the striatum. Distance of injection sites from bregma was determined using a mouse brain atlas (Franklin and Paxinos, 1997): anteroposterior +0.5 mm; mediolateral ±2.0 mm; dorsoventral −2.5 mm. The microinjection needle was left in position for five minutes prior to drug injection of 1.0 μl PBS (pH 7.4), 1.0 μl 4 ng/μl SAP (Advanced Targeting Systems, San Diego, CA), or 1.0μl 4ng/μl SSP-SAP (Advanced Targeting Systems, San Diego, CA). Drugs were injected over a ten-minute (~0.1μl/min) period and the needle was left in place for an additional five minutes before removal from the striatum.
TUNEL (terminal deoxyncleotidyl transferase-mediated dUTP nick end labeling) Histochemistry
Freshly frozen 20 μm coronal sections were taken between bregma 0.38 ± 0.1 mm and fixed in 4% paraformaldehyde for 30 minutes. After washing with PBS, sections were immersed in 0.4% Triton-X-100 in PBS for five to ten minutes at 70 °C. Sections were then washed and TUNEL reactions (Roche Applied Science, Indianapolis, IN) were applied directly onto sections and incubated for one hour in a humidified chamber. After TUNEL staining, sections were counterstained with DAPI (TUNEL and DAPI are both nuclear stains). Stained sections were then washed in PBS and coverslipped with Vectashield (Vector Laboratories, Burlingame, CA). Images were taken using the Leica TCS SP2 Spectral Confocal Microscope (Leica Microsystems).
Immunocytochemistry of somatostatin, choline acetyltransferase, Parvalbumin, DARRP-32, neurokinin-1 receptor and 3-NT
Staining of striatal 20 μm coronal sections from one, two, and three weeks post-intrastriatal microinjections were all processed by the free-floating method.
In staining for choline acetyltransferase (ChAT), non-specific binding sites were blocked with 5% normal rabbit serum in 0.2% Triton-X-100 in PBS for one hour. Sections were washed in PBS and incubated with goat anti-ChAT (1:500, Chemicon, Temecula, CA) with 2% normal rabbit serum in 0.2% Triton-X-100 overnight. After PBS wash, sections are incubated with Cy3-conjugated rabbit anti-goat (1:1000, Chemicon, Temecula, CA) with 1% normal rabbit serum in 0.2% Triton-X-100 in PBS for one hour and washed with PBS.
In staining for somatostatin (SST), dopamine and cAMP regulated phosophoprotein 32 kDa (DARPP-32), and the neurokinin-1 receptor, non-specific binding sites are blocked with 5% normal goat serum (Vector Laboratories, Burlingame, CA) in 0.2% Triton X-100 in PBS for 1 hour. Sections were then incubated with rabbit anti-SST (1:500, Chemicon, Temecula, CA), rabbit anti-DARPP-32 (1:250, Cell Signaling Technology, Beverly, MA), or rabbit anti-neurokinin-1 receptor (1:1000, Chemicon, Temecula, CA) with 2% normal goat serum in 0.1% Triton X-100 in PBS overnight at 4°C. After PBS wash, sections are incubated with Cy3-conjugated goat anti-rabbit (1:1000, Chemicon, Temecula, CA) or FITC-conjugated goat anti-rabbit (1:1000, Chemicon, Temecula, CA) in 1% normal goat serum in 0.2% Triton X-100 in PBS for one hour and washed with PBS.
In staining for parvalbumin, nonspecific binding sites were blocked with M.O.M. ® Mouse Ig Blocking Reagent (Vector Laboratories, Burlingame, CA) for one hour. Sections were washed and incubated with M.O.M. ® diluent for 15 minutes. Sections were then incubated mouse anti-parvalbumin (1:2000, Chemicon, Temecula, CA) in M.O.M.® diluent with 0.1% Triton X-100 in PBS overnight at 4 °C. Sections were next washed in PBS and blocked again with 5% normal goat serum in 0.1% Triton X-100 in PBS for 30 minutes. After washing with PBS, sections were incubated with Cy3-conjugated goat anti-mouse (1:1000, Chemicon, Temecula, CA) with 1% normal goat serum in 0.1% Triton X-100 in PBS for one hour and then washed again with PBS. Sections were then mounted on glass slides and overlaid with a coverslip using Vectashield (Vector Laboratories, Burlingame, CA). A set of sections was incubated with an antibody against 3-NT (1:500, SCBT, CA) followed with a biotinylated secondary antibody and color development with a DAB Substrate Kit (Vector Laboratories, CA).
No staining was observed when primary antibody was left out or when primary antibody was pre-absorbed with either SST or ChAT. All incubations and washes were performed at 25 °C unless otherwise stated. Sections were viewed and photographed with a Leica TCS SP2 Spectral Confocal Microscope.
Double-labeling: Immunocytochemistry of neurokinin-1 receptors and TUNEL staining
Dual staining of the neurokinin-1 receptor and TUNEL staining were processed on striatal 20 μm coronal sections from three weeks post-intrastriatal surgery. Sections were rinsed in PBS and then immersed in heated 0.4% Triton-X-100 in PBS for five minutes and immediately washed in PBS. Staining of neurokinin-1 receptors and TUNEL-labeling were then processed as described above. Sections were viewed and photographed with a Leica TCS SP2 Spectral Confocal Microscope (Leica Microsystems).
HPLC-EC detection of 3-NT and tyrosine concentration
Determination of 3-NT and tyrosine in mouse striatum was performed by HPLC-Coularry electrochemical detection method (Imam and Ali, 2001). Briefly, one striatum (alternating between left and right at Bregma 0.38 +/−0.2 mm) was completely dissected out from a 1.0 mm thick coronal section and sonicated in 400 μl of 10 mM sodium acetate NaOAc, pH 6.5. A 25 μl aliquot of the homogenate was used to determine protein concentration (BCA method). The remaining homogenate was centrifuged at 14,000 rpm (Eppendorf 5403 centrifuge) for 10 minutes at 4°C. The supernatant was removed and treated with 100 ml of 1 mg/ml pronase for 18 hours at 50°C. Enzymatic digests were then treated with 0.5 ml of 10% TCA and centrifuged at 14,000 rpm for 10 minutes at 4°C. Supernatants were then passed through a 0.2 μm PVDF filter before injection onto the HPLC instrument. Samples were analyzed on an ESA (Cambridge, MA, USA) CoulArray HPLC equipped with 8 electrochemical channels using platinum electrodes arranged in line and set to increasing specified potentials [channel (potential): 1 (320 mV); 2 (450 mV) ; 3 (490 mV) ;4 (610 mV) ; 5(670 mV) ; 6( 870 mV) ; 7( 900 mV) ; 8( 930mV)]. The analytical column was a Luna C18 column (3 micron, 2.1× 150mm, Phenomenex Co., Torrance, CA). The mobile phase was 50 mM NaAc, 5% (v/v) methanol, pH 4.8. HPLC was performed under isocratic conditions. 3-NT and tyrosine were quantified relative to known standards. 3-NT values were represented as 3-NT per 100 tyrosines. We divided 3-NT values in all groups by tyrosine to normalize the data (Imam and Ali, 2001).
Cell Counts and Quantification
Coronal sections of 20 μm thickness were taken from bregma 0.38 ± 0.1 mm. TUNEL-positive cells were quantified in areas of 0.26 mm2 for the dorsal-medial [DM], dorsal-lateral [DL], ventral-medial [VM], and ventral-lateral [VL]) regions of the CPu (See inset for Fig. 1).
Figure 1.
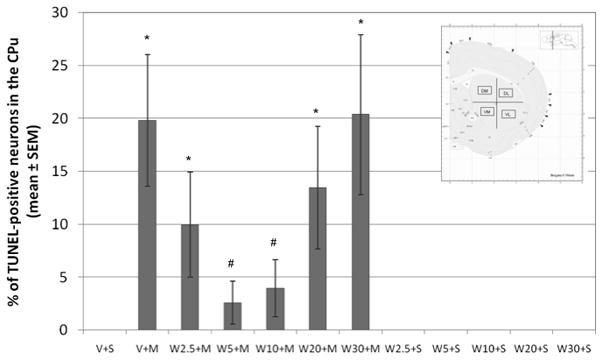
Pretreatment with the neurokinin-1 receptor antagonist, WIN 51,708, attenuates METH-induced cell death in the mouse striatum. Mice (10–12 per group) received a single intraperitoneal (i.p.) injection of the neurokinin-1 receptor antagonist, WIN 51, 708 (2.5, 5, 10, 20 or 30 mg/kg of body weight), 30 minutes prior to METH treatment (30 mg/kg of body weight, i.p.). Cell death was detected on striatal tissue sections by TUNEL 24 hours post-METH treatment. *p<0.05 compared to V + S. #p<0.05 compared to V + M. Antagonist alone did not induce TUNEL staining. N=12–18 animals per group. Vehicle (V), saline (S), METH (M), WIN-51,708 (W). Inset: Schematic of the striatum indicating the four regions of the CPu (caudate-putamen) where cell counts were conducted. Dorsal-medial (DM), ventral-medial (VM), dorsal-lateral (DL), and ventral-lateral (VL) regions of the CPu. Black boxes indicate the 0.26 mm2 area where TUNEL-positive cell counts were conducted within each region.
Neurons were labeled with NeuN (immunofluorescence) and average neuronal cell counts were done as previously described (Zhu et al., 2006) from six control animals as a baseline for quantification of the percentage of TUNEL-positive neurons in experimental animals. TUNEL cell counts were averaged from five 20 μm serial sections per animal.
Body Temperature
Core body temperature was determined using a BAT-12 thermometer coupled to RET-3 mouse rectal probe (Physitemp Instruments, Clifton, NJ). Ambient room temperature was maintained at 20–22 °C.
Statistical Analysis
Analysis was performed from mean ± SEM. The differences between groups were analyzed first by two-way ANOVA and then followed by post hoc comparison using Fisher’s protected least significance test. The significance criterion was set at p ≥0.05.
RESULTS
Attenuation of METH-induced Apoptosis
We assessed METH-induced apoptosis by TUNEL in the dorsal-medial (DM), dorsal-lateral (DL), ventral-lateral (VL) and ventral-medial (VM) aspects of the striatum as depicted in schematic form in the inset of Figure 1. Although the DM region of the striatum exhibited a trend of less TUNEL-positive neurons after METH exposure, no significant difference was found between regions (data not shown). METH was injected at a dose of 30 mg/kg (i.p.) of body weight because we had determined in a previous study that this dose induces significant levels of apoptosis and dopamine terminal toxicity (Zhu et al., 2005). In this previous study we also observed that the peak of apoptosis occurs 24 hours after METH administration. In a different study, we also determined that METH induces apoptosis in neurons but not glial cells (Zhu et al., 2006). METH induced TUNEL staining in 19.8+/−6.2% of striatal neurons (Figure 1). Pretreatment with the neurokinin-1 receptor antagonist WIN-51,708, 30 minutes before METH treatment, significantly attenuated the METH-induced apoptosis in the striatum. With 2.5 mg/kg of the drug prior to METH it attenuated 50+/−25% of the METH-induced TUNEL-positive neurons (9.9+/−5% of striatal neurons were TUNEL-positive) and with 5 and 10 mg/kg, it attenuated 87+/−10% (f (17,17)=9.381, p<0.0001) and 80+/−14% (f (17,14)=6.426, p<0.0001) of METH-induced TUNEL-positive neurons, respectively (2.6+/−2% and 4+/−2.7% of striatal neurons were TUNEL-positive). The F values are low but this may be due to the large interanimal variability in TUNEL staining. T-tests, ANOVA and posthoc ANOVA demonstrated significance. At higher doses of WIN-51,708, the drug became less or no longer protective (Figure 1). With 20 and 30 mg/kg of the drug prior to METH, 13.5+/−5.8% and 20.3+/−7.6%, respectively, of striatal neurons were TUNEL-positive.
The amount of apoptosis induced by a single injection of METH displays a high degree of inter-animal variability with some animals displaying as much as 70% TUNEL staining. However, pretreatment with WIN-51,708 consistently diminished METH-induced apoptosis in all animals tested (data not shown). Pretreatment with WIN-51,708 did not inhibit the METH-induced hyperthermia (data not shown).
Selective Ablation of Striatal Neurokinin-1 Receptor-Expressing Interneurons
As a complement to finding that a neurokinin-1 receptor antagonist attenuates METH-induced apoptosis in some striatal neurons, we investigated the effect of removal of cells expressing the striatal neurokinin-1 receptor (namely, cholinergic and somatostatin/NPY/NOS interneurons) on the METH-induced striatal apoptosis. To that end, a selective neurokinin-1 receptor agonist, [Sar9,Met(O2)11] substance P, conjugated to a ribosomal-inactivating toxin, saporin, (SSP-SAP) was employed (Wiley and Lappi, 1999). We first ensured that this neurotoxin was effective and selective in eliminating neurokinin-1 receptor-bearing neurons by immunohistochemical methods. SSP-SAP was injected into the ipsilateral striatum and saporin (SAP) into the contralateral striatum as control. In a separate experiment, we ascertained that intrastriatal injection of SAP did not affect neurokinin-1 receptor immunoreactivity in the striatum (data not shown).
There was a loss of neurokinin-1 receptor immunoreactivity in the ipsilateral striatum that received the SSP-SAP, but no loss of neurokinin-1 receptor staining in the contralateral striatum that was injected with SAP alone (Figure 2). There was complete loss of neurokinin-1 receptor staining in the ipsilateral striatum three weeks after intrastriatal injection with SSP-SAP (Figure 2). For this reason, we chose the three-week time point for all subsequent experiments. To emphasize that only the neurokinin-1 receptor-expressing interneurons were ablated in the core of the lesion, we showed that immnunostaining for selective markers of these interneurons, somatostatin (Figure 3a–d) and choline acetyltransferase (Figure 3e–h), were absent on the side receiving the injection of SSP-SAP. In sharp contrast, after a similar SSP-SAP injection, GABA-parvalbumin interneurons and the projection neurons (stained with DARPP-32 and representing approximately 90% of all striatal neurons) were spared (Figure 4a–d and e–h, respectively). After extensive microscopic examination and cell counts, we could not find any evidence supporting loss DARPP-32-positive projection neurons after SSP-saporin.
Figure 2.
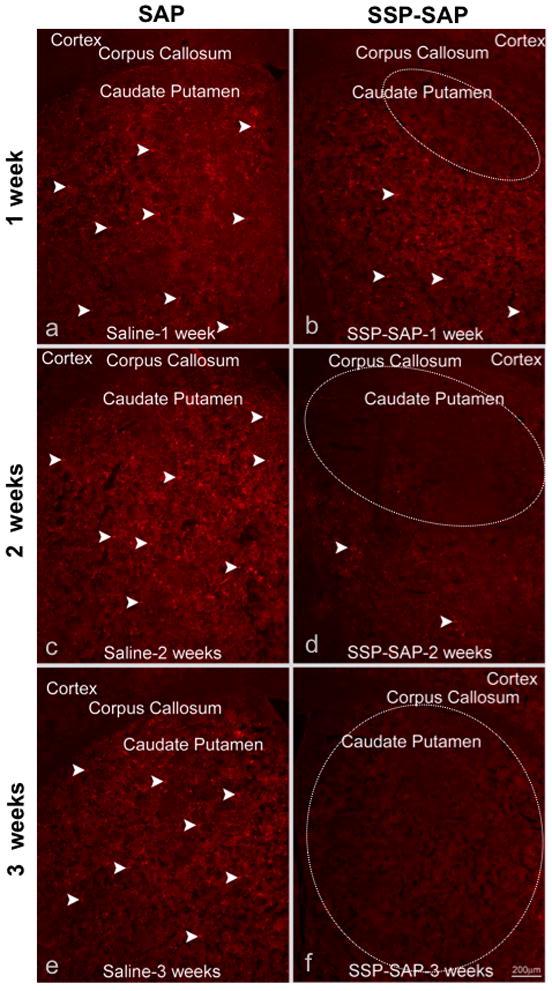
Ablation of striatal neurokinin-1 receptor-expressing neurons using SSP-SAP. Animals were intrastriatally injected with SSP-SAP [4ng, 1 ul volume] (b, d and f) or saporin (SAP) [4ng, 1 ul volume] ) on the contralateral side (a, c and e). Animals were sacrificed at 1, 2 or 3 weeks after the intrastriatal injections and sections of striatal tissue were immunostained with an antibody against the neurokinin-1 receptor. Microinjection of SAP alone had no effect on neurokinin-1 receptor immunostaining in the striatum. Note the lack of immunostaining for the neurokinin-1 receptor at 3 weeks after the injection with SSP-SAP (f). White dotted circle indicates the region lacking neurons that express the neurokinin-1 receptors. White arrows indicate immunostaining of representative neurokinin-1 receptor-bearing interneurons. Magnification bar is 200 μm.
Figure 3.
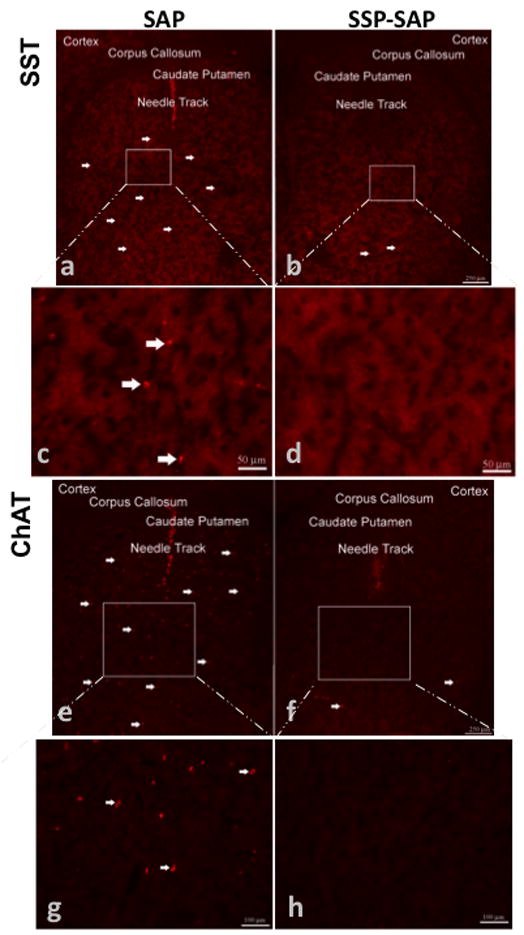
Striatal somatostatin (SST) and cholinergic (ChAT, choline acetyltransferase) interneurons were selectively ablated using SSP-SAP. Animals were intrastriatally injected with SSP-SAP [4ng, 1 ul volume] (b,d,f,h) and saporin (SAP) [4ng, 1 ul volume] on the contralateral side (a,c,e,g). Three weeks after microinjections, coronal sections of striatal tissue were immunostained with antibodies against SST or ChAT. White arrows indicate representative somatostatin (a–c) or ChAT staining (e–g). Note the absence of immunostaining for SST or ChAT interneurons on the side injected with SSP-SAP (d,h). Magnification bars indicate 250 μm for low magnification images (a,b,e,f) and 50 μm for high magnification images (c,d,g,h).
Figure 4.
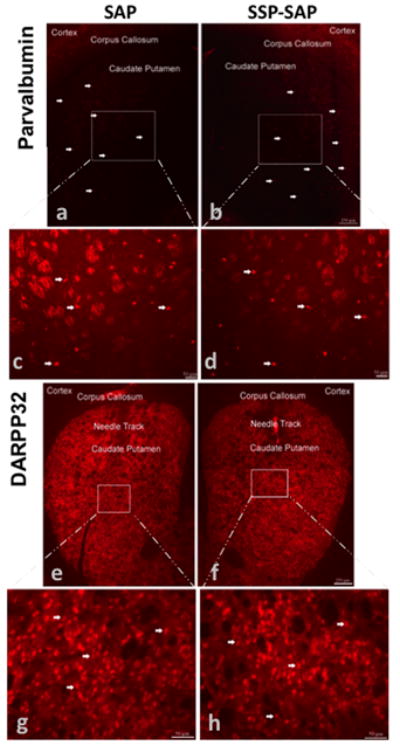
Parvalbumin-containing GABAergic interneurons and DARPP-32-containing projection neurons remained intact in the striatum after exposure to SSP-SAP. Animals were intrastriatally injected with SSP-SAP [4ng, 1 ul volume] (b,d,f,h) and saporin (SAP) [4ng, 1 ul volume] on the contralateral side (a,c,e,g). Coronal tissue sections were immunostained with antibodies against parvalbumin or dopamine and cyclic-AMP-regulated phosphoprotein, 32 kDa (DARPP-32) three weeks after microinjections. White arrows indicate representative parvalbumin (a–d) or DARPP-32 staining (e–h). Magnification bar indicates 250 μm for low magnification images (a,b,e,f) and 50 μm for high magnification images (c,d,g,h).
Ablation of Striatal Neurokinin-1 Receptor-Expressing Interneurons Prevents Apoptosis
To assess the role of the striatal neurokinin-1 receptors on METH-induced apoptosis, we gave a cohort of mice intrastriatal injection of SSP-SAP and SAP. Three weeks later, the mice were given METH (30 mg/kg, i.p.) and were sacrificed 24 hours after METH and used to assess apoptosis by TUNEL. The peak of METH-induced apoptosis occurs 24 hours after METH treatment (Zhu et al., 2005).
As shown in Figure 5, SSP-SAP effectively eliminated the neurokinin-1 receptor immunostaining in the striatum. More importantly, in the absence of neurokinin-1 receptor-positive immunostaining, there was no detectable METH-induced apoptosis. In contrast, on the contralateral side injected with SAP, while there was little loss of neurokinin-1 receptor immunostaining there was considerable apoptosis (Figure 5). These findings indicate that the removal of neurokinin-1 receptors of the striatum provides protection against METH-induced apoptosis. These results demonstrate the involvement of the striatal neurokinin-1 receptors in the METH-induced apoptosis of some striatal neurons.
Figure 5.
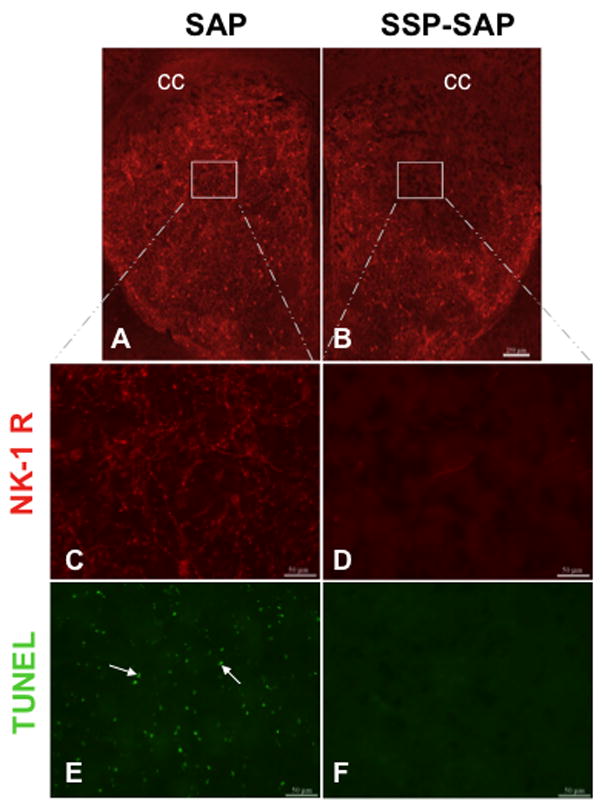
Ablation of striatal neurokinin-1 receptor-expressing interneurons prevented methamphetamine-induced cell death in the striatum. Animals received intrastriatal injections of SSP-SAP [4ng, 1 ul volume] into one striatum (B, D & F) and saporin (SAP) [4ng, 1 ul volume] into the contralateral side (A, C & E). Three weeks after the injections, the animals received a single intraperitoneal injection of METH (30 mg/kg). The animals were then sacrificed one day after METH treatment. Striatal sections were processed for immunofluorescence using TUNEL and an antibody against the neurokinin-1 receptor. White square indicates region devoid of neurokinin-1 receptor immunofluorescence. Local ablation of striatal neurokinin-1 receptor-expressing interneurons conferred protection from METH-induced cell death on the ipsilateral side (B, D and F). No protection was observed on the contralateral side receiving only SAP (A, C and E). White arrows show representative TUNEL-positive cells. Magnification bars are 250 μm (A & B) and 50 μm (C, D, E & F). cc = corpus collasum.
The Striatal Neurokinin-1 Receptor Modulates the Formation of 3-NT
In order to assess the effect of METH on nitric oxide formation we measured the levels of striatal 3-NT (an indirect index of nitric oxide formation) by HPLC in homogenates of tissue from one striatum. The mice (n=8) were injected with METH (30 mg/kg, i.p.) and were sacrificed at 1, 2, 8, 16 and 32 hours after METH. The striatal levels of 3-NT rose steadily between 1–16 hours in the METH treated groups. In the groups receiving METH and WIN-51,708 (5 mg/kg, i.p. 30 minutes before METH), the levels of striatal 3-NT were significantly lower compared to control (Figure 6). Saline or WIN-51,708 injections alone failed to alter the levels of striatal 3-NT at any time point (Figure 6). Pretreatment with WIN-51,708 caused the greatest inhibition of the METH-induced formation of 3-NT at two (p=0.0021, f=17) and eight (p=0.0128, f=8.5) hours after METH (Figure 6).
Figure 6.
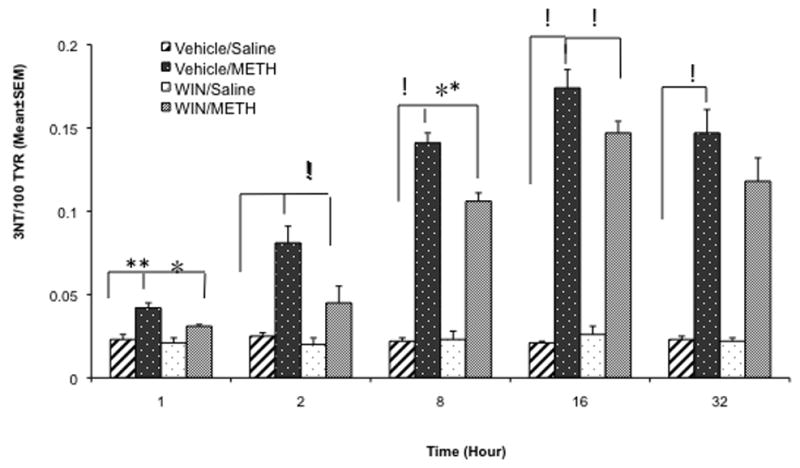
Pretreatment with the neurokinin-1 receptor antagonist WIN-51,708 attenuated the METH-induced increase of striatal 3-nitrotyrosine (3-NT). The mice (n=8) received an injection of WIN-51,708 (WIN, 5 mg/kg, i.p.) 30 minutes prior to METH (30 mg/kg, i.p.) and the animals were sacrificed at the times indicated under the x-axis. 3-NT levels were determined by HPLC from one striatum. Note that the neurokinin-1 receptor antagonist WIN-51,708 significantly attenuated the METH-induced augmentation of striatal 3-NT (an indirect index of nitric oxide synthesis) at all time points. TYR, tyrosine. The data were analyzed by 2-way ANOVA. *p<0.01, **p<0.001, !p<0.0001.
We also assessed the effect of WIN-51,708 on the METH-induced increase of striatal 3-NT formation by immunohistochemistry using an antibody against 3-NT coupled to diaminobenzidine staining. The mice (n=8) received an injection of METH (30 mg/kg, i.p.) and were sacrificed four hours after the injection. Cells staining positive for 3-NT were visible in the striatum four hours after METH. In contrast, the mice receiving METH and WIN-51,708 (5 mg/kg, i.p. 30 minutes before METH) failed to display 3-NT positive cells (Figure 7). No staining was observed in the METH group when the antibody was left out or with the addition of 3-NT (data not shown). We did not observe 3-NT-positive cells in the absence of METH suggesting that the levels of 3-NT in normal brain are below the level of detection of our histological assay. Taken together, the biochemical and histological data support the hypothesis that the METH-induced increase of striatal 3-NT can be modulated by the neurokinin-1 receptor.
Figure 7.
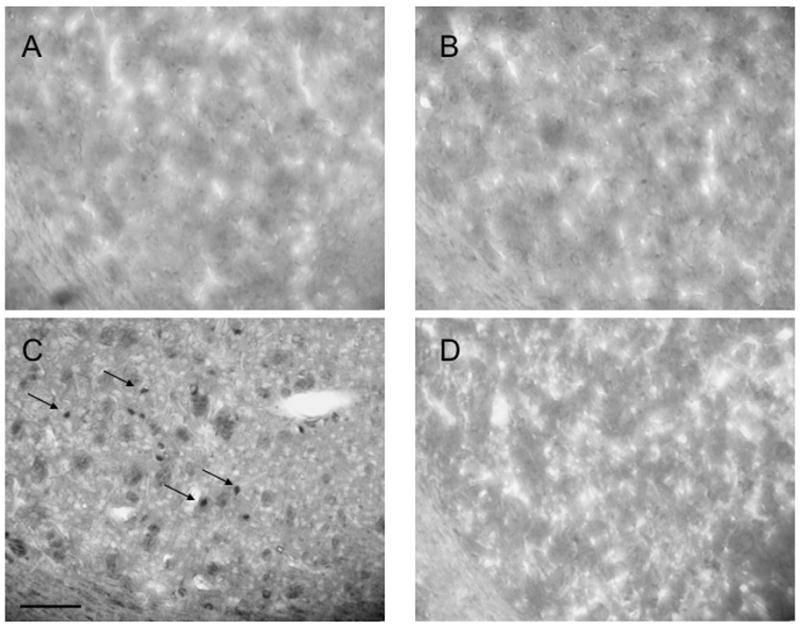
Histological evidence demonstrating that pharmacological blockade of the neurokinin-1 receptor with WIN-51,708 (WIN) attenuates the METH-induced augmentation of striatal 3-NT, an indirect index of nitric oxide production. A, vehicle/saline; B, WIN-/saline; C, vehicle/METH; D, WIN/METH. 3-NT was detected by immunohistochemistry using diaminobenzidine staining in the dorsal-medial aspect of the striatum (see inset of Figure 1, DM). The mice (n=8) were pre-treated with WIN-51,708 (5 mg/kg, i.p.) 30 minutes prior to METH (30 mg/kg, i.p.) and were sacrificed four hours after the METH injection. Arrows in C indicate the position of 3-NT-positive cells from an animal treated with METH. Scale bar = 100μm.
DISCUSSION
In the present study we demonstrate that pharmacological blockade of the neurokinin-1 receptor with a selective antagonist that enters the brain, WIN-51,708, significantly attenuated the apoptosis of some striatal neurons induced by METH. In addition, we demonstrate that ablation of the striatal interneurons that express the neurokinin-1 receptors (cholinergic and somatostatin/NPY/NOS) with the selective neurotoxin SSP-SAP prevented the METH-induced apoptosis of some striatal neurons. Because a plethora of studies link the METH-induced neural damage with nitric oxide, we assessed the levels of striatal 3-NT (an indirect index of nitric oxide formation). METH induced a steady increase of striatal 3-NT from 1–16 hours. Interestingly, pretreatment with WIN-51,708 attenuated the METH-induced augmentation of striatal 3-NT. Taken together with previous work from our laboratory demonstrating that the same neurokinin-1 receptor antagonist attenuated METH-induced depletion of striatal tyrosine hydroxylase, dopamine transporters, dopamine tissue content, and the induction of glial fibrillary acidic protein in astrocytes (Yu et al., 2002, 2004), we propose the hypothesis that the striatal neuropeptide substance P (the natural ligand of the neurokinin-1 receptor) plays a role in the METH-induced neural damage at both pre- and post-synaptic sites.
Two immediate consequences of METH on the striatal dopamine terminals are the release of vesicular dopamine stores and the reversal of the plasmalemmal dopamine transporters. The latter event is responsible for the excessively high levels of extracellular dopamine in the presence of METH (Fischer and Cho, 1979; Sulzer et al., 1995; Jones et al., 1998). Dopamine receptor antagonists lessen the METH-induced depletions of dopamine terminal markers (Sonsalla et al., 1986) and the apoptosis of some striatal neurons (Xu et al., 2005), linking dopamine receptor signaling with neural damage. This property of METH has a direct impact on the striatal neurons, most of which express more than one neuropeptide (Hokfelt et al., 2000). There is significant evidence demonstrating that pharmacological agonists or antagonists of the dopamine receptors modulate the levels of expression of striatal neuropeptides (For review see Angulo and McEwen, 1994). For example, high doses of METH increase striatal substance P (Hanson et al., 1986) and preprotachykinin-A mRNA levels (Adams et al., 2001). Substance P has been found to contribute to the rewarding effects of opiates (Ripley et al., 2002; Gadd et al., 2003). Moreover, antagonists of the substance P receptor display anti-depressant activity (Kramer et al., 1998; Rupniak et al., 2001), and modulation of behavior induced by activation of the striatal dopamine D1 receptor (Krolewski et al., 2005). Recently, we postulated that increased signaling by substance P through the neurokinin-1 receptor of the striatum constitutes one mechanism contributing to the METH-induced toxicity of the dopamine terminals (Yu et al., 2002, 2004). Here we demonstrate that pharmacological blockade of the neurokinin-1 receptor with WIN-51,708, or ablation of the striatal interneurons bearing the neurokinin-1 receptor with the selective neurotoxin SSP-SAP, attenuates METH-induced apoptosis of some striatal neurons. We propose that the excessively high levels of extracellular dopamine induced by METH causes excessive release of the neuropeptide substance P, which in turn causes excessive production of the diffusible second messenger nitric oxide. METH-induced dopamine overflow may be the primary causative agent because dopamine overflow occurs minutes after METH whereas glutamate overflow occurs after the third injection of METH in a schedule involving four injections at two-hour intervals (Nash and Yamamoto, 1992). In support of this model, we rely on the observation that exposure to METH causes the preferential internalization of the substance P/neurokinin-1 receptor complex in striatal somatostatin/NPY/NOS interneurons. Moreover, this METH-induced internalization of the peptide/receptor complex can be attenuated with selective antagonists of the dopamine D1 or D2 receptors (Wang and Angulo, 2009). Taken together, these results suggest that there is a causal relationship between METH and the somatostatin/NPY/NOS interneuron of the striatum via substance P and the neuroknin-1 receptor.
In the present study we have found that ablation of the striatal neurokinin-1 receptor-expressing interneurons with the selective neurotoxin SSP-SAP prevented the METH-induced apoptosis of some striatal neurons, suggesting that the participation of this receptor is required for cell death. The neurokinin-1 receptor is expressed by two populations of striatal interneurons: the cholinergic and the somatostatin/NPY/NOS (Kawaguchi et al., 1995). Based on our results, we hypothesize that of these two populations of interneurons, the somatostatin/NPY/NOS interneuron plays a predominant role in METH-induced apoptosis through the production of the diffusible second messenger nitric oxide. In a separate study, we observed that 30 minutes after a single systemic injection of METH (30 mg/kg), the substance P/neurokinin-1 receptor complex was internalized into endosomes primarily in the somatostatin/NPY/NOS interneurons (Wang and Angulo, 2009). Moreover, we also found that METH kills significant numbers of striatal projection neurons, cholinergic and GABA-parvalbumin interneurons, but spares the somatostatin/NPY/NOS interneurons (Zhu et al., 2006). These observations, taken together with the finding that ablation of the neurokinin-1 receptor-expressing interneurons prevented METH-induced apoptosis, suggest that the striatal neurokinin-1 receptors play a role in the cell death induced by METH in this brain region.
The neurotoxic cascade activated by nitric oxide has been implicated in the degeneration of various brain regions (Dawson and Dawson, 1996). For example, pharmacological inhibition of neuronal nitric oxide synthase by 7-nitroindazole attenuated striatal lesions produced by intrastriatal injection of NMDA (Schulz et al., 1995). Furthermore, the striatum of mice lacking the neuronal nitric oxide synthase gene was significantly more resistant to the excitotoxic lesions produced by intrastriatal injections of NMDA than wild-type mice (Ayata et al., 1997). In the present study we have observed that METH increases the levels of striatal 3-NT up to 32 hours after a bolus injection of METH. Moreover, these increases are attenuated by pre-treatment with the selective neurokinin-1 receptor antagonist WIN-51,708 (see Figures 6 & 7). The neurokinin-1 receptor antagonist did not completely abolish the production and accumulation of striatal 3-NT suggesting that other mechanisms are involved. However, it is interesting to note that attenuation of 3-NT (indirect index of nitric oxide production) suffices to protect the striatum from the apoptosis induced by METH. The magnitude of protection afforded by the neurokinin-1 receptor antagonist WIN-51,708 is dependent on the dose of the compound used (see Figure 1). At higher doses (20 and 30 mg/kg) this compound may display agonist properties at the neurokinin-1 receptor (Quartara and Maggi, 1997).
In parallel with the activation of nitric oxide synthesis via the neurokinin-1 receptor, glutamate can also increase striatal nitric oxide synthesis. For example, in cultured neurons the stimulation of NMDA receptors leads to the activation of neuronal nitric oxide synthase and production of nitric oxide (Dawson et al., 1998; Yun et al., 1998). Moreover, several studies connect the neurotransmitter glutamate with both METH-induced toxicity and damage in the striatum (Sonsalla et al., 1991; O’Dell et al., 1992). METH treatment releases glutamate in the striatum (Nash and Yamamoto, 1992). It has also been established that neurotoxic regimens of METH induce sustained release of glutamate and oxidative stress (Yamamoto et al., 1998; Yamamoto and Zhu, 1998). Thus, it is conceivable that the pharmacological blockade of the neurokinin-1 receptors may attenuate the METH-induced overflow of striatal glutamate that in turn would attenuate the production of 3-NT. This possibility is supported by the observation that the striatal overflow of glutamate induced by infusion of NMDA is dependent on endogenous striatal substance P and dopamine (Marti et al., 2005).
In conclusion, the present study demonstrates the role of the striatal neurokinin-1 receptor in METH-induced apoptosis. Our data suggest that an intrastriatal mechanism participates in the METH-induced apoptosis of some striatal neurons involving the modulation of nitric oxide production by the neurokinin-1 receptor on the somatostatin/NPY/NOS interneurons. Work in progress in this laboratory is investigating the mechanism by which the striatal neurokinin-1 receptors contribute to the apoptosis induced by METH.
Acknowledgments
This work was supported by R01 DA020142 from the National Institute on Drug Abuse to JAA. Support for infrastructure came from the Research Centers in Minority Institutions grant awarded to Hunter College by NCRR/NIH.
Abbreviations used
- METH
methamphetamine
- 3-NT
3-nitrotyrosine
- SAP
saporin
- SSP-SAP
[Sar9,Met(O2)11] substance P-saporin
- TUNEL
terminal deoxyncleotidyl transferase-mediated dUTP nick end labeling
- WIN-51,708
17-ϐ-Hydroxy-17-a-ethynyl-5-a-androstano[3,2-ϐ]pyrimido[1,2-a]benzimidazole
References
- Adams DH, Hanson GR, Keefe KA. Differential effects of cocaine and methamphetamine on neurotensin/neuromedin N and preprotachykinin messenger RNA expression in unique regions of the striatum. Neuroscience. 2001;102:843–851. doi: 10.1016/s0306-4522(00)00530-3. [DOI] [PubMed] [Google Scholar]
- Angulo JA, McEwen BS. Molecular aspects of neuropeptide regulation and function in the corpus striatum and nucleus accumbens. Brain Res Rev. 1994;19:1–28. doi: 10.1016/0165-0173(94)90002-7. [DOI] [PubMed] [Google Scholar]
- Ayata C, Ayata G, Hara H, et al. Mechanisms of reduced striatal NMDA excitotoxicity in type I nitric oxide synthase knock-out mice. J Neurosci. 1997;17:6908–6917. doi: 10.1523/JNEUROSCI.17-18-06908.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baucum AJ, 2nd, Rau KS, Riddle EL, Hanson GR, Fleckenstein AE. Methamphetamine increases dopamine transporter higher molecular weight complex formation via a dopamine- and hyperthermia-associated mechanism. J Neurosci. 2004;24:3436–3443. doi: 10.1523/JNEUROSCI.0387-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowyer JF, Robinson B, Ali S, Schmued LC. Neurotoxic-related changes in tyrosine hydroxylase, microglia, myelin, and the blood-brain barrier in the caudate-putamen from acute methamphetamine exposure. Synapse. 2008;62:193–204. doi: 10.1002/syn.20478. [DOI] [PubMed] [Google Scholar]
- Dawson TM, Sasaki M, Gonzalez-Zulueta M, Dawson VL. Regulation of neuronal nitric oxide synthase and identification of novel nitric oxide signaling pathways. Prog Brain Res. 1998;118:3–11. doi: 10.1016/s0079-6123(08)63196-9. [DOI] [PubMed] [Google Scholar]
- Dawson VL, Dawson TM. Nitric oxide neurotoxicity. J Chem Neuroanat. 1996;10:179–190. doi: 10.1016/0891-0618(96)00148-2. [DOI] [PubMed] [Google Scholar]
- Deng X, Wang Y, Chou J, Cadet JL. Methamphetamine causes widespread apoptosis in the mouse brain: evidence from using an improved TUNEL histochemical method. Brain Res Mol Brain Res. 2001;93:64–69. doi: 10.1016/s0169-328x(01)00184-x. [DOI] [PubMed] [Google Scholar]
- De Vito MJ, Wagner GC. Methamphetamine-induced neuronal damage: a possible role for free radicals. Neuropharmacol. 1989;28:1145–1150. doi: 10.1016/0028-3908(89)90130-5. [DOI] [PubMed] [Google Scholar]
- Eisch AJ, Marshall JF. Methamphetamine neurotoxicity: dissociation of striatal dopamine terminal damage from parietal cortical cell body injury. Synapse. 1998;30:433–445. doi: 10.1002/(SICI)1098-2396(199812)30:4<433::AID-SYN10>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
- Ernst T, Chang L, Leonido-Yee M, Speck O. Evidence for long-term neurotoxicity associated with methamphetamine abuse: A 1H MRS study. Neurology. 2000;54:1344–1349. doi: 10.1212/wnl.54.6.1344. [DOI] [PubMed] [Google Scholar]
- Fischer JF, Cho AK. Chemical release of dopamine from striatal homogenates: evidence for an exchange diffusion model. J Pharmacol Exp Ther. 1979;208:203–209. [PubMed] [Google Scholar]
- Fleckenstein AE, Metzger RR, Wilkins DG, Gibb JW, Hanson GR. Rapid and reversible effects of methamphetamine on dopamine transporters. J Pharmacol Exp Ther. 1997;282:834–838. [PubMed] [Google Scholar]
- Fornai F, Lenzi P, Gesi M, et al. Methamphetamine produces neuronal inclusions in the nigrostriatal system and in PC12 cells. J Neurochem. 2004;88:114–123. doi: 10.1046/j.1471-4159.2003.02137.x. [DOI] [PubMed] [Google Scholar]
- Franklin KBJ, Paxinos G. The Mouse Brain in Stereotaxic Coordinates. New York: The Academic Press; 1997. [Google Scholar]
- Freese TE, Miotto K, Reback CJ. The effects and consequences of selected club drugs. J Subst Abuse Treat. 2002;23:151–156. doi: 10.1016/s0740-5472(02)00267-2. [DOI] [PubMed] [Google Scholar]
- Fridovich I. Biological effects of the superoxide radical. Arch Biochem Biophys. 1986;247:1–11. doi: 10.1016/0003-9861(86)90526-6. [DOI] [PubMed] [Google Scholar]
- Gadd CA, Murtra P, De Felipe C, Hunt SP. Neurokinin-1 receptor-expressing neurons in the amygdala modulate morphine reward and anxiety behaviors in the mouse. J Neurosci. 2003;23:8271–8280. doi: 10.1523/JNEUROSCI.23-23-08271.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanson GR, Letter AA, Merchant K, Gibb JW. Comparison of responses by striatonigral substance P and neurokinin A systems to methamphetamine treatment. Peptides. 1986;7:983–987. doi: 10.1016/0196-9781(86)90125-7. [DOI] [PubMed] [Google Scholar]
- Hokfelt T, Broberger C, Xu ZQ, Sergeyev V, Ubink R, Diez M. Neuropeptides--an overview. Neuropharmacology. 2000;39:1337–1356. doi: 10.1016/s0028-3908(00)00010-1. [DOI] [PubMed] [Google Scholar]
- Imam SZ, Newport GD, Itzhak Y, Cadet JL, Islam F, Slikker W, Jr, Ali SF. Peroxynitrite plays a role in methamphetamine-induced dopaminergic neurotoxicity: evidence from mice lacking neuronal nitric oxide synthase gene or overexpressing copper-zinc superoxide dismutase. J Neurochem. 2001a;76:745–749. doi: 10.1046/j.1471-4159.2001.00029.x. [DOI] [PubMed] [Google Scholar]
- Imam SZ, Ali SF. Aging increases the susceptibility to methamphetamine-induced dopaminergic neurotoxicity in rats: correlation with peroxynitrite production and hyperthermia. J Neurochem. 2001b;78:952–959. doi: 10.1046/j.1471-4159.2001.00477.x. [DOI] [PubMed] [Google Scholar]
- Itzhak Y, Ali SF. The neuronal nitric oxide synthase inhibitor, 7-nitroindazole, protects against methamphetamine-induced neurotoxicity in vivo. J Neurochem. 1996;67:1770–1773. doi: 10.1046/j.1471-4159.1996.67041770.x. [DOI] [PubMed] [Google Scholar]
- Johnston LD, O’Malley PM, Bachman JG, Schulenberg JE. Monitoring the Future National Survey Results on Drug Use, 1975–2006: Volume II, College Students and Adults Ages 19–45. Bethesda, MD: National Institute on Drug Abuse; 2007. NIH Publication No. 07–6206. [Google Scholar]
- Jones SR, Gainetdinov RR, Wightman RM, Caron MG. Mechanisms of amphetamine action revealed in mice lacking the dopamine transporter. J Neurosci. 1998;18:1979–1986. doi: 10.1523/JNEUROSCI.18-06-01979.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawaguchi Y, Wilson CJ, Augood SJ, Emson PC. Striatal interneurones: chemical, physiological and morphological characterization. Trends Neurosci. 1995;18:527–535. doi: 10.1016/0166-2236(95)98374-8. [DOI] [PubMed] [Google Scholar]
- Kramer MS, Cutler N, Feighner J, et al. Distinct mechanism for antidepressant activity by blockade of central substance P receptors. Science. 1998;281:1640–1645. doi: 10.1126/science.281.5383.1640. [DOI] [PubMed] [Google Scholar]
- Krolewski DM, Bishop C, Walker PD. Intrastriatal dopamine D1 receptor agonist-mediated motor behavior is reduced by local neurokinin-1 receptor antagonism. Synapse. 2005;57:1–7. doi: 10.1002/syn.20148. [DOI] [PubMed] [Google Scholar]
- Liu H, Mazarati AM, Katsumori H, Sankar R, Wasterlain CG. Substance P is expressed in hippocampal principal neurons during status epilepticus and plays a critical role in the maintenance of status epilepticus. Proc Natl Acad Sci U S A. 1999a;96:5286–5291. doi: 10.1073/pnas.96.9.5286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H, Cao Y, Basbaum AI, Mazarati AM, Sankar R, Wasterlain CG. Resistance to excitotoxin-induced seizures and neuronal death in mice lacking the preprotachykinin A gene. Proc Natl Acad Sci U S A. 1999b;96:12096–12101. doi: 10.1073/pnas.96.21.12096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marti M, Manzalini M, Fantin M, Bianchi C, Della Corte L, Morari M. Striatal glutamate release evoked in vivo by NMDA is dependent upon ongoing neuronal activity in the substantia nigra, endogenous striatal substance P and dopamine. J Neurochem. 2005;93:195–205. doi: 10.1111/j.1471-4159.2005.03015.x. [DOI] [PubMed] [Google Scholar]
- McCann UD, Wong DF, Yokoi F, Villemagne V, Dannals RF, Ricaurte GA. Reduced striatal dopamine transporter density in abstinent methamphetamine and methcathinone users: evidence from positron emission tomography studies with [11C]WIN-35,428. J Neurosci. 1998;18:8417–8422. doi: 10.1523/JNEUROSCI.18-20-08417.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nash JF, Yamamoto BK. Methamphetamine neurotoxicity and striatal glutamate release: comparison to 3,4-methylenedioxymethamphetamine. Brain Res. 1992;581:237–243. doi: 10.1016/0006-8993(92)90713-j. [DOI] [PubMed] [Google Scholar]
- O’Callaghan JP, Miller DB. Neurotoxicity profiles of substituted amphetamines in the C57BL/6J mouse. J Pharmacol Exp Ther. 1994;270:741–751. [PubMed] [Google Scholar]
- O’Dell SJ, Weihmuller FB, Marshall JF. MK-801 prevents methamphetamine-induced striatal dopamine damage and reduces extracellular dopamine overflow. Ann N Y Acad Sci. 1992;648:317–319. doi: 10.1111/j.1749-6632.1992.tb24567.x. [DOI] [PubMed] [Google Scholar]
- O’Dell SJ, Weihmuller FB, Marshall JF. Methamphetamine-induced dopamine overflow and injury to striatal dopamine terminals: attenuation by dopamine D1 or D2 antagonists. J Neurochem. 1993;60:1792–1799. doi: 10.1111/j.1471-4159.1993.tb13405.x. [DOI] [PubMed] [Google Scholar]
- O’Shaughnessy MC, Vetsika EK, Inglis JJ, Carlesson J, Haigh R, Kidd BL, Winyard PG. The effect of substance P on nitric oxide release in a rheumatoid arthritis model. Inflamm Res. 2006;55:236–240. doi: 10.1007/s00011-006-0079-8. [DOI] [PubMed] [Google Scholar]
- Pu C, Broening HW, Vorhees CV. Effect of methamphetamine on glutamate-positive neurons in the adult and developing rat somatosensory cortex. Synapse. 1996;23:328–334. doi: 10.1002/(SICI)1098-2396(199608)23:4<328::AID-SYN11>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- Ralevic V, Khalil Z, Helme RD, Dusting GJ. Role of nitric oxide in the actions of substance P and other mediators of inflammation in rat skin microvasculature. Eur J Pharmacol. 1995;284:231–239. doi: 10.1016/0014-2999(95)00321-b. [DOI] [PubMed] [Google Scholar]
- Ricaurte GA, Schuster CR, Seiden LS. Long-term effects of repeated methylamphetamine administration on dopamine and serotonin neurons in the rat brain: a regional study. Brain Res. 1980;193:153–163. doi: 10.1016/0006-8993(80)90952-x. [DOI] [PubMed] [Google Scholar]
- Ricaurte GA, Guillery RW, Seiden LS, Schuster CR, Moore RY. Dopamine nerve terminal degeneration produced by high doses of methylamphetamine in the rat brain. Brain Res. 1982;235:93–103. doi: 10.1016/0006-8993(82)90198-6. [DOI] [PubMed] [Google Scholar]
- Ricaurte GA, Seiden LS, Schuster CR. Further evidence that amphetamines produce long-lasting dopamine neurochemical deficits by destroying dopamine nerve fibers. Brain Res. 1984;303:359–364. doi: 10.1016/0006-8993(84)91221-6. [DOI] [PubMed] [Google Scholar]
- Ripley TL, Gadd CA, De Felipe C, Hunt SP, Stephens DN. Lack of self-administration and behavioural sensitisation to morphine, but not cocaine, in mice lacking NK1 receptors. Neuropharmacology. 2002;43:1258–1268. doi: 10.1016/s0028-3908(02)00295-2. [DOI] [PubMed] [Google Scholar]
- Rupniak NM, Carlson EJ, Webb JK, et al. Comparison of the phenotype of NK1R−/− mice with pharmacological blockade of the substance P (NK1 ) receptor in assays for antidepressant and anxiolytic drugs. Behav Pharmacol. 2001;12:497–508. doi: 10.1097/00008877-200111000-00011. [DOI] [PubMed] [Google Scholar]
- Schmidt CJ, Ritter JK, Sonsalla PK, Hanson GR, Gibb JW. Role of dopamine in the neurotoxic effects of methamphetamine. J Pharmacol Exp Ther. 1985;233:539–544. [PubMed] [Google Scholar]
- Schulz JB, Matthews RT, Jenkins BG, et al. Blockade of neuronal nitric oxide synthase protects against excitotoxicity in vivo. J Neurosci. 1995;15:8419–8429. doi: 10.1523/JNEUROSCI.15-12-08419.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seiden LS, MacPhail RC, Oglesby MW. Catecholamines and drug-behavior interactions. Fed Proc. 1975;34:1823–1831. [PubMed] [Google Scholar]
- Sonsalla PK, Gibb JW, Hanson GR. Roles of D1 and D2 dopamine receptor subtypes in mediating the methamphetamine-induced changes in monoamine systems. J Pharmacol Exp Ther. 1986;238:932–937. [PubMed] [Google Scholar]
- Sonsalla PK, Riordan DE, Heikkila RE. Competitive and noncompetitive antagonists at N-methyl-D-aspartate receptors protect against methamphetamine-induced dopaminergic damage in mice. J Pharmacol Exp Ther. 1991;256:506–512. [PubMed] [Google Scholar]
- Stephans SE, Yamamoto BK. Methamphetamine-induced neurotoxicity: roles for glutamate and dopamine efflux. Synapse. 1994;17:203–209. doi: 10.1002/syn.890170310. [DOI] [PubMed] [Google Scholar]
- Sulzer D, Chen TK, Lau YY, Kristensen H, Rayport S, Ewing A. Amphetamine redistributes dopamine from synaptic vesicles to the cytosol and promotes reverse transport. J Neurosci. 1995;15:4102–4108. doi: 10.1523/JNEUROSCI.15-05-04102.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas DM, Walker PD, Benjamins JA, Geddes TJ, Kuhn DM. Methamphetamine neurotoxicity in dopamine nerve endings of the striatum is associated with microglial activation. J Pharmacol Exp Ther. 2004;311:1–7. doi: 10.1124/jpet.104.070961. [DOI] [PubMed] [Google Scholar]
- Thomas DM, Francescutti-Verbeem DM, Kuhn DM. Methamphetamine-induced neurotoxicity and microglial activation are not mediated by fractalkine receptor signaling. J Neurochem. 2008;105:605–616. doi: 10.1111/j.1471-4159.2008.05421.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volkow ND, Chang L, Wang GJ, et al. Higher cortical and lower subcortical metabolism in detoxified methamphetamine abusers. Am J Psychiatry. 2001a;158:383–389. doi: 10.1176/appi.ajp.158.3.383. [DOI] [PubMed] [Google Scholar]
- Volkow ND, Chang L, Wang GJ, et al. Association of dopamine transporter reduction with psychomotor impairment in methamphetamine abusers. Am J Psychiatry. 2001b;158:377–382. doi: 10.1176/appi.ajp.158.3.377. [DOI] [PubMed] [Google Scholar]
- Wang J, Angulo JA. Methamphetamine induces striatal neurokinin-1 receptor endocytosis primarily in somatostatin/NPY/NOS interneurons in mice and the role of dopamine receptors. Synapse. 2009 doi: 10.1002/syn.20848. submitted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiley RG, Lappi DA. Targeting neurokinin-1 receptor-expressing neurons with [Sar9,Met(O2)11 substance P-saporin. Neurosci Lett. 1999;277:1–4. doi: 10.1016/s0304-3940(99)00846-0. [DOI] [PubMed] [Google Scholar]
- Wilson JM, Kalasinsky KS, Levey AI, et al. Striatal dopamine nerve terminal markers in human, chronic methamphetamine users. Nat Med. 1996;2:699–703. doi: 10.1038/nm0696-699. [DOI] [PubMed] [Google Scholar]
- Xu W, Zhu JP, Angulo JA. Induction of striatal pre- and postsynaptic damage by methamphetamine requires the dopamine receptors. Synapse. 2005;58:110–121. doi: 10.1002/syn.20185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto BK, Zhu W. The effects of methamphetamine on the production of free radicals and oxidative stress. J Pharmacol Exp Ther. 1998;287:107–114. [PubMed] [Google Scholar]
- Yamamoto BK, Gudelsky GA, Stephans SE. Amphetamine neurotoxicity: roles for dopamine, glutamate and oxidative stress. In: Qureshi GA, editor. Neurochemical Markers of Degenerative Nervous Diseases and Drug Addiction. Utrecht, Netherlands: VSP Press; 1998. pp. 223–244. [Google Scholar]
- Yu J, Cadet JL, Angulo JA. Neurokinin-1 (NK-1) receptor antagonists abrogate methamphetamine-induced striatal dopaminergic neurotoxicity in the murine brain. J Neurochem. 2002;83:613–22. doi: 10.1046/j.1471-4159.2002.01155.x. [DOI] [PubMed] [Google Scholar]
- Yu J, Wang J, Cadet JL, Angulo JA. Histological evidence supporting a role for the striatal neurokinin-1 receptor in methamphetamine-induced neurotoxicity in the mouse brain. Brain Res. 2004;1007:124–131. doi: 10.1016/j.brainres.2004.01.077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yun HY, Gonzalez-Zulueta M, Dawson VL, Dawson TM. Nitric oxide mediates N-methyl-D-aspartate receptor-induced activation of p21ras. Proc Natl Acad Sci U S A. 1998;95:5773–5778. doi: 10.1073/pnas.95.10.5773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu JP, Xu W, Angulo JA. Disparity in the temporal appearance of methamphetamine-induced apoptosis and depletion of dopamine terminal markers in the striatum of mice. Brain Res. 2005;1049:171–181. doi: 10.1016/j.brainres.2005.04.089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu JP, Xu W, Angulo JA. Methamphetamine-induced cell death: selective vulnerability in neuronal subpopulations of the striatum in mice. Neuroscience. 2006;140:607–622. doi: 10.1016/j.neuroscience.2006.02.055. [DOI] [PMC free article] [PubMed] [Google Scholar]