

Abstract
Summary
TEAD2, one of the first transcription factors expressed at the beginning of mammalian development, appears to be required during neural development. For example, Tead2 expression is greatest in the dorsal neural crest where it appears to regulate expression of Pax3, a gene essential for brain development. Consistent with this hypothesis, we found that inactivation of the Tead2 gene in mice significantly increased the risk of exencephaly (a defect in neural tube closure). However, none of the embryos exhibited spina bifida, the major phenotype of Pax3 nullizygous embryos, and expression of Pax3 in E11.5 Tead2 nullizygous embryos was normal. Thus, Tead2 plays a role in neural tube closure that is independent of its putative role in Pax3 regulation. In addition, the risk of exencephaly was greatest with Tead2 nullizygous females, and could be suppressed either by folic acid or pifithrin-α. These results reveal a maternal genetic contribution to neural tube closure, and suggest that Tead2-deficient mice provide a model for anencephaly, a common human birth defect that can be prevented by folic acid. genesis 45:577–587, 2007.
Keywords: exencephaly, anencephaly, TEAD, Pax3, fetal brain
INTRODUCTION
Mammals express four closely related and evolutionarily conserved transcription factors, the prototype for which was isolated from human cells as TEF1 (Davidson et al., 1988) and later renamed TEAD1 by the human genome project. A mouse homologue was subsequently identified (TEAD1/TEF1; Blatt and DePamphilis, 1993; Shimizu et al., 1993), as well as three additional members of the TEAD family (TEAD2/TEF4; Jacquemin et al., 1996; Kaneko et al., 1997; Yasunami et al., 1995, TEAD3/TEF5; Kaneko et al., 1997; Yasunami et al., 1996, and TEAD4/TEF3; Jacquemin et al., 1996; Yasunami et al., 1996; Yockey et al., 1996). These transcription factors are defined by the presence of the 72 amino acid TEA DNA binding domain in their N-terminal half that is virtually identical among TEAD proteins and by the conspicuous homology among their C-terminal halves (Kaneko and DePamphilis, 1998). This conservation of structure is reflected in the ability of these proteins to bind the same transcriptional coactivator proteins (Mahoney et al., 2005; Vassilev et al., 2001) and to substitute for TEAD proteins in other organisms (Deshpande et al., 1997). Nevertheless, despite their structural similarities, the four mammalian TEAD proteins are not functionally redundant, because mice lacking TEAD1 fail to develop a proper heart and die between embryonic day 11 (E11) and E12 (Chen et al., 1994), whereas mice lacking TEAD4 arrest development before E3.5 (Yagi et al., 2007). Here we address the role of TEAD2.
The mouse Tead2 gene is expressed first as a maternal RNA that is recruited to polysomes and presumably translated (Wang and Latham, 2000) and then subsequently transcribed at the onset of zygotic gene expression during the 2-cell stage of development (Kaneko et al., 1997). Tead2 is the most abundantly expressed TEAD protein in embryos during their first 7 days of development (Kaneko et al., 1997), and its postimplantation embryonic expression pattern suggests that it plays a critical role in neural development (Milewski et al., 2004; Sawada et al., 2005).
Mouse Tead2 is expressed at basal levels in most adult tissues (Jacquemin et al., 1996; Kaneko et al., 1997; Yasunami et al., 1995), but it appears to play a critical role during development. Originally isolated as a gene that is expressed preferentially in fetal neural precursor cells (Yasunami et al., 1995), Tead2 was shown later to be expressed in proliferating fetal neuroepithelial cells, but not in postmitotic neurons (Jacquemin et al., 1996; Yasunami et al., 1995). In fact, Tead2 expression decreases in perinatal brain (postnatal day 0) relative to fetal brain (E12), revealing that Tead2 is downregulated during neural differentiation (Matsuki et al., 2005). This finding is consistent with the role proposed for Tead2 as a regulator of Pax3 expression, a gene that is essential for cell migration and differentiation in the neural crest (Milewski et al., 2004). Tead2 may be involved in the production or maintenance of the stem cells responsible for embryonic development, because it is expressed in embryonic, neural, and hematopoietic stem cells (Ramalho-Santos et al., 2002). Moreover, since these analyses compared transcripts expressed preferentially in proliferating stem cells relative to terminally differentiated cells, a common thread ties together all of the Tead2 expression studies: Tead2 appears to be expressed specifically in proliferating cells, although the absence of Tead2 expression in proliferating lymphoma and myeloma cell lines (Davidson et al., 1988; Kaneko et al., 2004) suggests that TEAD2 protein is not required for cells to proliferate per se but facilitates the process.
In an effort to identify a specific requirement for TEAD2 protein during mammalian development, the Tead2 gene was inactivated in the mouse. Consistent with its strong expression during neural development in postimplantation embryos, embryos lacking a functional TEAD2 protein were discovered to be at risk for exencephaly, a rare and fatal defect in which the brain protrudes into the amniotic cavity as a consequence of the failure of the neural tube to close during embryonic development. Moreover, the risk of TEAD2-dependent exencephaly was linked to TEAD2 nullizygous mothers, but not to Pax3 gene expression. Tead2 nullizygous embryos that escaped exencephaly produced viable, fertile adults with no visible abnormalities. These results reveal that TEAD2 plays an important role during closure of the embryonic neural tube, although not necessarily through regulation of Pax3 expression. Furthermore, TEAD2 is not required to sustain adult animals, although it may facilitate tissue regeneration (Ramalho-Santos et al., 2002; Zhao et al., 2006).
RESULTS
Construction of a Tead2 Conditional Knock-Out Allele
To create Tead2 heterozygous mutant mice, the Tead2 allele (Fig. 1A) was mutated by homologous recombination with a genetically altered targeting vector in mouse embryonic stem cells. The targeting vector consisted of upstream loxP site in intron 1 and downstream loxP site and a neomycin resistance gene in intron 3 (Fig. 1B; Materials and Methods). The targeting vector was linearized and then electroporated into 129/SvJ embryonic stem cells (contain genes for agouti coat color) and screened for clones that had undergone homologous recombination. They were then injected into C57BL/6J blastocysts (black coat color), transferred to foster mothers, and chimeric pups were obtained and genotyped (data not shown). The resulting chimeras containing the mutant allele were mated with C57BL/6J females to obtain agouti-colored pups, indicative of germline transmission. Genomic DNA prepared from tails of agouti mice was screened for the presence of the mutant allele by PCR (data not shown), and the results confirmed by digesting genomic DNA with either Xho1 or Nhe1 restriction endonuclease (Fig. 1B) and then identifying the appropriate DNA fragment by Southern blotting-hybridization (Fig. 2A) using either a 5′- or 3′-specific DNA probe (Fig. 1B). Both assays confirmed that homologously recombined mutant allele was transmitted through the germline. These mice were then mated with Ella-Cre+ mice (Lakso et al., 1996) in order to delete exons 2 and 3 from the mutant allele by Cre-dependent homologous recombination between the two loxP sites (Fig. 1B,C). EIIa promoter activity in mice is restricted to oocytes and preimplantation embryos (Dooley et al., 1989). Genomic DNA was prepared from tails of the progeny, and subjected to Southern blotting hybridization by digesting it with EcoRV restriction endonuclease and identifying the appropriate DNA fragment using the “KO probe” (Fig. 1C).
FIG. 1.

Construction of a Tead2 conditional KO mouse. The mouse Tead2 gene is located at 23 cM on chromosome 7 in ~30 kb region that includes the Dkkl1 and CD37 genes. (A) Schematic depiction of the features of the Tead2 gene that are relevant for construction of the knockout allele. Locations of cleavage sites for EcoRV (RV), Nhe I (N), and Xho I (X) restriction endonucleases are indicated. Locations of the 5′ and 3′-sequence specific DNA probes used in Southern blotting-hybridization analysis are indicated. Locations of primers A, B, and C used for multiplex genomic DNA PCR assays are indicated. Primer sequences are in Table 4. A putative Tead2 gene enhancer (“E”) is located within intron 1. (B) The targeting vector spanned the region from just upstream of the Tead2 promoter through part of intron 7. LoxP sites (closed triangles) were inserted within introns 1 and 3. A neomycin resistance gene (Neo) driven by the phosphoglycerol kinase (PGK) gene promoter, flanked by Flip recombinase target (FRT) sites (closed ovals), was inserted into intron 3 just upstream of 3′-loxP site. Newly created restriction sites are indicated with an asterisk. (C) Exposure of the targeted allele to Cre recombinase results in excision of the region between the two loxP sites. The deleted allele was screened by muliplex genomic DNA PCR (primer A/C) as well as by Southern blotting-hybridization analysis after digesting with EcoRV enzyme and probing with KO probe.
FIG. 2.

Genomic DNA analysis confirmed the presence of the Tead2 mutant allele. (A) Genomic DNA was extracted from the tails of pups born from matings between chimeric males and C57BL/6J females. DNA was digested with the indicted restriction endonuclease, subjected to Southern blotting-hybridization, and then detected with the indicated probe (Fig. 1A, B). Lanes 2–4 contain DNA from agouti pups. Lane 1 contains DNA from a black littermate. XhoI-5′ probe gives rise to a 19.9 kb DNA fragment from the wild type (Wt) allele, and a 4.6 kb DNA fragment from the mutant allele (Mt). NheI-3′ probe gives rise to 8.4 kb fragment from the Wt allele, and a 9.5 kb fragment from the mutant allele. Genomic DNA from WT ES cells was included as a positive control. (B) Mice containing the mutant allele were mated to EllaCre+ mice, and resulting pups were assayed for the deleted/KO allele. Genomic DNA was digested with EcoRV restriction endonuclease, subjected to Southern blotting-hybridization, and the DNA detected with “KO probe” (Fig. 1C). The Wt allele generates a 7.2 kb DNA fragment (Wt). The knockout allele generates a 8.5 kb DNA fragment (KO). Result shown is an example from such matings (“parents”). These Tead2 heterozygous parents (male and female) were then mated to each other and their litters of five pups were similarly assayed for the presence of KO allele. (C) The DNA samples (“litter”) in panel B were also subjected to multiplex genomic DNA PCR analysis. Primers A and B (Fig. 1B) amplified a 165 bp fragment from the Wt allele. Primers A and C (Fig. 1C) amplified a 210 bp fragment from the KO allele.
The net result was that exons 2 and 3, encoding the N-terminal 99 amino acids of the 445 amino acid TEAD2 protein, were deleted in the Tead2 knock-out (KO) allele (Fig. 1C). This deletion removed the translation start codon, 82% of the TEA DNA binding domain, and the putative Tead2 enhancer element that resides within 3′-half of intron 1 (Zhao et al., 2006; data not shown). The Tead2 transcription start site lies within exon 1, ~1.4 kb upstream of exon 2 (Kaneko et al., 2004). Therefore, this deletion should reduce the overall level of Tead2 mRNA, prevent translation of full-length Tead2 protein, and in the event that some Tead2 protein is produced, insure that it is not functional.
Inactivation of the Tead2 Gene Impedes, but does not Prevent, Development
Mice that were heterozygous for the Tead2 KO allele (Fig. 2B, “parents”) were mated. Their offspring were genotyped both by PCR analysis (Fig. 2C) and by Southern blotting hybridization analysis (Fig. 2B, “litter”). Both assays confirmed Cre-mediated deletion. The genotypic ratio of Tead2+/+, Tead2+/− , and Tead2−/− mice from these heterozygous matings revealed a skewed ratio in which the number of Tead2-nullizygous mice was less than expected from simple Mendelian segregation (Table 1), suggesting that embryonic development was hindered in the absence of TEAD2 but not completely blocked. The size, morphology, behavior, and reproduction of Tead2 nullizygous mice were indistinguishable from wild-type (WT) mice with the same mixed genetic background, suggesting that maintenance of adult functions does not require Tead2.
Table 1.
Progeny from Tead2+/− X Tead2+/− Matings
| Male | Female | Total | |
|---|---|---|---|
| Total Mice | 111 | 118 | 229 |
| Genotype | Male | Female | Sum | % Total | Theoretical |
|---|---|---|---|---|---|
| Wild-type | 24 | 38 | 62 | 27.1 | 25 |
| Heterozygous | 66 | 56 | 122 | 53.3 | 50 |
| Nullizygous | 21 | 24 | 45 | 19.7 | 25 |
Summary of genotyped offspring obtained from heterozygous intercross. Tail samples from pups born from heterozygous inter-cross were used to analyze for their genotype. The numbers indicate compilation of 32 litters.
Tead2 Nullizygous Mice do not Contain Tead2 WT mRNA
Previous studies have reported that Tead2 mRNA is present in embryonic stem cells (Kaneko et al., 2004; Ramalho-Santos et al., 2002), in fetal neural tissue from postimplantation embryos (Yasunami et al., 1995), and in adult lung tissue (Kaneko et al., 1997), but it is virtually undetectable in adult brain (Yasunami et al., 1995). Northern blotting hybridization analysis confirmed that Tead2 mRNA was absent from adult brain tissue (Fig. 3A, lane 1), although abundant in embryos undergoing brain development (Fig. 3A, lane 2), in embryonic stem cells (Fig. 3A, lane 4), and in adult lung (Fig. 3A, lane 5). In contrast, Tead2 mRNA was greatly reduced in these tissues from mice lacking one or both Tead2 genes (Fig. 3A, lanes 3, 6, 7). These results were consistent with the fact that the Tead2 KO allele retains the native Tead2 promoter but lacks the Tead2 enhancer (Fig. 1A).
FIG. 3.

Full-length Tead2 mRNA was absent in Tead2 nullizygous mice. Tead2 heterozygous mice were mated, and DNA extracted from the tails of littermates was genotyped. (A) Total RNA (15 μg) was prepared from adult forebrain (lane 1) and fetal brain (E13.5; lane 2) from Tead2 WT mouse, and fetal brain (E13.5) from a Tead2 nullizygous mouse (lane 3), fractionated by agarose gel electrophoresis, transferred to nylon membrane, and hybridized with radiolabeled Tead2 cDNA (Kaneko et al., 1997). Similarly, total RNA (15 μg) was prepared from the lungs of a Tead2 homozygous (lane 5), a heterozygous (lane 6) and a nullizygous (lane 7) littermate. Samples were subjected to Northern blotting-hybridization analysis. Total RNA (15 μg) isolated from WT CCE ES cells (lane 4), which abundantly express Tead2 (Kaneko et al., 2004), was used as a positive control. Ethidibum bromide stained 28S rRNA was used as loading control. 0.24–9.5 kb RNA ladder from Invitrogen was used as marker. (B, C) Tead2 KO mice produce no detectable level of Tead2 mRNA, but still express the Fgfr4 and Tead4 genes. Total RNA from lung (B) or ovary (C) tissue was isolated from mice that were WT, heterozygous, or nullizygous at Tead2 allele and then used as a template for cDNA synthesis. PCR was performed for the indicated number of cycles using Tead2 primers that amplified the region from exons 2 to 6 (Kaneko et al., 1997). The same aliquot of RNA was used to amplify Fgfr4-specific and Tead4-specific RNA. DNA marker used was 1 kb ladder from Invitrogen. Gapdh control primers confirmed that relatively equivalent RNA samples were amplified (data not shown).
To determine whether or not Tead2 exons 2 and 3 had been deleted from Tead2 mRNA produced in Tead2 nullizygous mice (Fig. 1C), total RNA prepared from lung or ovarian tissue was assayed for Tead2 exons 2 through 6 (Fig. 1A) by reverse-transcription/polymerase chain reaction (RT-PCR) (Kaneko et al., 1997). As expected, the region from exons 2 through 6 was detected from either Tead2 WT or heterozygous mice, but not from RNA in Tead2 nullizygous mice (Fig. 3B,C). Hence, none of the mRNA produced from the Tead2 mutant allele could express a functional TEAD2 protein.
TEAD2 is not Required for Expression of Fgfr4, Tead4, or Pax3 Genes
TEAD2 has been hypothesized to regulate expression of the Fgfr4 gene in the lung (Weinstein et al., 1998; Zhao et al., 2006), and perhaps also in granulosa cells of developing follicles where the same cells express both Fgfr4 (Puscheck et al., 1997; Weinstein et al., 1998) and Tead2 genes (Kaneko et al., 1997). However, no change in Fgfr4 expression was detected either in lung (Fig. 3B) or in ovaries (Fig. 3C) from Tead2 WT, heterozygous or nullizygous females.
Loss of TEAD2 may result in compensatory expression of other TEAD proteins. For example, Tead4 and Tead2 are both expressed in preimplantation embryos (Yagi et al., 2007) and in adult lung (Yockey et al., 1996). However, expression of Tead4 (Fig. 3B,C) was not dependent on Tead2 expression. Similar results were obtained with Tead1 (data not shown). Therefore, TEAD2 does not appear to be involved in regulating expression of other Tead genes.
TEAD2 also has been hypothesized to regulate expression of Pax3 (Milewski et al., 2004). Hence, loss of TEAD2 would be expected to downregulate expression of the Pax3 gene. However, when RNA was prepared from individual E11.5 embryos from a single heterozygous intercross and examined for Pax3 expression, Pax3 mRNA levels were the same, regardless of the presence or absence of TEAD2 (Fig. 4A).
FIG. 4.

Exencephalic embryos were detected as early as E11.5, although Pax3 expression was not altered by the absence of TEAD2. (A) Total RNA was prepared from 11 embryos at E11.5 that were obtained from a single heterozygous female mated to a heterozygous male. This RNA was assayed for Pax3 sequences using RT-PCR. RNA from E13.5 fetal brain served as a positive control, while RNA from ES cells served as a negative control. One embryo that exhibited exencephaly (asterisk) is shown in panel C. (B) Multiplex PCR analysis was used to determine the genotype of each embryo. (C) One normal (Tead2+/−) and one exencephalic (Tead2−/−) embryo from the same litter at E11.5 are shown using bright-field illumination. Arrows indicate sites where neural tube failed to close.
TEAD2 is not Required for Follicular Development
Tead2 expression in ovary is restricted primarily to mural granulosa cells, suggesting that it may play a role in ovarian function (Kaneko et al., 1997). To test this hypothesis, immature females that were either heterozygous or nullizygous for Tead2 were injected with pregnant mare’s serum (PMS) to mimic the action of follicle stimulating hormone. PMS induces expression of several genes in immature females, including aromatase (Cyp19) and leutenizing hormone/choriogonadotropin receptor (Lhcgr) from ovarian somatic cells (Couse et al., 2005). However, contrary to this hypothesis, these enzymatic changes were not dependent on a functional Tead2 gene either in unstimulated or PMS stimulated ovaries (data not shown).
TEAD2 is Involved in Neural Tube Closure
The fraction of Tead2 nullizygous pups born from heterozygous intercrosses was less than expected (Table 1). This deviation from Mendelian segregation was not due simply to genetic background effects, because heterozygous intercrosses of a conditional KO allele in Dkkl1 (located 3 kb upstream of Tead2 gene; Kaneko and DePamphilis, 2000) that was constructed in the same manner as the conditional KO allele in Tead2 and in the same mouse strain yielded normal Mendelian segregation ratios (Kaneko et al., manuscript in preparation). Deviation from Mendelian genetics could result from an increased risk for exencephaly, a developmental abnormality wherein parts of the fetal brain lie outside the neural tube (Harris and Juriloff, 2007). The “natural” incidence of exencephaly in many inbred strains is less than 0.5% (Juriloff et al., 2001, and references therein). Two of 32 embryos from Tead2 heterozygous intercrosses that were examined between E11.5 and E13.5 exhibited exencephaly (Fig. 4, data not shown). For example, as early as E11.5, a nullizygous embryo exhibited clear abnormalities in the cranial region when compared to a normal heterozygous littermate (Fig. 4A,C).
This diagnosis was confirmed by mating Tead2 nullizygous females with heterozygous males so that ~50% of the embryos would be nullizygous for Tead2. Pregnant females were euthanized, and each litter was examined for abnormally developing embryos. Of 54 embryos examined, 19% were exencephalic (Table 2A). Moreover, 30% of the Tead2 nullizygous embryos were exencephalic, and surprisingly 7% of the Tead2 heterozygous embryos were exencephalic. For example, several exencephalic embryos were found among litters at E13.5 (Fig. 5). Another litter at E16.5 contained an exencephalic embryo that was clearly alive (as evidenced by its beating heart; Fig. 6A), a dead exencephalic embryo that was in the process of being resorbed (Fig. 6B; as evidenced by lack of beating heart, absence of blood vessels, and skin disintegration), as well as normal embryos (Fig. 6C).
Table 2.
Tead2-Dependent Exencephaly
| Tead2+/− | Tead2−/− | Total | |
|---|---|---|---|
| (A) ♂ Tead2+/− X ♀Tead2−/− | |||
| Total mice | 27 | 27 | 54 |
| Exencephalic mice | 2 | 8 | 10 |
| % Exencephalic | 7 | 30 | 19 |
| (B) ♂ Tead2−/− X ♀ Tead2+/− | |||
| Total mice | 30 | 22 | 52 |
| Exencephalic mice | 0 | 2 | 2 |
| % Exencephalic | 0 | 9 | 4 |
(A) Tead2 nullizygous females were mated with heterozygous males, and embryos at E11.5 to E17.5 were examined for neural tube defects and genotyped. Results from seven litters were combined. (B) Tead2 nullizygous males were mated with heterozygous females, and postimplantation stage embryos were examined for NTDs and genotyped. Results from six litters were combined. The difference between “% Execenphalic (Total)” in panels A and B was statistically significant (P < 0.02). The data were analyzed by testing the difference in proportions using normal approximation to the binomial.
FIG. 5.
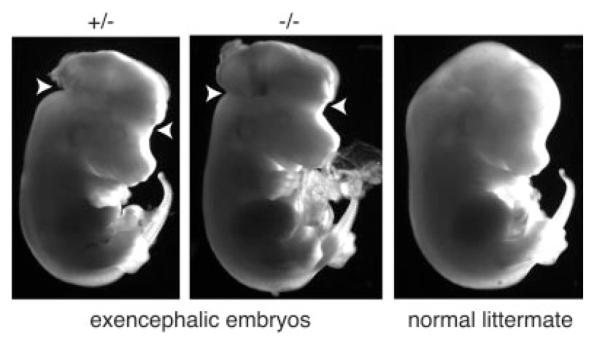
Exencephalic embryos were clearly evident by E13.5. Tead2 nullizygous females were mated to a heterozygous male, and embryos were harvested at day E13.5. Two exencephalic and one normal embryo from the same litter are shown using dark-field illumination.
FIG. 6.

By E16.5, some exencephalic embryos are alive while others are being resorbed. Tead2 nullizygous females were mated to a heterozygous male, and embryos were harvested at day E16.5. (A) Tead2−/− exencephalic embryo. (B) Tead2−/− exencephalic embryo undergoing resorption. (C) Tead2+/− littermate.
Folic acid has been reported to prevent 70% of human neural tube defects (NTDs; Fleming and Copp, 1998). To determine whether or not folic acid can suppress Tead2-dependent exencephaly, nullizygous pregnant mothers were injected with folic acid on E8.5 and E9.5, and the embryos were examined on E13.5. Folic acid reduced the risk of Tead2-dependent exencephaly about threefold (Table 3A) when compared to control treatment (Table 3C).
Table 3.
Suppression of Exencephaly by Either Folic Acid or Pifithrin-α
| Tead2+/− | Tead2−/− | Total | |
|---|---|---|---|
| (A) ♂ Tead2+/− X ♀ Tead2−/− (+ folic acid) | |||
| Total mice | 8 | 8 | 16 |
| Exencephalic mice | 0 | 1 | 1 |
| % Exencephalic | 0 | 13 | 6 |
| (B) ♂ Tead2+/− X ♀ Tead2−/− (+ pifithrin-α) | |||
| Total mice | 9 | 11 | 20 |
| Exencephalic mice | 0 | 1 | 1 |
| % Exencephalic | 0 | 9 | 5 |
| (C) ♂ Tead2+/− X ♀ Tead2−/− (control) | |||
| Total mice | 11 | 13 | 24 |
| Exencephalic mice | 0 | 4 | 4 |
| % Exencephalic | 0 | 31 | 17 |
Tead2 nullizygous females were mated with heterozygous males, injected with supplements, and embryos at E13.5 were examined for neural tube defects and genotyped. (A) At E8.5 and E9.5, pregnant females were injected intraperitoneally with 10 mg folic acid per kg weight (Fleming and Copp, 1998), and the embryos were examined at E13.5 for NTDs and genotyped. Results from three litters were combined. (B) At E8.5 and E9.5, pregnant females were injected intraperitoneally with pifithrin-α (Pani et al., 2002), and the embryos were examined at E13.5 for NTDs and genotyped. Results from three litters were combined. (C) Mice were mated in parallel with (A) and (B) but treated with saline instead of either folic acid or pifithrin-α.
Pifithrin-α, an inhibitor of p53-dependent apoptosis (Murphy et al., 2004), has been reported to rescue NTDs in Pax3-deficient embryos (Pani et al., 2002). Similarly, pifithrin-α also prevented Tead2-dependent exencephaly to the same extent as did folic acid (Table 3B), suggesting that TEAD2 may regulate neural tube closure by inhibiting p53-dependent apoptosis, rather than by inducing neural tube-specific gene expression.
If the risk for exencephaly rested solely on Tead2 genotype of the conceptus, the risk for exencephaly would be the same regardless of which parent was nullizygous for Tead2 gene. To test this hypothesis, nullizygous males were mated to heterozygous females. The result was a substantial reduction in the incidence of exencephaly. Of 54 embryos examined, only 4% were exencephalic, and both of these were Tead2 nullizygous (Table 2B). The frequency of exencephaly was fivefold greater when the mother was nullizygous for Tead2 rather than when the father was nullizygous. Thus, the risk for Tead2-dependent exencephaly is much greater when the mother is missing both copies of a functional Tead2 gene.
DISCUSSION
Exencephaly in mice is a defect in neural tube closure that is equivalent to anencephaly in humans. NTD, which includes anencephaly, appears in one out of one thousand births making it the most common form of birth abnormality in humans. Although specific mutations in mice can cause NTDs, multiple genes are thought to be involved in human NTDs (Juriloff and Harris, 2000). Therefore, the mouse models most relevant for clinical implications are those that exhibit incomplete penetrance (Juriloff et al., 1989).
Neural tube closure begins when the epithelial cells lining the neural folds “elevate,” and fusion begins at four separate sites from the cranial to the caudal regions (Juriloff and Harris, 2000). Exencephaly results from failure of the cranial neuroepithelial cells to elevate at or near Closure 2. The extent of penetrance (risk) for exencephaly in Tead2 nullizygous embryos was consistent with that observed for other murine genetic defects that give rise to exencephaly (Harris and Juriloff, 2007). For example, only 30% of mice that completely lack Closure 2 develop exencephaly even though one would expect 100% penetrance (MacDonald et al., 1989). However, genetic background can also greatly affect penetrance in exencephaly (Juriloff et al., 1991). For example, only 30% of Pax2 mutants develop exencephaly in C57BL/6J mice, but 100% do so in 129/Sv mice (Schwarz et al., 1997). Therefore, it is possible that penetrance of Tead2-dependent exencephaly can be increased by backcrossing to another strain.
Studies both in humans and in mice have shown that administration of a variety of metabolites to pregnant females can suppress NTDs (Blom et al., 2006; Harris and Juriloff, 2007). However, little is known regarding the mechanism by which these supplements work, since either the mother and/or the conceptus could be the primary target. Moreover, whether or not neural tube closure requires a unique maternal genetic contribution is not yet clear. In this regard, it is noteworthy that Tead2-dependent exencephaly was dependent on the maternal genotype (Table 2). The incidence of exencephaly was 5-times greater when the mother was Tead2−/− compared to when the father was Tead2−/−. Moreover, when the mother was Tead2 nullizygous, a fraction of exencephalic embryos were Tead2 heterozygous, revealing that, under these conditions, even a single Tead2 mutant allele could increase the incidence of exencephaly. In contrast, when the mother was Tead2 heterozygous, all of the excencephalic embryos were Tead2 nullizygous, regardless of whether the Tead2+/− females had been mated to Tead2−/− (Table 2B) or Tead2+/− (Fig. 4, data not shown) males. The fact that embryos produced by Tead2 nullizygous females have a greater incidence of exencephaly than those from heterozygous females reveals the existence of a maternal contribution for neural tube closure. To our knowledge, only one other study has examined the maternal genetic contribution to exencephaly, and they concluded that exencephaly is controlled largely by genes active in the pregnant female (Beck, 1999). Finally, the risk of exencephaly was reduced about threefold by intraperitoneal administration of folic acid to pregnant females (Table 3), suggesting that the maternal effect in Tead2-dependent exencephaly is linked to folic acid metabolism, as previously reported for anencephaly in humans (Fleming and Copp, 1998). Hence, the maternal genetic effect observed in Tead2 nullizygous females might provide an important model for human NTDs.
Previous studies strongly implicated TEAD2 as a regulator of Pax3 expression in the neural crest (Milewski et al., 2004). This is consistent with the fact that Tead2 is expressed exclusively in mitotic neuroepithelial cells during fetal brain development (Jacquemin et al., 1996; Yasunami et al., 1995). Moreover, as previously reported for Pax3 mutants (Pani et al., 2002), injection of pregnant females with pifithrin-α, an inhibitor of p53 signaling (Murphy et al., 2004), suppressed exencephaly in Tead2 nullizygous females (Table 3B), suggesting that inhibition of apoptosis can rescue this NTD. However, the NTD seen in Tead2 nullizygous mice contrasts with that observed in Pax3 nullizygous mice. Pax3 mutants not only exhibit complete penetrance of a NTD, but they exhibit both exencephaly and spina bifida aperta (Harris and Juriloff, 2007; Juriloff and Harris, 2000). The absence of a more severe phenotype in Tead2 nullizygous embryos reflects the fact that expression of Pax3 at E11.5 was not suppressed in embryos lacking Tead2 (Fig. 4A,B), revealing that alternative mechanisms are available for maintaining Pax3 expression. Nevertheless, the fact that only some Pax3 mutants show exencenphaly is consistent with low penentrance by Tead2 inactivation. Perhaps the exencephalic defect in Tead2 nullizygous embryos is mediated through reduction in Pax3 expression specifically at areas around neural crest cells at or near Closure 2.
The results presented here confirm that TEAD2 plays a role in brain development through its role in neural tube closure, but reveal that TEAD2 is indispensable neither for this process nor for the production and maintenance of viable, fertile adult animals. Two explanations are possible. First, TEAD2 is simply one of several factors that determine the level of an effector gene, and therefore the absence of TEAD2 reduces, but does not eliminate, production of the effector gene product. Second, other transcription factors, possibly other members of the TEAD family, can substitute for TEAD2 function. Furthermore, although Tead2 mRNA is detectable in adult tissues (Jacquemin et al., 1996; Kaneko et al., 1997), the levels are much lower in comparison to Tead2 mRNA in embryonic cells (Fig. 3). Thus, once embryonic development is completed, the need for TEAD2 in adult tissues may be minimal. Nevertheless, given the high level of Tead2 expression in adult neural stem cells (Ramalho-Santos et al., 2002; data not shown) and in regenerating muscle (Zhao et al., 2006), defects in Tead2 nullizygous mice may only manifest themselves with age.
Finally, it is notable that of the four Tead genes in mammals, the biological roles for three of them (Tead1, Tead2, and Tead4) have now been identified through genetic mutations. Remarkably, despite the fact that all four genes have identical DNA binding domains and bind the same transcriptional coactivators, each of them plays a unique role in mammalian development. TEAD1 is involved in cardiovascular development (Chen et al., 1994). TEAD4 is involved in preimplantation development (Yagi et al., 2007), and TEAD2 is involved in brain development (this report).
METHODS
Conditional Tead 2 Knockout Allele
The targeting vector (Fig. 1B) was constructed as described using recombineering methods (Liu et al., 2003; described in detail at http://recombineering.ncifcrf.gov/). Briefly, BAC clone 75.6i (Genome Systems; Kaneko and DePamphilis, 2000) was introduced into bacterial strain EL350 by electroporation. Region upstream of Tead2 promoter (nucleotides 1055501–1056417; Genbank NW_000319.1) was amplified using primers D and E (Table 4), Pfu Taq polymerase (Strata-gene) and BAC clone 75.6i as template. This amplicon was digested with SpeI and HindIII. Region within Tead2 intron 7 (nucleotides 1065309–1065555; Genbank NW_000319.1) was amplified using primers F and G, and the amplicon was digested with HindIII and XbaI. These two amplicons were combined and then ligated into pL253 that had been digested with XbaI and SpeI. The resulting plasmid was digested with HindIII, purified with a Qiagen miniprep spin column, and electroporated into EL350 containing BAC clone 75.6i, previously treated at 42°C for 15 min to obtain “gap repaired plasmid.”
Table 4.
PCR Primers and Inserted Sequences
| Name | Sequence |
|---|---|
| A | tgtctctttgtctggccacatg |
| B | cccaaagttctgagtcccatc |
| C | ccccacaacttagagaaaagcc |
| D | ggactagtcttctggcctctgtaggtatac |
| E | gtcaagcttaactcacaaaccccaagcagcatcccgattc |
| F | gtcaagcttagggctcacatcttgatctg |
| G | aagcccatctctagaccttg |
| H | gatcccgactacaaggacgacgatgacaagtacccctacgac gtgcccgactacgcc |
| I | gatcggcgtagtcgggcacgtcgtaggggtacttgtcatcgt cgtccttgtagtcgg |
| J | ggaattcagatctttgttcacctaacctctctc |
| K | gtccttgtagtcgggatcccccatctgggcctg |
| L | acgcgtcgactcaccatacatcttgccctc |
| M | ataagcggccgcaaccctacccagtccttagatgg |
| N | gtcgaattcatctcaagcccagcccagtgtcg |
| O | ataagcggccgctttggaactttggtctgggg |
| P | gtcgaattcacacaacaaggccagaaagacg |
| Q | ggaattcggatccggcttttctctaagttgtgggg |
| R | acgcgtcgactcttccacaggtcacacagc |
| S | ctggcaaagctccttgccaaaa |
| T | aatgatgcagagggtgtatggagc |
| U | gagaactctctgggtagcattc |
| V | tatactctcctgcatcctcagc |
| W | atagtggagatggcccaccatg |
| X | tcatctgactgaggtgcagagg |
Oligonucleotides encoding the FLAG-HA polypeptide (primers H and I; Table 4) were annealed, phosphorylated, and ligated to gap repaired plasmid that had been digested with BamHI. This placed the FLAG-HA sequence just downstream of the first ATG (methionine) codon within exon 2 and in the same reading frame as the Tead2 gene.
A loxP site was introduced into intron 1 (Fig. 1B) using a mini-targeting vector that contained phosphoglycerol kinase promoter driving a neomycin resistance gene (PGK-Neo) flanked by loxP sites. The “right-arm” was constructed by combinatorial PCR. PCR was done by either using primers J and K (Table 4), and BAC 75i6 as template, or using primers H and L, and FLAG-HA containing gap repaired plasmid as template. There was an 18-nucleotide overlap between primers H and K. After purification through Qiagen miniElute PCR purification kit, the two amplicons were combined, and PCR was performed using primers J and L. The PCR product was digested with SalI and EcoRI.
The “left-arm” was constructed by PCR using primers M and N, and BAC 75i6 as template. This amplicon was then digested with EcoRI and NotI, mixed with digested “right-arm” amplicon, and ligated to pBluescript (Strata-gene) that had been digested previously with NotI and SalI. LoxP flanked PGK-Neo cassette was released from pL452 by digesting the plasmid with EcoRI and BamHI, purified by agarose gel electrophoresis, and then ligated to a mini-targeting vector that had been digested with BlgII and EcoRI. This mini-targeting vector was then digested with NotI and SalI, and the targeting cassette was purified by agarose gel electrophoresis. This targeting cassette and FLAG-HA containing gap repaired plasmid were electroporated into EL350 bacteria that had been treated previously at 42°C for 15 min. The resulting plasmid contained a loxP-flanked PGK-Neo cassette within intron 1 together with FLAG-HA epitope tag immediately downstream of the ATG translation start site within exon 2. This plasmid was then electroporated into EL350 bacteria, previously induce to produce Cre recombinase in the presence of 10% L-(+)-arabinose (Liu et al., 2003), in order to excise the PGK-Neo cassette, thereby leaving a single loxP site within intron 1.
A second mini-targeting vector was constructed to introduce a PGK-Neo/loxP cassette flanked by FRT sites into intron 3 (Fig. 1B). “Left-arm” was constructed by PCR using primers O and P, and BAC 75i6 as template. This fragment was digested with NotI and EcoRI. The “right-arm” was constructed by PCR using primers Q and R, and BAC 75i6 as template. This fragment was digested with EcoRI and SalI. The two fragments were combined and ligated into pBluescript that had been digested previously with NotI and SalI. The FRT flanked loxP/PGK-Neo cassette was purified from pL451 after digesting the plasmid with EcoRI and BamHI. Purified fragment was then ligated to the second mini-targetting vector that had been digested previously with BamHI and EcoRI. This mini-targetting vector was digested with NotI and SalI, and the targeting cassette was purified by agarose gel electrophoresis. This targeting cassette and loxP-FLAG-HA containing gap repaired plasmid were electroporated into heated-treated EL350 to produce the final targeting vector (Fig. 1B). Altered sequences and additions in the targeting vector were confirmed by sequencing.
The targeting vector was linearized with KpnI, electroporated into GSI-1 ES line (Genome Systems; derived from 129/SvJ strain mice), and selected with G418 and gancyclovir (Ohtsubo et al., 2004). Genomic DNA prepared from surviving colonies was screened by restriction digestion and Southern blotting hybridization using probes outside of the targeting vector (data not shown; Fig. 1B, see below). Positive clones were injected into C57BL/6J blastocysts and surgically transferred to foster mother. Chimeric mice containing the mutant allele were then mated with C57BL/6J females to obtain germline-transmitted agouti-coat color pups.
Mice
For experiments involving embryo isolation, genotyped males and females (6–10 weeks old) were mated and the females were checked for the presence of a vaginal plug (designated E0.5). Pregnant females were euthanized at the indicated day, uteri were dissected and placed in PBS. Decidua were dissected and placed in separate dishes containing PBS. Embryos were dissected out of placenta/decidua tissue, and visually examined under the microscope for presence of exencephaly or other abnormalities. Part of either embryonic tail or amnion was saved for genomic DNA analysis. For total RNA preparation, the entire embryo or dissected telencephalon of E13.5 embryos was frozen in liquid nitrogen and stored at −80°C until RNA isolation (see below). All animal care and procedures were done according to guidelines provided by the IACUC.
Genomic DNA Analysis
Genomic DNA was prepared from mouse tail clippings or embryonic tail/chorion using Puregene DNA purification kit (Gentra), and the precipitated DNA was hydrolyzed in 30 μl 10 mM Tris—EDTA, pH 8.0 and analyzed as follows.
Multiplex genomic DNA PCR analysis
1/30 of each sample was assayed using 0.2 μM primer A + C (Table 4), 0.4 μM primer B, and Platinum Taq polymerase (Invitrogen) according to the manufacturer’s instructions. PCR reactions of 25 μl were incubated for 5 min at 95°C followed by 40 cycles of 30 s at 95°C, 30 s at 60°C, and 30 s at 72°C with a final extension of 7 min at 72°C. Aliquots of 10 μl were subjected to electrophoresis in 5% polyacrylamide gels containing Tris—borate—EDTA buffer (BioRad Ready Gel). DNA was visualized with ethidium bromide.
Southern blotting hybridization analysis
To confirm germline transmission of the recombined allele (Figs. 1B and 2A), 1/3 of the genomic DNA sample was digested with 120 U of either XhoI or NheI (Roche Biochemicals). To confirm the Cre-mediated deletion allele (Figs. 1C and 2B), 1/3 of the genomic DNA sample was digested with 120 U EcoRV (Roche Biochemicals). Digested DNA was fractionated by electrophoresis in 0.8% agarose gel containing Tris—Borate—EDTA buffer. XhoI digested DNA was hybridized with radiolabeled 5′-probe (nucleotides 1052380–1053246; Genbank NW_000319.1), NheI digested DNA with 3′-probe (nucleotides 1065550–1066861, Fig. 1B), and EcoRV digested DNA with “KO” probe (nucleotides 1059178–1059702, Fig. 1C). DNA probes were prepared by standard PCR technology using either BAC 75i6 or subclone fragment isolated from BAC 75i6, and radiolabeled using Ready-To-Go DNA labeled Beads and [32]P-dCTP (Amersham) (Kaneko and DePamphilis, 2000).
Total RNA Analyses
Embryos, as well as adult and fetal tissues, were frozen in liquid nitrogen upon isolation. Total RNA was isolated as previously described (Kaneko et al., 1997). After addition of RNAgents denaturation solution (Promega), samples were homogenized before proceeding with the manufacturer’s protocol. Total RNA (15 μg) was analyzed by Northern blotting hybridization as previously described (Kaneko et al., 1997). Alternatively, a semiquantitative reverse transcription-polymerase chain reaction (RT-PCR) was carried out, as previously described (Kaneko et al., 2004). After the initial cycle and at every two cycles thereafter as indicated, a 5 μl aliquot was saved before PCR was continued. Aliquots were then fractionated simultaneously by electrophoresis in 5% polyacrylamide gels containing Tris—borate—EDTA buffer. DNA primers for amplification of Tead2 exons 2–6 and Gapdh have been described (Kaneko et al., 2004). Primers for other genes were: Tead4 (S and T), Fgfr4 (U and V), Pax3 (W and X). Tead4 primers amplify either a 396 bp WT gene segment, or a 267 bp splice variant gene segment (Yockey et al., 1996). Primers for Fgfr4 and Pax3 amplify 316 bp and 434 bp DNA fragments, respectively.
ACKNOWLEDGMENTS
We thank N. Copeland for generous gift of recombineering reagents and Mark N. Boldrin for assistance with statistical analyses.
Contract grant sponsor: Intramural Research Program, NIH.
Footnotes
Publisher's Disclaimer: Published 2007 Wiley-Liss, Inc.
LITERATURE CITED
- Beck SL. Contributions of dam and conceptus to differences in sensitivity to valproic acid among C57 black and SWV mice. Reprod Toxicol. 1999;13:353–360. doi: 10.1016/s0890-6238(99)00038-6. [DOI] [PubMed] [Google Scholar]
- Blatt C, DePamphilis ML. Striking homology between mouse and human transcription enhancer factor-1 (TEF-1) Nucleic Acids Res. 1993;21:747–748. doi: 10.1093/nar/21.3.747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blom HJ, Shaw GM, den Heijer M, Finnell RH. Neural tube defects and folate: Case far from closed. Nat Rev Neurosci. 2006;7:724–731. doi: 10.1038/nrn1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z, Friedrich GA, Soriano P. Transcriptional enhancer factor 1 disruption by a retroviral gene trap leads to heart defects and embryonic lethality in mice. Genes Dev. 1994;8:2293–2301. doi: 10.1101/gad.8.19.2293. [DOI] [PubMed] [Google Scholar]
- Couse JF, Yates MM, Deroo BJ, Korach KS. Estrogen receptor-beta is critical to granulosa cell differentiation and the ovulatory response to gonadotropins. Endocrinology. 2005;146:3247–3262. doi: 10.1210/en.2005-0213. [DOI] [PubMed] [Google Scholar]
- Davidson I, Xiao JH, Rosales R, Staub A, Chambon P. The HeLa cell protein TEF-1 binds specifically and cooperatively to two SV40 enhancer motifs of unrelated sequence. Cell. 1988;54:931–942. doi: 10.1016/0092-8674(88)90108-0. [DOI] [PubMed] [Google Scholar]
- Deshpande N, Chopra A, Rangarajan A, Shashidhara LS, Rodrigues V, Krishna S. The human transcription enhancer factor-1, TEF-1, can substitute for Drosophila scalloped during wingblade development. J Biol Chem. 1997;272:10664–10668. doi: 10.1074/jbc.272.16.10664. [DOI] [PubMed] [Google Scholar]
- Dooley TP, Miranda M, Jones NC, DePamphilis ML. Transactivation of the adenovirus EIIa promoter in the absence of adenovirus E1A protein is restricted to mouse oocytes and preimplantation embryos. Development. 1989;107:945–956. doi: 10.1242/dev.107.4.945. [DOI] [PubMed] [Google Scholar]
- Fleming A, Copp AJ. Embryonic folate metabolism and mouse neural tube defects. Science. 1998;280:2107–2109. doi: 10.1126/science.280.5372.2107. [DOI] [PubMed] [Google Scholar]
- Harris MJ, Juriloff DM. Mouse mutants with neural tube closure defects and their role in understanding human neural tube defects. Birth Defects Res A: Clin Mol Teratol. 2007;79:187–210. doi: 10.1002/bdra.20333. [DOI] [PubMed] [Google Scholar]
- Jacquemin P, Hwang JJ, Martial JA, Dolle P, Davidson I. A novel family of developmentally regulated mammalian transcription factors containing the TEA/ATTS DNA binding domain. J Biol Chem. 1996;271:21775–21785. doi: 10.1074/jbc.271.36.21775. [DOI] [PubMed] [Google Scholar]
- Juriloff DM, Gunn TM, Harris MJ, Mah DG, Wu MK, Dewell SL. Multifactorial genetics of exencephaly in SELH/Bc mice. Teratology. 2001;64:189–200. doi: 10.1002/tera.1064. [DOI] [PubMed] [Google Scholar]
- Juriloff DM, Harris MJ. Mouse models for neural tube closure defects. Hum Mol Genet. 2000;9:993–1000. doi: 10.1093/hmg/9.6.993. [DOI] [PubMed] [Google Scholar]
- Juriloff DM, Harris MJ, Tom C, MacDonald KB. Normal mouse strains differ in the site of initiation of closure of the cranial neural tube. Teratology. 1991;44:225–233. doi: 10.1002/tera.1420440211. [DOI] [PubMed] [Google Scholar]
- Juriloff DM, MacDonald KB, Harris MJ. Genetic analysis of the cause of exencephaly in the SELH/Bc mouse stock. Teratology. 1989;40:395–405. doi: 10.1002/tera.1420400412. [DOI] [PubMed] [Google Scholar]
- Kaneko KJ, Cullinan EB, Latham KE, DePamphilis ML. Transcription factor mTEAD-2 is selectively expressed at the beginning of zygotic gene expression in the mouse. Development. 1997;124:1963–1973. doi: 10.1242/dev.124.10.1963. [DOI] [PubMed] [Google Scholar]
- Kaneko KJ, DePamphilis ML. Regulation of gene expression at the beginning of mammalian development and the TEAD family of transcription factors. Dev Genet. 1998;22:43–55. doi: 10.1002/(SICI)1520-6408(1998)22:1<43::AID-DVG5>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- Kaneko KJ, DePamphilis ML. Soggy, a spermatocyte-specific gene, lies 3.8 kb upstream of and antipodal to TEAD-2, a transcription factor expressed at the beginning of mouse development. Nucleic Acids Res. 2000;28:3982–3990. doi: 10.1093/nar/28.20.3982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaneko KJ, Rein T, Guo ZS, Latham K, DePamphilis ML. DNA methylation may restrict but does not determine differential gene expression at the Sgy/Tead2 locus during mouse development. Mol Cell Biol. 2004;24:1968–1982. doi: 10.1128/MCB.24.5.1968-1982.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lakso M, Pichel JG, Gorman JR, Sauer B, Okamoto Y, Lee E, Alt FW, Westphal H. Efficient in vivo manipulation of mouse genomic sequences at the zygote stage. Proc Natl Acad Sci USA. 1996;93:5860–5865. doi: 10.1073/pnas.93.12.5860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu P, Jenkins NA, Copeland NG. A highly efficient recombineering-based method for generating conditional knockout mutations. Genome Res. 2003;13:476–484. doi: 10.1101/gr.749203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacDonald KB, Juriloff DM, Harris MJ. Developmental study of neural tube closure in a mouse stock with a high incidence of exencephaly. Teratology. 1989;39:195–213. doi: 10.1002/tera.1420390211. [DOI] [PubMed] [Google Scholar]
- Mahoney WM, Jr., Hong JH, Yaffe MB, Farrance IK. The transcriptional co-activator TAZ interacts differentially with transcriptional enhancer factor-1 (TEF-1) family members. Biochem J. 2005;388:217–225. doi: 10.1042/BJ20041434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuki T, Hori G, Furuichi T. Gene expression profiling during the embryonic development of mouse brain using an oligonucleotide-based microarray system. Brain Res Mol Brain Res. 2005;136:231–254. doi: 10.1016/j.molbrainres.2005.02.008. [DOI] [PubMed] [Google Scholar]
- Milewski RC, Chi NC, Li J, Brown C, Lu MM, Epstein JA. Identification of minimal enhancer elements sufficient for Pax3 expression in neural crest and implication of Tead2 as a regulator of Pax3. Development. 2004;131:829–837. doi: 10.1242/dev.00975. [DOI] [PubMed] [Google Scholar]
- Murphy PJ, Galigniana MD, Morishima Y, Harrell JM, Kwok RP, Ljungman M, Pratt WB. Pifithrin-alpha inhibits p53 signaling after interaction of the tumor suppressor protein with hsp90 and its nuclear translocation. J Biol Chem. 2004;279:30195–30201. doi: 10.1074/jbc.M403539200. [DOI] [PubMed] [Google Scholar]
- Ohtsubo T, Rovira II, Starost MF, Liu C, Finkel T. Xanthine oxidoreductase is an endogenous regulator of cyclooxygenase-2. Circ Res. 2004;95:1118–1124. doi: 10.1161/01.RES.0000149571.96304.36. [DOI] [PubMed] [Google Scholar]
- Pani L, Horal M, Loeken MR. Rescue of neural tube defects in Pax-3-deficient embryos by p53 loss of function: implications for Pax-3- dependent development and tumorigenesis. Genes Dev. 2002;16:676–680. doi: 10.1101/gad.969302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puscheck EE, Patel Y, Rappolee DA. Fibroblast growth factor receptor (FGFR)-4, but not FGFR-3 is expressed in the pregnant ovary. Mol Cell Endocrinol. 1997;132:169–176. doi: 10.1016/s0303-7207(97)00131-7. [DOI] [PubMed] [Google Scholar]
- Ramalho-Santos M, Yoon S, Matsuzaki Y, Mulligan RC, Melton DA. “Stemness”: transcriptional profiling of embryonic and adult stem cells. Science. 2002;298:597–600. doi: 10.1126/science.1072530. [DOI] [PubMed] [Google Scholar]
- Sawada A, Nishizaki Y, Sato H, Yada Y, Nakayama R, Yamamoto S, Nishioka N, Kondoh H, Sasaki H. Tead proteins activate the Foxa2 enhancer in the node in cooperation with a second factor. Development. 2005;132:4719–4729. doi: 10.1242/dev.02059. [DOI] [PubMed] [Google Scholar]
- Schwarz M, Alvarez-Bolado G, Urbanek P, Busslinger M, Gruss P. Conserved biological function between Pax-2 and Pax-5 in midbrain and cerebellum development: evidence from targeted mutations. Proc Natl Acad Sci USA. 1997;94:14518–14523. doi: 10.1073/pnas.94.26.14518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimizu N, Smith G, Izumo S. Both a ubiquitous factor mTEF-1 and a distinct muscle-specific factor bind to the M-CAT motif of the myosin heavy chain beta gene. Nucleic Acids Res. 1993;21:4103–4110. doi: 10.1093/nar/21.17.4103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vassilev A, Kaneko KJ, Shu H, Zhao Y, DePamphilis ML. TEAD/TEF transcription factors utilize the activation domain of YAP65, a Src/Yes-associated protein localized in the cytoplasm. Genes Dev. 2001;15:1229–1241. doi: 10.1101/gad.888601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, Latham KE. Translation of maternal messenger ribonucleic acids encoding transcription factors during genome activation in early mouse embryos. Biol Reprod. 2000;62:969–978. doi: 10.1095/biolreprod62.4.969. [DOI] [PubMed] [Google Scholar]
- Weinstein M, Xu X, Ohyama K, Deng CX. FGFR-3 and FGFR-4 function cooperatively to direct alveogenesis in the murine lung. Development. 1998;125:3615–3623. doi: 10.1242/dev.125.18.3615. [DOI] [PubMed] [Google Scholar]
- Yagi R, Kohn M, Karavanova I, Kaneko KJ, Buonanno A, DePamphilis ML. TEAD4 Is Required Specifically For Establishment Of The Trophectoderm Lineage During Mammalian Preimplantation Development. 2007. submitted. [DOI] [PubMed]
- Yasunami M, Suzuki K, Houtani T, Sugimoto T, Ohkubo H. Molecular characterization of cDNA encoding a novel protein related to transcriptional enhancer factor-1 from neural precursor cells. J Biol Chem. 1995;270:18649–18654. doi: 10.1074/jbc.270.31.18649. [DOI] [PubMed] [Google Scholar]
- Yasunami M, Suzuki K, Ohkubo H. A novel family of TEA domain-containing transcription factors with distinct spatiotemporal expression patterns. Biochem Biophys Res Commun. 1996;228:365–370. doi: 10.1006/bbrc.1996.1667. [DOI] [PubMed] [Google Scholar]
- Yockey CE, Smith G, Izumo S, Shimizu N. cDNA cloning and characterization of murine transcriptional enhancer factor-1-related protein 1, a transcription factor that binds to the M-CAT motif. J Biol Chem. 1996;271:3727–3736. doi: 10.1074/jbc.271.7.3727. [DOI] [PubMed] [Google Scholar]
- Zhao P, Caretti G, Mitchell S, McKeehan WL, Boskey AL, Pachman LM, Sartorelli V, Hoffman EP. Fgfr4 is required for effective muscle regeneration in vivo. Delineation of a MyoD—Tead2—Fgfr4 transcriptional pathway. J Biol Chem. 2006;281:429–438. doi: 10.1074/jbc.M507440200. [DOI] [PMC free article] [PubMed] [Google Scholar]