

Abstract
Oxidative DNA damage has been implicated in a number of central nervous system pathologies. The base excision repair (BER) pathway is one of the most important cellular protection mechanisms that respond to oxidative DNA damage. Human apurinic (apyrimidinic) endonuclease/redox effector factor (APE1/Ref-1 or APE1) is an essential enzyme in the BER pathway and is expressed in both mitotic and post-mitotic cells in humans. In neurons, a reduction of APE1 expression increases chemotherapy-induced cytotoxicity, while overexpression of APE1 protects cells against the cytotoxicity. However, given the multiple functions of APE1, knockdown of total APE1 is not completely informative of whether it is the redox or DNA repair activity, or interactions with other proteins. Therefore, the use of selective small molecules that can block each function independent of the other is of great benefit in ascertaining APE1 function in post-mitotic cells. In this study, we chose differentiated SH-SY5Y cells as our post-mitotic cell line model to investigate whether a drug-induced decrease in APE1 DNA repair or redox activity contributes to the growth and survival of post-mitotic cells under oxidative DNA damaging conditions. Here, we demonstrate that overexpression of WT-APE1 or C65-APE1 (repair competent) results in significant increase in cell viability after exposure to H2O2. However, the 177/226-APE1 (repair deficient) did not show a protective effect. This phenomenon was further confirmed by the use of methoxyamine (MX), which blocks the repair activity of APE1 that results in enhanced cell killing and apoptosis in differentiated SH-SY5Y cells and in neuronal cultures after oxidative DNA damaging treatments. Blocking APE1 redox function by a small molecule inhibitor, BQP did not decrease viability of SH-SY5Y cells or neuronal cultures following oxidative DNA damaging treatments. Our results demonstrate that the DNA repair function of APE1 contributes to the survival of nondividing post-mitotic cells following oxidative DNA damage.
Keywords: Base excision repair (BER), oxidative stress, APE1/Ref-1, small molecule inhibitors, post-mitotic cells
1. Introduction
A number of environmental agents or metabolites induce neuronal damage through oxidative stress [1–3] that occurs secondary to the generation of reactive oxygen species (ROS) in the central nervous system (CNS). This can result in permanent damage given that the brain is especially vulnerable to ROS [4] and since neurons do not readily regenerate [5]. Oxidative stress is increasingly being implicated as an important causative agent in neuronal degenerative diseases such as amyotrophic lateral sclerosis (ALS), Parkinson’s disease (PD) and Alzheimer’s disease (AD) [6,7] as well as in aging. Recent data also have implicated oxidative stress and DNA damage in peripheral neuropathy and cognitive dysfunction that follows chemotherapy and ionizing radiation (IR) treatments of cancer patients [8,9].
The base excision repair (BER) pathway is one of the most important cellular protection mechanisms that responds to oxidative DNA damage, having a more prominent role in nondividing tissue [1]. The BER pathway including human apurinic (apyrimidinic) endonuclease/redox effector factor (APE1/Ref-1 or APE1) is required for repair of single-base modifications in DNA caused by ROS or spontaneous depurinations [1]. Removal of the damaged base by a DNA glycosylase creates an apurinic/apyrimidinic (AP) site. The endonuclease, APE1, a critical component in this pathway, hydrolyzes the phosphodiester backbone 5’ to the AP site. Failure to repair AP sites or impairing the function of APE1 leads to DNA strand breaks, enhanced oxidative stress-induced cytotoxicity, and increased apoptosis [10]. In addition to its DNA repair function, APE1 also acts as a reduction-oxidation (redox) factor for a variety of downstream transcription factors which impact upon a number of varied signaling pathways including those involved in neuronal function and survival [1].
Since the early 1990s, many studies have highlighted APE1’s multiple functions, the complexity of studying its multiple functions, and the importance of regulating APE1 activity [11]. APE1 has received growing attention in neuronal and cardiovascular physiology and pathophysiology in recent years [11]. Several laboratories, including ours, have demonstrated that overexpression of APE1 protects cells against chemotherapeutic agents [1]. Previous work from our laboratory has demonstrated that APE1 promotes survival of rat hippocampal and sensory neuronal cells after exposed to H2O2 and cisplatin [8,9], primarily through studies overexpressing or reducing expression of APE1. The question remains, however, whether the neuroprotective effects of APE1 in post-mitotic cells are due to its ability to repair DNA or its ability to alter the oxidative state of other molecules, or both. To address this question, we chose to examine the effects of APE1 mutants that have only repair or only redox function on oxidative stress-induced cytotoxicity and apoptosis in the SH-SY5Y cell line. These cells are derived from a neural crest tumor of early childhood, predominantly composed of undifferentiated neuroblast-like cells. Because these cells can be differentiated by exposure to retinoic acid (RA) [12,13], we chose these cells as our model system to study the post-mitotic phenotype. To delineate which function of APE1 was critical in the response to oxidative damage, we overexpressed wild type APE1 (WT-APE1), redox deficient/repair competent APE1 (C65-APE1), and repair deficient/redox competent APE1 (177/226-APE1) in differentiated SH-SY5Y cells. The cysteine at position 65 is required for full redox function of APE1 [14]. The redox deficient APE1 contains an alanine at position 65, and can no longer stimulate transactivation of a number of downstream targets such as nuclear factor-κB (NF-κB) or AP-1, and others [3]. The repair deficient APE1 used in these experiments contains two amino acid substitutions: R177A and N226A. This double mutant has dramatically decreased endonuclease activity and decreased binding to damaged DNA [15]. However, in this study, we also used for the first time, specific small molecule APE1 DNA repair inhibitor (MX) or a redox inhibitor (BQP) as another means of manipulating the various functions of APE1. In this report, we demonstrate using both genetic manipulation and small molecules that the DNA repair function of APE1 and not its redox activity is critical for post-mitotic cell survival and function.
2. Materials and methods
2.1. Cell culture
SH-SY5Y human neuroblastoma cells were obtained from ATCC (Manassas, VA), and were cultured in Dulbecco’s MEM/Ham’s F12 medium (1:1 mixture, ATCC) supplemented with 10% heat-inactivated fetal calf serum and 100U/ml penicillin and 100ug/ml streptomycin in a humidified 5% CO2, 95% air incubator at 37°C. The medium was changed every other day. To induce differentiation, the cells were treated with 10µM All trans-retinoic acid (RA) in the dark as described previously [12]. Adult rat dorsal root ganglion (DRG) and hippocampal cell cultures were prepared as previously described [9,16].
2.2. Drugs
All trans-retinoic acid (RA), hydrogen peroxide (H2O2), 2-methyl-1,4-naphthoquinone (menadione), hypoxanthine (HX), xanthine oxidase (XO), and methoxyamine hydrochloride (MX) were purchased from Sigma (St. Louis, MO). 3-[5-(2,3-dimethoxy-6-methyl-1,4-benzoquinoyl)]-2-nonyl-2-propionic acid (BQP) was custom synthesized [3]. MX was dissolved in PBS, and stored as a 1mol/L stock at −20°C for 1 month. BQP was dissolved in pure ethanol, stored as a 40mmol/L stock at −80°C for 2 weeks. Hypoxanthine (HX) was dissolved in 1M NaOH, with the stock solution being 25mg/ml by adjusting the solution pH to 7.4 using HCl. RA was prepared fresh with pure ethanol as a 10mmol/L, and then diluted to 10µM with culture medium; menadione was prepared fresh with culture medium before use. All stock solutions were appropriated diluted with buffer prior to use.
2.3. Adenoviral infection
Differentiated SH-SY5Y cells were infected with either the wild type Ad5 HA-APE1-IRES2-EGFP (WT-APE1), the redox deficient/DNA repair competent Ad5 HA-C65A-IRES2-EGFP (C65-APE1), DNA repair deficient/redox competent Ad5 HA-R177A + N226A-IRES2-EGFP (177/226-APE1), or vector control Ad5 IRES2-EGFP (Vector) adenovirus at 25 plaque-forming unitper cell for 24hrs by adding the virus directly to the growth medium, as previously described [8,9]. An HA tag was added to the amino terminus of APE1 and APE1 mutants to distinguish exogenous transgene overexpression from endogenous APE1 protein levels. After 24hrs, the medium containing adenovirus was aspirated and replaced with culture medium. Infection efficiencies of transduced cells were determined using fluorescence microscopy (Nikon, Tokyo, Japan) by measuring enhanced green fluorescent protein expression levels and Western blotting to detect APE1 and HA-tagged proteins.
2.4. Cell viability assay
Trypan blue exclusion analysis was performed as previously described [9]. Briefly, cells were detached from the plate using a 0.05% trypsin-EDTA solution and media was added. Equal volumes of the cell suspension and 0.4% (w/v) trypan blue in PBS were mixed, and the cells were scored under phase contrast microscope using a hemacytometer. Percent survival was calculated as percent of live cells divided by total cell number (including dead and live cells).
2.5. 3-(4-5-Dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium salt (MTS) assay
Cell viability was also measured using the MTS assay (Celltiter 96 Aqueous One solution cell proliferation assay, Promega Corp., Madison, WI) on differentiated SH-SY5Y cells grown in 96-well plates. Briefly, 24hrs after various drug treatments, the MTS solution was added to each well and the plates were incubated for 4hrs. The absorbance at 490nm was measured using a multiwell plate reader. The values were standardized to wells containing media alone.
2.6. Cell apoptosis analysis
Flow cytometric detection of apoptosis was performed using the Annexin V Apoptosis Detection kit (BD Biosciences, Rockville, MD) according to manufacturer's instructions. Cells were harvested using Trypsin-EDTA (Invitrogen Corp., Carlsbad, California). After washing twice with PBS, the cells were resuspended in 100µl binding buffer and stained with Annexin V-FITC/propidium iodide (PI). The cell suspension was incubated for 15min in the dark at 4°C. The percentage of cells undergoing apoptosis was determined using flow cytometry. Apoptotic cells were defined as those positive for Annexin V.
2.7. Western blot analysis
Western blots were performed as described previously [8,17]. Briefly, protein was quantified and electrophoresed in a 12% SDS-polyacrylamide gel. After electrophoresis, the gel was transferred to a nitrocellulose membrane. The membranes were separately incubated with a primary antibody against GAP-43 (Novus Biologicals, Inc., Littleton, CO), APE1 [8,9], HA (Roche Diagnostics Corp., Indianapolis, IN) and β-Actin (NeoMarkers, Inc., Fremont, CA). Antibody binding was detected following appropriate secondary antibody using chemiluminescence (Roche Diagnostics Corp., Indianapolis, IN), and equal loading was confirmed by probing with β-Actin monoclonal antibody.
2.8. qRT-PCR assay
qRT-PCR was used to measure the mRNA expression levels of TNF-α (tumor necrosis factor-alpha) gene. Adult sensory neuronal cultures were pretreated with or without 10 or 20µM BQP 3 days, then treated with 1mM H2O2 1h, then total RNA was extracted from cells following 1h and 5hrs culture using the combination of Qiagen QiaShredder kit and Qiagen RNeasy Mini kit (Valencia, CA) according to the manufacturer's instructions. The extracted RNA was quantified by a SmartSpec spectrophotometer (Bio-Rad Laboratories, Hercules, CA, USA) at a wavelength of 260 nm. cDNA was synthesized by High-Capacity cDNA Reverse Transcription Kit according to the protocol provided by the manufacture (Part No.: 4368814 Applied Biosystems). The primers for TNF-α were 5’-AGGCTGCCCCGACTATGTG-3’ and 5’-AGGAGGCTGACTTTCTCCTGGTA-3’; the primers for GAPDH were 5’-GTATGACTCTAC CCACGGCAAGT-3’ and 5’-TCTCGCTCCTGGAAGATGGT-3’. qRT-PCR was conducted on an ABI PRISM 7500 Sequence Detection System (Applied Biosystems) using RT2-SYBR Green /ROX qPCR Master Mix (SABiosciences, Frederick, MD) in a 25ul final reaction mixture. The cycling conditions were an incubation at 50°C for 2 min, 95°C for 10min and 40 cycls of 15s at 95°C, and 1 min at 60°C. Experiments were performed in triplicate for each sample. TNF-α was normalized to GAPDH, fold difference calculated by 2−ΔΔCt as described before [18].
2.9. Statistical analysis
Quantitative data were obtained from three independent experiments and expressed as the mean +/− SD. Statistical significances of differences between two groups were determined by using Student’s t test. Statistical significance was taken as p<0.05 in all experiments.
3. Results
3.1. Post-mitotic cell line model: differentiated SH-SY5Y cells
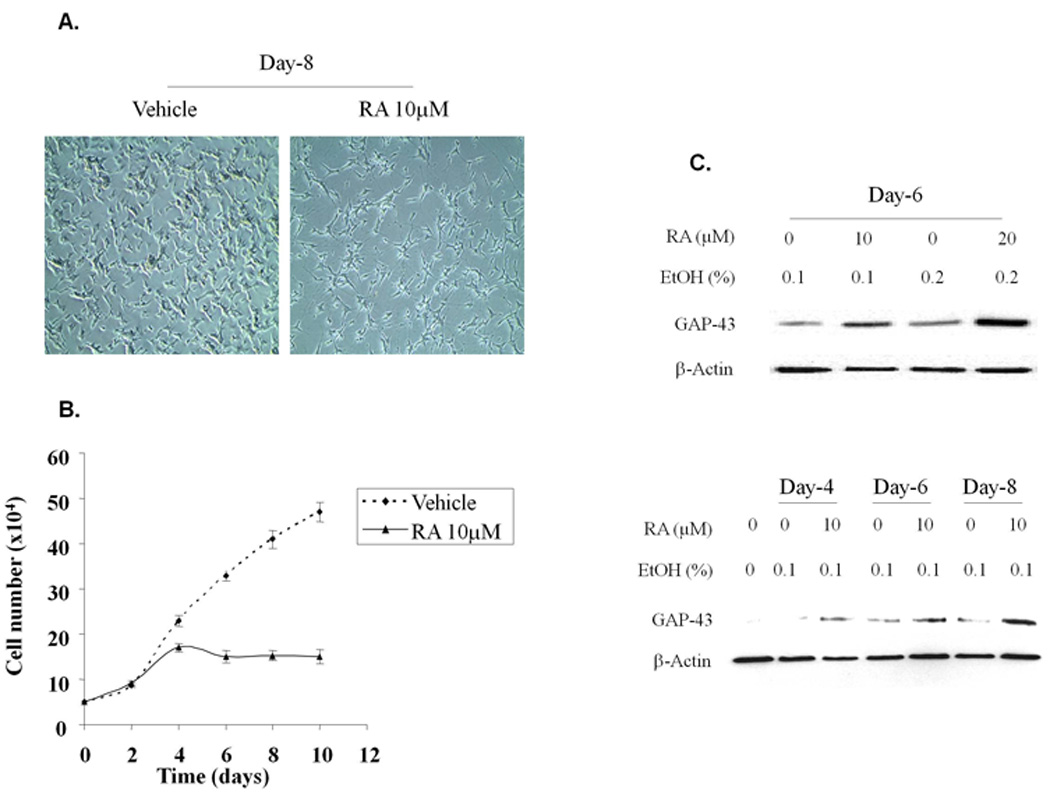
SH-SY5Y cells differentiate into a post-mitotic phenotype after exposure to retinoic acid [19,20]. To characterize the effects of RA on differentiation of SH-SY5Y cells, cells were treated with 10µM RA, changed every two days and the RA was removed after 6 days of treatment. During the time course, cell morphology was observed, cells were counted and expression of GAP-43 was examined on various days during and after treatment. GAP-43 expression was used as a marker of differentiation since it is expressed by developing and regenerating neurons [1] and its levels are highest during periods of neurite outgrowth [21]. RA treatment resulted in differentiation of SH-SY5Y cells, >90% of the cells displaying long neurites on day 8 (Fig. 1A). Proliferation of the SH-SY5Y cells was dramatically decreased following treatment with RA, as expected (Fig. 1B). There also was a dramatic increase in GAP-43 expression following RA treatment for six days (Fig. 1C, upper panel). Both 10µM and 20µM RA induced expression of GAP-43 by day 6. In addition, 10µM RA induced expression of GAP-43 over time (days 4 – 8) (Fig. 1C, bottom panel). Based on these results and previous publications [12,20], in all further experiments, we examined post-mitotic SH-SY5Y cells 8 days after initiating RA treatment at 10µM.
Fig. 1.
3.2. Effect of APE1 overexpression on viability of differentiated SH-SY5Y following H2O2 treatment
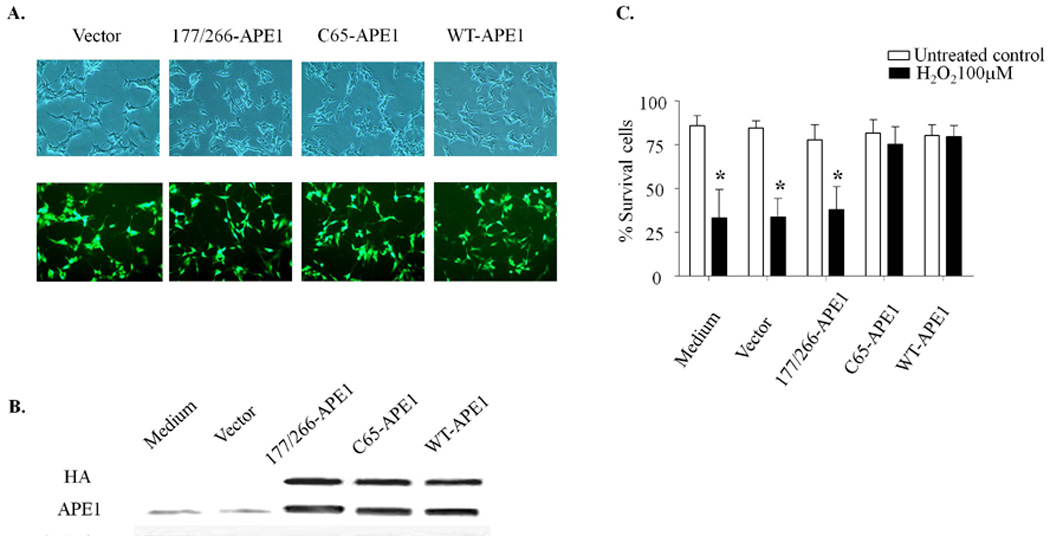
Previous studies in our laboratory have demonstrated that a reduction in APE1 expression in central and peripheral neurons increases cytotoxicity, while overexpression of APE1 decreases cytotoxicity following treatment with both laboratory and chemotherapeutic agents that produce oxidative stress [8,9]. To ascertain whether overexpression of APE1 effects survival of differentiated SH-SY5Y cells, we infected the cells with HA tagged WT-APE1, C65-APE1, 177/226-APE1 or vector control adenoviral constructs containing an IRES site, and EGFP. Adenovirus infection efficiency was determined using fluorescence microscopy with over 80% of the cells showing green fluorescence after 48hrs exposure to virus (Fig. 2A). APE1 overexpression was confirmed using western blot analysis with HA or APE1 antibodies to distinguish endogenous and transgene APE1 protein levels. As can be seen in representative western blot (Fig. 2B), APE1 levels were significantly increased over endogenous levels in cells infected with WT-APE1 and the two mutant APE1s. To ascertain whether overexpression of WT-APE1 or APE1 mutants protect differentiated SH-SY5Y cells, the infected cells expressing WT-APE1, C65-APE1, 177/226-APE1 or vector control were treated with 100µM H2O2 for 24hrs, and cell viability was measured using trypan blue exclusion. As observed in Fig. 2C, overexpression of WT-APE1 and C65-APE1 rescued differentiated SH-SY5Y cells from cell killing by H2O2. In contrast, overexpression of the repair deficient 177/226-APE1 did not provide any protective effect (Fig. 2C). Since the C65-APE1 mutant has DNA repair but not redox activity and the 177/226-APE1 has decreased DNA repair activity [15], these results suggest that the repair, but not the redox function of APE1 is necessary for protection against H2O2-induced cell death in post-mitotic SH-SY5Y cells.
Fig. 2.
3.3. Effect of MX on cytotoxicity induced by agents producing oxidative DNA damage in the differentiated SH-SY5Y cells, or DRG and hippocampal neuronal cultures
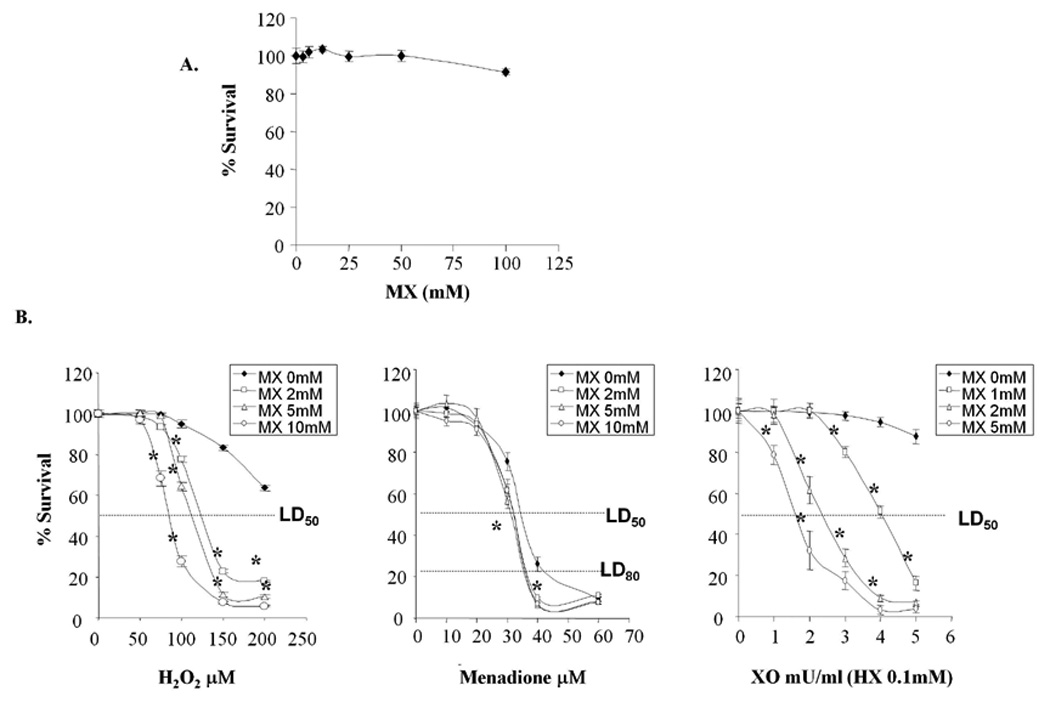
In view of the fact that APE1 has multiple functions and interacts with many other proteins [1], the overexpression of APE1 protein may result in multiple effects, thereby making it difficult to ascertain which of the functions of APE1 result in protection [3,22]. Consequently to further substantiate that the repair function of APE1 is important in protecting post-mitotic cells, we used MX to modify and stabilize the AP site, block the ability of APE1 to cut the sugar-phosphate backbone, and thus prevent the completion of the repair process [23,24]. To determine whether APE1 repair activity was linked to loss in cell viability, differentiated SH-SY5Y cells were exposed to various concentrations of MX in the absence or presence of oxidative stress and cell viability measured using the MTS assay. In cells treated with MX alone, little to no reduction in cell viability was observed up to 100mM MX (Fig. 3A). As a result, in subsequent studies, we used concentrations of MX up to 10mM. When differentiated SH-SY5Y cells were exposed to MX in combination with increasing concentrations of H2O2, MX enhanced H2O2-induced cell killing at all concentrations tested (Fig. 3B, left panel). We also measured the effects of MX on cell killing induced by two additional compounds that generate ROS, menadione and xanthine oxidase. Menadione, (2-methyl-1,4-naphthoquinone), is a quinone compound that undergoes redox cycling and generates reactive oxygen species [25,26]. Xanthine oxidase (XO) is an enzyme that catalyzes the sequential breakdown of hypoxanthine (HX) to xanthine and subsequently to uric acid. In this oxidation process, XO concomitantly generates several ROS by reducing molecular oxygen [27,28]. When cells were exposed to increasing concentrations of menadione in the presence of MX, cell killing was enhanced at concentrations of menadione greater than 30µM, albeit not as dramatically as H2O2 or XO (Fig. 3B, middle panel). Similar to H2O2 and menadione, when APE1’s DNA repair function was blocked by MX, there was a significant enhancement of XO/HX-induced cell killing (Fig. 3B, right panel). The LD50 of H2O2 and XO/HX in the presence or absence of MX treatment are shown in Table 1. The LD80 of menadione is included because the effect of MX was more significant at the LD80. MX significantly decreased the LD50 or LD80 of the three oxidizing agents compared to agent alone in a concentration-dependent manner. However, the effect of MX with menadione, while statistically significant was not as dramatic.
Fig. 3.
Tabel 1.
Addition of MX to Oxidizing Agents Results in a Decrease in Cell Survival in differentiated SH-SY5Y cells
| Oxidizing agent | MX 0mM | MX 1mM | Mx 2mM | MX 5mM | MX 10mM |
|---|---|---|---|---|---|
| H2O2 (uM) LD50 | 220 ± 7 | 125 ± 2* | 112 ± 2*# | 85 ± 4*# | |
| §menadiones (uM) LD80 | 44 ± 3 | 36 ± 2* | 36 ± 2* | 36 ± 1* | |
| XO (mU/ml)/HX LD50 | 17 ± 3 | 4 ± 0.2* | 2.4 ± 0.1*# | 1.6 ± 0.2*# |
Note: The values are LD50 of H2O2 and XO/HX and LD80 of menadione or without MX treatment for 24hrs (n=3).
Abbreviations: H2O2, hydrogen peroxide; XO, Xanthine Oxidase; HX, hypoxanthine; MX, methoxyamine.
P< 0.05, all compared each +/− MX to oxidant alone at LD50 dose.
P< 0.05, all compared each + higher dose MX to oxidant + lower MX dose at LD50 dose.
LD80menadione
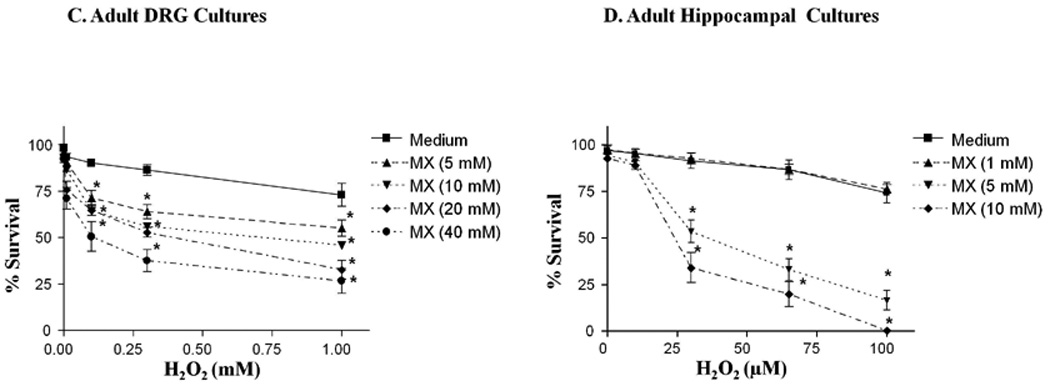
To ascertain whether the cytotoxicity of primary neurons to oxidizing agents is enhanced when the DNA repair activity of APE1 is blocked, we treated adult rat DRG cultures and adult rat hippocampal cultures with various concentrations of H2O2 for 1hr in the presence or absence of MX and assessed cellular viability using trypan blue exclusion at 24hrs. As observed in the SH-SY5Y cells, MX enhanced H2O2-induced cytotoxicity in both neuronal cultures. The enhanced cell killing induced by MX was MX concentration-dependent (Fig. 3C, D). The LD50 of H2O2 +/− MX treatment in DRG and hippocampal neuronal are shown in Table 2.
Table 2.
LD50 of the H2O2 +/− MX treatment in the DRG and Hippocampal Cell cultures
| MX 0mM | MX 1mM | MX 5mM | MX 10mM | MX 20mM | MX 40mM | |
|---|---|---|---|---|---|---|
| Hippocampal | ||||||
| H2O2 (uM) | 341 ± 12 | 354 ± 6 | 42 ± 6*# | 24 ± 3*# | ||
| DRG | ||||||
| H2O2 (uM) | 3413 ± 63 | 2310 ± 45*# | 861 ± 51*# | 478 ± 27*# | 110 ± 29*# |
Note: The values are LD50 of H2O2 with or without MX pretreatment for 24hrs (n=3).
Abbreviations: H2O2, hydrogen peroxide; MX, methoxyamine. Drg, dorsal root ganglion.
P< 0.05, all compared each H2O2 +/− MX to oxidant alone at LD50 dose.
P< 0.05, all compared each H2O2 +higher dose MX to H2O2 + lower MX dose at LD50 dose.
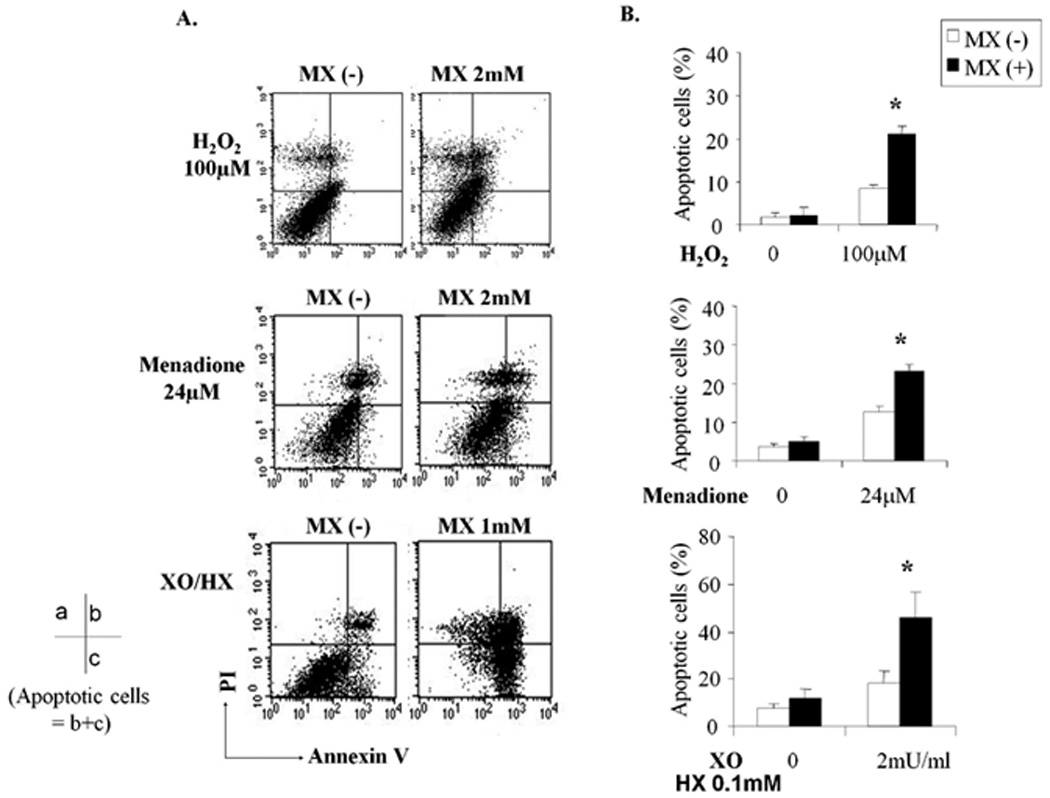
3.4. MX increases apoptosis of differentiated SH-SY5Y cells induced by oxidative DNA damaging agents
ROS are potent inducers of apoptosis [29]. To determine whether blocking the DNA repair function of APE1 with MX affects oxidative stress-induced apoptosis in the differentiated SH-SY5Y cells and confirm the cytotoxicity data in Fig. 3, we incubated the cells with either H2O2, menadione or XO/HX in the absence or presence of MX for 24hrs. Apoptosis was determined using Annexin V and propidium iodide (PI) staining. As shown in the representative experiment in Fig. 4A, addition of MX to oxidative DNA damaging agents resulted in a significant increase in Annexin V positive cells (lower and upper right quadrants) indicating apoptotic cells. In three separate experiments, the addition of MX caused a 9% to 21% increase in apoptosis induced by H2O2 treatment, a 12% to 23% increase in apoptosis induced by menadione treatment, and an 18% to 44% increase in apoptotic cells in cultures treated with XO/HX (Fig. 4B). The addition of MX to H2O2, menadione, and XO/HX treatment dramatically reduced cell survival both by killing and apoptosis-based assays.
Fig. 4.
3.5. Effect of BQP on cytotoxicity in differentiated SH-SY5Y cells induced by oxidative DNA damaging agents
BQP is a specific, small molecule inhibitor of the redox function of APE1 that does not affect its DNA repair function [3,30]. To further confirm that the redox function of APE1 was not as critical following oxidative stress, we treated cells with oxidative DNA damaging agents in combination with APE1 redox inhibitor, BQP. The MTS assay was used to evaluate cell survival following increasing doses of BQP in the absence or presence of oxidative DNA damaging agents. At BQP concentrations of 50µM or greater, cell survival was significantly decreased in SH-SY5Y cells compared to vehicle control, with a resulting LD50 of 80µM (Fig. 5A). As noted in Fig. 5A, the viability of SH-SY5Y cells was not affected with BQP concentrations of 25µM or less within 24hrs treatment. Blocking APE1’s redox function using BQP at 15µM and 25µM did not significantly alter cytotoxicity induced by H2O2, menadione, or XO/HX (Fig. 5B). To ascertain whether primary cultures of neuronal cells are responding in a similar manner to differentiated SH-SY5Y cells, we treated rat DRG and hippocampal cultures with H2O2 in the absence or presence of 10, 20 or 40µM BQP. In contrast to differentiated SHSY5Y cells, BQP significantly reduced H2O2-induced cytotoxicity in DRG or hippocampal neuronal cell cultures (Fig. 6A, B), suggesting it has a protective effect in these cultures.
Fig. 5.
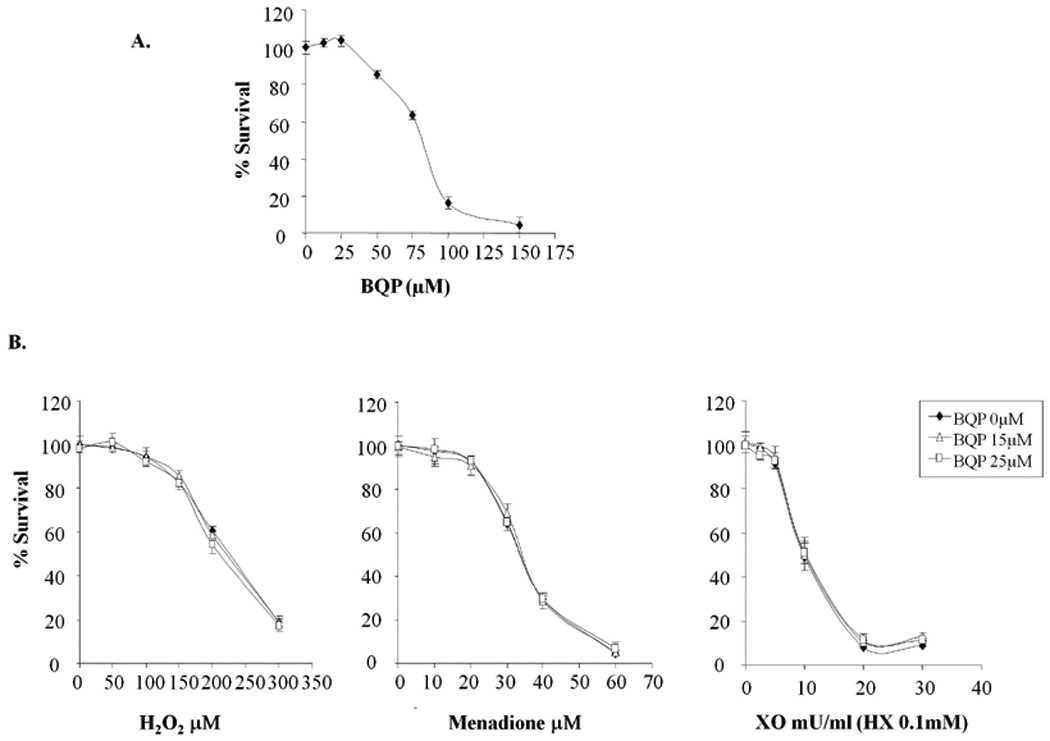
Fig. 6.

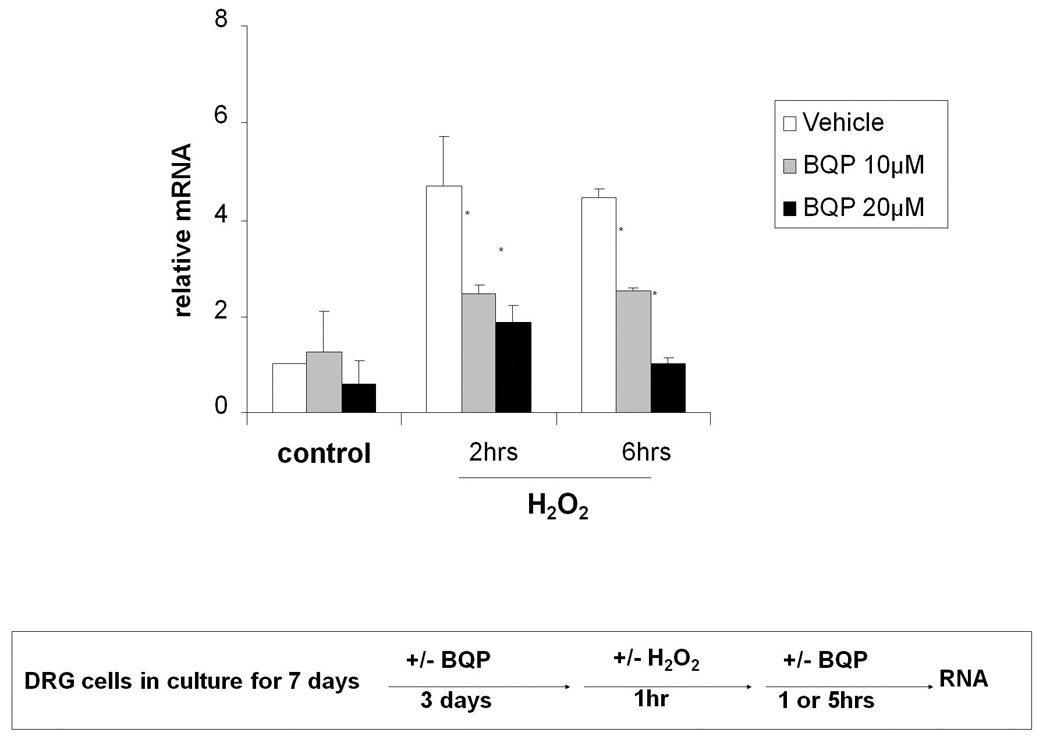
3.6. Effect of BQP on H2O2-induced TNF-α mRNA expression in DRG neuronal cell cultures
Some previous studies have demonstrated that BQP inhibits the production of TNF-α through inhibition of NF-κB, a redox signaling target of APE1, at the transcriptional level [31–33]. TNF-α is also known to induce neuronal damage in mitochondrial respiration [34]. To ascertain whether BQP protects DRG cell cultures against H2O2-induced cytotoxity through a NF-κB mediated pathway producing TNF-α production, we pretreated DRG cell cultures with BQP for 3 days. We then challenged the cells with 1mM H2O2 for 1hr and analyzed TNF-α mRNA levels using qRT-PCR assay. We used DRG cells for these studies as we could not detect TNF-α in the SH-SY5Y cells and they represent a more real life model. As predicted, 10µM and 20µM BQP significantly reduced H2O2-induced TNF-α mRNA expression in DRG cultures. Using 10µM BQP, H2O2-induced TNF-α mRNA level was reduced 40% at the two and six hour time points, while 20µM BQP, H2O2-induced TNF-α mRNA level was reduced 60% at the two hour time point and and 80% at the six hour time point compared to no BQP vehicle control (Fig. 7). Because inhibition of mRNA for TNF-α serves as a biomarker for NF-κB production [31,32], these results suggest that inhibition of APE1’s redox function by BQP contributed to the protective effect in neuronal cell cultures through APE1 down-regulated activity of NF-κB and subsequent downstream events such as TNF-α reduction [33].
Fig. 7.
4. Discussion
Based on the results presented here, it is clear that the DNA repair function of APE1 is critical for protection of post-mitotic cells against agents that produce oxidative DNA damage. The SH-SY5Y cell line is derived from a neural crest tumor of early childhood, predominantly composed of undifferentiated neuroblast-like cells. These neuroblastoma cells respond to retinoic acid (RA) by differentiating into cells of neuronal, post-mitotic phenotype [19,35]. The SH-SY5Y system allows us to prepare large numbers of differentiated cells for use in our neuronal studies [35] and to compare and contrast with studies in primary rat DRG and hippocampal neuronal cell cultures. Overexpression of WT-APE1 in differentiated SH-SY5Y cells protected cells from H2O2-induced cytotoxicity. This finding is analogous to our previous work demonstrating that WT-APE1 is protective when overexpressed in neuronal cells grown in culture [8, 9]. To further examine which function of APE1, DNA repair or redox provided this protective effect, we overexpressed APE1 mutants in SH-SY5Y cells. The C65-APE1 mutant contains a mutation at the Cys at position 65 which abolishes APE1’s redox function but does not affect its repair capability [3,14]. Overexpression of C65-APE1 provides the same degree of protection as WT-APE1. In contrast, overexpression of the DNA repair deficient mutant, 177/226-APE1, did not protect cells from H2O2-induced cell death.
We also used small molecule inhibitors of the DNA repair or redox function of APE1 to ascertain which functions of APE1 are critical for neuroprotection. Exposing SH-SY5Y cells to BQP, did not enhance cell death induced by oxidative DNA damage, whereas exposing primary neuronal cultures to BQP reduced rather than enhanced cell death induced by H2O2. Blocking APE1’s repair activity with MX reduced cell viability and increased apoptosis in response to oxidative DNA damaging agents in differentiated SH-SY5Y cells and in primary neuronal cultures. These results expand on our previous work in hippocampal and DRG cell cultures showing that overexpression of APE1 is neuroprotective against H2O2 and cisplatin, while reducing APE1 expression is neurotoxic [8,9]. In those studies, we surmised that in post-mitotic neuronal cells, APE1’s DNA repair function was critical to survival. This notion was based on overexpression of constructs containing similar to the ones described here. However we could not completely rule out the redox component of APE1 since overexpression of C65-APE1 provided partial protective effect in hippocampal and DRG cell cultures against H2O2 and cisplatin [8,9]. By using small molecule inhibitors of either the repair or redox function of APE1 in the current study, we can conclude that the DNA repair function of APE1 is the critical function in protecting post-mitotic cells against oxidative DNA damage. Previous studies have reported that knocking down APE1 leads to cell death [10,36] and is most likely due to the DNA repair function of APE1 rather than the redox activity [2,37–39]. Additionally, the role of APE1’s redox and repair functions, as well as APE1’s protein-protein interactions may be varied in post-mitotic cells, such as DRG cultures [9,17], as well as neuronal cell line models such as the extensively used SH-SY5Y cells. However, the results presented in this manuscript demonstrating that the DNA repair function of APE1 is important for cellular function and survival in post-mitotic cells is a novel finding and supports our previous studies using overexpression of APE1 repair or redox mutants. These findings also explain why inhibition of APE1’s redox function had no effect on the survival of the post-mitotic cells under oxidative DNA damaging conditions.
Our results implicating the DNA repair function of APE1 as crucial in the survival of post-mitotic cells following oxidative stress is not overly surprising. In light of data demonstrating that mice lacking APE1 are embryonic lethal and the need for adequate DNA repair early on in development [40,41], it is clear that the DNA repair function is critical to the survival of animals and, presumably, humans [2,3,17,22,30,42,43]. As a major member of the BER pathway, APE1 acts on AP sites in DNA, which, if left unrepaired, can result in a block to DNA replication, cytotoxicity, mutations, and genetic instability [22,44]. In embryonic development robust DNA repair is required to sustain DNA integrity during accelerated DNA transactions, which support rapid cell proliferation. However, knockdown or out studies remove both the DNA repair and redox functions of APE1.
In previous studies to reveal whether APE1 was essential to normal and/or cancer cells [1–3,8,17,22,30,38,39,42,43,45,46], antisense to APE1 or APE1 siRNA was utilized. These types of experimental conditions remove not only the DNA repair function of APE1, but also its redox function and all protein–protein interactions mediated by APE1 [22]. Thus, using antisense RNA or similar technology, it is not possible to determine precisely the role of the endonuclease or redox function of APE1 in cancer or normal cells without specific inhibitors of each function independently. As demonstrated by the results presented here, the use of specific small-molecule inhibitors such as MX and BQP allowed us to delineate the true role of APE1 in post-mitotic cells following oxidative stress [3,30]. Additional reports by others also demonstrate that overexpression of APE1 inhibits superoxide and hydrogen peroxide production [47] and that overexpression of APE1 in cells deficient for XRCC1 compensates for the deficient single-strand break repair in these cells following oxidative DNA damage and stress [48].
This is the first report using APE1 specific small molecule repair or redox inhibitors in a post-mitotic system, differentiated SH-SY5Y cells, or in primary neuronal cells: rat DRG and hippocampal cells. The APE1 repair inhibitor, MX, binds to AP sites in DNA and blocks the ability of APE1 to incise the sugar-phosphate backbone and prevents the completion of the repair process [22,23,49,50]. MX has previously been studied as a structural modulator of AP sites that enhances the therapeutic effect of chemotherapeutic agents, such as temozolomide (TMZ), through its ability to block the repair of AP sites [22,23,49,50]. Clinical trials with MX in combination with TMZ are currently being pursued [17,23,30,38,42,50,51]. As shown in Fig. 3 and Fig. 4, MX is not significantly cytotoxic by itself in post-mitotic cells, however when APE1’s DNA repair function is blocked with MX, we demonstrate a dramatic enhancement of oxidant-induced cytotoxicity and apoptosis in differentiated SH-SY5Y cells and primary DRG and hippocampal neuronal cell cultures. Interestingly, the effect was most dramatic when combining H2O2 and XO with MX and less so with menadione, although statistically significant (Fig. 3). This could be explained by the fact that menadione is a redox-cycling drug that creates large amounts of intracellular superoxide anions and lesser amounts of H2O2 [29]. In the XO system, a significant amount of H2O2 is produced as well as hydroxyl radicals which are both strong damagers of DNA [29]. Since menadione undergoes redox cycling there is a more gradual increase in ROS levels [52,53]. This could explain the differences between the results with agents that induce oxidative DNA damage in different ways.
Since APE1 stimulates the transcriptional activity of numerous transcription factors that are involved in a multitude of diverse physiological functions such as cell cycle control, apoptosis, angiogenesis, cellular growth, cellular differentiation, neuronal excitation, hematopoiesis and development through its redox function, it was possible that the redox function could be important in post-mitotic cells [44]. Our data (Fig. 5), however, indicate that inhibition of APE1’s redox function by BQP did not increase the killing effect of the oxidative damage analyzed. In fact, when BQP was used on primary DRG and hippocampal neuronal cell cultures in combination with H2O2, the cells were less sensitive to the effects of oxidative DNA damage by H2O2. This supports our hypothesis that the redox role is independent of the APE1 DNA repair function. The APE1 redox inhibitor, BQP, was initially identified as a small molecule that inhibited TNF-α production in vivo [32,54] using hepatitis models and LPS-stimulated macrophages including mononuclear cells and Kupffer cells. TNF-α is known to induce apoptosis in neurodegenerative diseases [55]. Although the mechanism by which BQP is protective in primary neuronal cultures remains unknown, our observations show that BQP reduced H2O2-induecd TNF-α mRNA levels in primary rat DRG, providing a possible mechanism by which the effect of BQP on TNF-α generation contributes to the neuroprotective effect. BQP was also shown to suppress DNA-binding of NF-κB [56]. Therefore, although BQP does not bind to NF-κB, but APE1, one possible explanation for its protective effect observed in the presence of H2O2 is it blocks NF-κB function by blocking NF-κB’s ability to bind to various promoter’s such as the TNF-α promoter and, therefore, reduce TNF-α expression. Clearly, this is just one pathway that could be affected and additional experiments are ongoing to determine additional pathways under APE1 redox control that could contribute to our observed results.
The question remains whether APE1 repair function is critical in DNA repair in the nucleus or in the mitochondria, or both. Thus, further studies are needed to determine the potential role of mitochondria, and specifically APE1 in mitochondria, in post-mitotic cells response to ROS. APE1 has been demonstrated to function as a DNA repair enzyme in mitochondrial DNA repair [17]. Given our findings that it is primarily the DNA repair and not the redox activity of APE1 that is important for post-mitotic cellular survival and response to oxidative DNA damage, the role of APE1 in mitochondrial function is of great interest. These studies will lead us to new avenues of research to determine if we can therapeutically block the effect of post-mitotic cellular killing and dysfunction following cancer treatments in order to decrease the side-effects that are often damaging and debilitating to the patients undergoing cancer treatments.
In summary, to our knowledge, this is the first report of using small molecule inhibitors of APE1’s DNA repair or redox function and the consequences of such inhibition on nondividing, post-mitotic cells. The use of small molecule APE1 redox or repair inhibitors confirms our analyses using mutant redox or repair APE1 transgene overexpression studies. These studies all conclude that the repair and not the redox function of APE1 is the most important activity of APE1 following oxidative stress in post-mitotic cells as typified by SH-SY5Y differentiated cells.
Acknowledgements
Financial support for this work was provided by the National Institutes of Health NS048565 to M.R.V., National Cancer Institute CA121168 to M.R.V., National Cancer Institute CA094025, CA106298, CA114571 and CA121168 to M.R.K. and the Riley Children’s Foundation (M.R.K) and National Cancer Institute CA122298 (M.L.F.). Some of these studies were conducted in a facility constructed with the support from Research Facilities Improvement Program Gant Number C06 RR015481-01 from the National Centre for Research Resources, National Institutes of Health (M.R.V.).
The abbreviations used are
- APE1
human apurinic (apyrimidinic) endonuclease/redox effector factor
- WTAPE1
wild type APE1
- C65-APE1
repair competent/redox deficient APE1
- 177/226-APE1
repair deficient/redox competent APE1
- MX
methoxyamine
- BQP
3-[5-(2,3-dimethoxy-6-methyl-1,4-benzoquinoyl)]-2-nonyl-2-propionic acid
- HX
hypoxanthine
- XO
xanthine oxidase
- RA
retinoic acid
- IR
ionizing radiation
- ROS
reactive oxygen species
- BER
base excision repair
- DRG
dorsal root ganglion
- ALS
amyotrophic lateral sclerosis
- PD
Parkinson’s disease
- AD
Alzheimer’s disease
- HA
hemagglutinin epitope
- EGFP
enhanced green fluorescent protein
- IRES
internal ribosome entry sites
- PI
propidium iodide
- FITC
fluorescein isothiocyanate
- TNF-α
tumor necrosis factor-alpha
- NF-κB
nuclear factor-κB
- TMZ
temozolomide
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest:
No conflicts of interest were disclosed.
References
- 1.Fishel ML, Vasko MR, Kelley MR. DNA repair in neurons: so if they don't divide what's to repair? Mutat Res. 2007;614:24–36. doi: 10.1016/j.mrfmmm.2006.06.007. [DOI] [PubMed] [Google Scholar]
- 2.Georgiadis MM, Luo M, Gaur RK, Delaplane S, Li X, Kelley MR. Evolution of the redox function in mammalian apurinic/apyrimidinic endonuclease. Mutat Res. 2008;643:54–63. doi: 10.1016/j.mrfmmm.2008.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Luo M, Delaplane S, Jiang A, Reed A, He Y, Fishel M, Nyland Ii RL, Borch RF, Qiao X, Georgiadis MM, Kelley MR. Role of the Multifunctional DNA Repair and Redox Signaling Protein Ape1/Ref-1 in Cancer and Endothelial Cells: Small-Molecule Inhibition of the Redox Function of Ape1. Antioxid Redox Signal. 2008 doi: 10.1089/ars.2008.2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wilson DM, 3rd, Mattson MP. Neurodegeneration: nicked to death. Curr Biol. 2007;17:R55–R58. doi: 10.1016/j.cub.2006.12.012. [DOI] [PubMed] [Google Scholar]
- 5.Mura CV, Delgado R, Aguirre P, Bacigalupo J, Nunez MT. Quiescence induced by iron challenge protects neuroblastoma cells from oxidative stress. J Neurochem. 2006;98:11–19. doi: 10.1111/j.1471-4159.2006.03798.x. [DOI] [PubMed] [Google Scholar]
- 6.Bains JS, Shaw CA. Neurodegenerative disorders in humans: the role of glutathione in oxidative stress-mediated neuronal death. Brain Res Brain Res Rev. 1997;25:335–358. doi: 10.1016/s0165-0173(97)00045-3. [DOI] [PubMed] [Google Scholar]
- 7.Olivieri G, Baysang G, Meier F, Muller-Spahn F, Stahelin HB, Brockhaus M, Brack C. Nacetyl-L-cysteine protects SHSY5Y neuroblastoma cells from oxidative stress and cell cytotoxicity: effects on beta-amyloid secretion and tau phosphorylation. J Neurochem. 2001;76:224–233. doi: 10.1046/j.1471-4159.2001.00090.x. [DOI] [PubMed] [Google Scholar]
- 8.Jiang Y, Guo C, Vasko MR, Kelley MR. Implications of apurinic/apyrimidinic endonuclease in reactive oxygen signaling response after cisplatin treatment of dorsal root ganglion neurons. Cancer Res. 2008;68:6425–6434. doi: 10.1158/0008-5472.CAN-08-1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vasko MR, Guo C, Kelley MR. The multifunctional DNA repair/redox enzyme Ape1/Ref-1 promotes survival of neurons after oxidative stress. DNA Repair (Amst) 2005;4:367–379. doi: 10.1016/j.dnarep.2004.11.006. [DOI] [PubMed] [Google Scholar]
- 10.Fung H, Demple B. A vital role for Ape1/Ref1 protein in repairing spontaneous DNA damage in human cells. Mol Cell. 2005;17:463–470. doi: 10.1016/j.molcel.2004.12.029. [DOI] [PubMed] [Google Scholar]
- 11.Jeon BH, Irani K. APE1/Ref-1: Versatility in Progress. Antioxid Redox Signal. 2008 doi: 10.1089/ars.2008.2223. [DOI] [PubMed] [Google Scholar]
- 12.Sharma M, Sharma P, Pant HC. CDK-5-mediated neurofilament phosphorylation in SHSY5Y human neuroblastoma cells. J Neurochem. 1999;73:79–86. doi: 10.1046/j.1471-4159.1999.0730079.x. [DOI] [PubMed] [Google Scholar]
- 13.Pahlman S, Mamaeva G, Meyerson G, Mattsson ME, Bjelfman C, Ortoft E, Hammerling U. Human neuroblastoma cells in culture: a model for neuronal cell differentiation and function. Acta Physiol Scand Suppl. 1990;592:25–37. [PubMed] [Google Scholar]
- 14.Tell G, Quadrifoglio F, Tiribelli C, Kelley MR. The Many Functions of APE1/Ref-1: Not Only a DNA Repair Enzyme. Antioxid Redox Signal. 2008 doi: 10.1089/ars.2008.2194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Izumi T, Schein CH, Oezguen N, Feng Y, Braun W. Effects of backbone contacts 3' to the abasic site on the cleavage and the product binding by human apurinic/apyrimidinic endonuclease (APE1) Biochemistry. 2004;43:684–689. doi: 10.1021/bi0346190. [DOI] [PubMed] [Google Scholar]
- 16.Brewer GJ, Torricelli JR, Evege EK, Price PJ. Optimized survival of hippocampal neurons in B27-supplemented Neurobasal, a new serum-free medium combination. J Neurosci Res. 1993;35:567–576. doi: 10.1002/jnr.490350513. [DOI] [PubMed] [Google Scholar]
- 17.Fishel ML, He Y, Smith ML, Kelley MR. Manipulation of base excision repair to sensitize ovarian cancer cells to alkylating agent temozolomide. Clin Cancer Res. 2007;13:260–267. doi: 10.1158/1078-0432.CCR-06-1920. [DOI] [PubMed] [Google Scholar]
- 18.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 19.Adem A, Mattsson ME, Nordberg A, Pahlman S. Muscarinic receptors in human SH-SY5Y neuroblastoma cell line: regulation by phorbol ester and retinoic acid-induced differentiation. Brain Res. 1987;430:235–242. doi: 10.1016/0165-3806(87)90156-8. [DOI] [PubMed] [Google Scholar]
- 20.Pahlman S, Ruusala AI, Abrahamsson L, Mattsson ME, Esscher T. Retinoic acid-induced differentiation of cultured human neuroblastoma cells: a comparison with phorbolester-induced differentiation. Cell Differ. 1984;14:135–144. doi: 10.1016/0045-6039(84)90038-1. [DOI] [PubMed] [Google Scholar]
- 21.Karns LR, Ng SC, Freeman JA, Fishman MC. Cloning of complementary DNA for GAP-43, a neuronal growth-related protein. Science. 1987;236:597–600. doi: 10.1126/science.2437653. [DOI] [PubMed] [Google Scholar]
- 22.Bapat A, Fishel M, Kelley MR. Going Ape as an Approach to Cancer Therapeutics. Antioxid Redox Signal. 2008 doi: 10.1089/ars.2008.2218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liu L, Taverna P, Whitacre CM, Chatterjee S, Gerson SL. Pharmacologic disruption of base excision repair sensitizes mismatch repair-deficient and -proficient colon cancer cells to methylating agents. Clin Cancer Res. 1999;5:2908–2917. [PubMed] [Google Scholar]
- 24.Fortini P, Calcagnile A, Vrieling H, van Zeeland AA, Bignami M, Dogliotti E. Mutagenic processing of ethylation damage in mammalian cells: the use of methoxyamine to study apurinic/apyrimidinic site-induced mutagenesis. Cancer Res. 1993;53:1149–1155. [PubMed] [Google Scholar]
- 25.Shi MM, Godleski JJ, Paulauskis JD. Regulation of macrophage inflammatory protein-1alpha mRNA by oxidative stress. J Biol Chem. 1996;271:5878–5883. doi: 10.1074/jbc.271.10.5878. [DOI] [PubMed] [Google Scholar]
- 26.Comporti M. Three models of free radical-induced cell injury. Chem Biol Interact. 1989;72:1–56. doi: 10.1016/0009-2797(89)90016-1. [DOI] [PubMed] [Google Scholar]
- 27.Kooij A. A re-evaluation of the tissue distribution and physiology of xanthine oxidoreductase. Histochem J. 1994;26:889–915. [PubMed] [Google Scholar]
- 28.Colpaert EE, Lefebvre RA. Interaction of hypoxanthine/xanthine oxidase with nitrergic relaxation in the porcine gastric fundus. Br J Pharmacol. 2000;130:359–366. doi: 10.1038/sj.bjp.0703317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kulkarni A, McNeill DR, Gleichmann M, Mattson MP, Wilson DM., 3rd XRCC1 protects against the lethality of induced oxidative DNA damage in nondividing neural cells. Nucleic Acids Res. 2008;36:5111–5121. doi: 10.1093/nar/gkn480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fishel ML, Kelley MR. The DNA base excision repair protein Ape1/Ref-1 as a therapeutic and chemopreventive target. Mol Aspects Med. 2007;28:375–395. doi: 10.1016/j.mam.2007.04.005. [DOI] [PubMed] [Google Scholar]
- 31.Kimura T, Sakaida I, Terai S, Matsumura Y, Uchida K, Okita K. Inhibition of tumor necrosis factor-alpha production retards liver regeneration after partial hepatectomy in rats. Biochem Biophys Res Commun. 1997;231:557–560. doi: 10.1006/bbrc.1997.6135. [DOI] [PubMed] [Google Scholar]
- 32.Nagakawa J, Hishinuma I, Miyamoto K, Hirota K, Abe S, Yamanaka T, Katayama K, Yamatsu I. Protective effects of (2E)-3-[5-(2,3-dimethoxy-6-methyl-1,4- benzoquinoyl)]-2-nonyl-2-propenoic acid on endotoxin-mediated hepatitis in mice. J Pharmacol Exp Ther. 1992;262:145–150. [PubMed] [Google Scholar]
- 33.O'Hara AM, Bhattacharyya A, Bai J, Mifflin RC, Ernst PB, Mitra S, Crowe SE. Tumor necrosis factor (TNF)-alpha-induced IL-8 expression in gastric epithelial cells: role of reactive oxygen species and AP endonuclease-1/redox factor (Ref)-1. Cytokine. 2009;46:359–369. doi: 10.1016/j.cyto.2009.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tangpong J, Cole MP, Sultana R, Joshi G, Estus S, Vore M, St Clair W, Ratanachaiyavong S, St Clair DK, Butterfield DA. Adriamycin-induced, TNF-alpha-mediated central nervous system toxicity. Neurobiol Dis. 2006;23:127–139. doi: 10.1016/j.nbd.2006.02.013. [DOI] [PubMed] [Google Scholar]
- 35.LoPresti P, Poluha W, Poluha DK, Drinkwater E, Ross AH. Neuronal differentiation triggered by blocking cell proliferation. Cell Growth Differ. 1992;3:627–635. [PubMed] [Google Scholar]
- 36.Mitra S, Izumi T, Boldogh I, Bhakat KK, Chattopadhyay R, Szczesny B. Intracellular trafficking and regulation of mammalian AP-endonuclease 1 (APE1), an essential DNA repair protein. DNA Repair (Amst) 2007;6:461–469. doi: 10.1016/j.dnarep.2006.10.010. [DOI] [PubMed] [Google Scholar]
- 37.Vascotto C, Cesaratto L, Zeef LA, Deganuto M, D'Ambrosio C, Scaloni A, Romanello M, Damante G, Taglialatela G, Delneri D, Kelley MR, Mitra S, Quadrifoglio F, Tell G. Genome-wide analysis and proteomic studies reveal APE1/Ref-1 multifunctional role in mammalian cells. Proteomics. 2009;9:1058–1074. doi: 10.1002/pmic.200800638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fishel ML, He Y, Reed AM, Chin-Sinex H, Hutchins GD, Mendonca MS, Kelley MR. Knockdown of the DNA repair and redox signaling protein Ape1/Ref-1 blocks ovarian cancer cell and tumor growth. DNA Repair (Amst) 2008;7:177–186. doi: 10.1016/j.dnarep.2007.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zou GM, Luo MH, Reed A, Kelley MR, Yoder MC. Ape1 regulates hematopoietic differentiation of embryonic stem cells through its redox functional domain. Blood. 2007;109:1917–1922. doi: 10.1182/blood-2006-08-044172. [DOI] [PubMed] [Google Scholar]
- 40.Laposa RR, Cleaver JE. DNA repair on the brain. Proc Natl Acad Sci U S A. 2001;98:12860–12862. doi: 10.1073/pnas.241519498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Friedberg EC, Meira LB. Database of mouse strains carrying targeted mutations in genes affecting biological responses to DNA damage. Version 5. DNA Repair (Amst) 2003;2:501–530. doi: 10.1016/s1568-7864(03)00005-3. [DOI] [PubMed] [Google Scholar]
- 42.Kelley MR, Fishel ML. DNA repair proteins as molecular targets for cancer therapeutics. Anticancer Agents Med Chem. 2008;8:417–425. doi: 10.2174/187152008784220294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pines A, Perrone L, Bivi N, Romanello M, Damante G, Gulisano M, Kelley MR, Quadrifoglio F, Tell G. Activation of APE1/Ref-1 is dependent on reactive oxygen species generated after purinergic receptor stimulation by ATP. Nucleic Acids Res. 2005;33:4379–4394. doi: 10.1093/nar/gki751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Evans AR, Limp-Foster M, Kelley MR. Going APE over ref-1. Mutat Res. 2000;461:83–108. doi: 10.1016/s0921-8777(00)00046-x. [DOI] [PubMed] [Google Scholar]
- 45.Harrison JF, Rinne ML, Kelley MR, Druzhyna NM, Wilson GL, Ledoux SP. Altering DNA base excision repair: use of nuclear and mitochondrial-targeted N-methylpurine DNA glycosylase to sensitize astroglia to chemotherapeutic agents. Glia. 2007;55:1416–1425. doi: 10.1002/glia.20556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tell G, Damante G, Caldwell D, Kelley MR. The intracellular localization of APE1/Ref-1: more than a passive phenomenon? Antioxid Redox Signal. 2005;7:367–384. doi: 10.1089/ars.2005.7.367. [DOI] [PubMed] [Google Scholar]
- 47.Park WS, Ko EA, Jung ID, Son YK, Kim HK, Kim N, Park SY, Hong KW, Park YM, Choi TH, Han J. APE1/Ref-1 promotes the effect of angiotensin II on Ca2+ -activated K+ channel in human endothelial cells via suppression of NADPH oxidase. Arch Pharm Res. 2008;31:1291–1301. doi: 10.1007/s12272-001-2109-y. [DOI] [PubMed] [Google Scholar]
- 48.Sossou M, Flohr-Beckhaus C, Schulz I, Daboussi F, Epe B, Radicella JP. APE1 overexpression in XRCC1-deficient cells complements the defective repair of oxidative single strand breaks but increases genomic instability. Nucleic Acids Res. 2005;33:298–306. doi: 10.1093/nar/gki173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yan L, Bulgar A, Miao Y, Mahajan V, Donze JR, Gerson SL, Liu L. Combined treatment with temozolomide and methoxyamine: blocking apurininc/pyrimidinic site repair coupled with targeting topoisomerase IIalpha. Clin Cancer Res. 2007;13:1532–1539. doi: 10.1158/1078-0432.CCR-06-1595. [DOI] [PubMed] [Google Scholar]
- 50.Liu L, Gerson SL. Therapeutic impact of methoxyamine: blocking repair of abasic sites in the base excision repair pathway. Curr Opin Investig Drugs. 2004;5:623–627. [PubMed] [Google Scholar]
- 51.Liu L, Yan L, Donze JR, Gerson SL. Blockage of abasic site repair enhances antitumor efficacy of 1,3-bis-(2-chloroethyl)-1-nitrosourea in colon tumor xenografts. Mol Cancer Ther. 2003;2:1061–1066. [PubMed] [Google Scholar]
- 52.Shi M, Gozal E, Choy HA, Forman HJ. Extracellular glutathione and gamma-glutamyl transpeptidase prevent H2O2-induced injury by 2,3-dimethoxy-1,4-naphthoquinone. Free Radic Biol Med. 1993;15:57–67. doi: 10.1016/0891-5849(93)90125-e. [DOI] [PubMed] [Google Scholar]
- 53.Chang M, Shi M, Forman HJ. Exogenous glutathione protects endothelial cells from menadione toxicity. Am J Physiol. 1992;262:L637–L643. doi: 10.1152/ajplung.1992.262.5.L637. [DOI] [PubMed] [Google Scholar]
- 54.Nagakawa J, Hirota K, Hishinuma I, Miyamoto K, Sonoda J, Yamanaka T, Katayama K, Yamatsu I. Protective effect of E3330, a novel quinone derivative, in galactosamine-induced hepatitis in rats. J Pharmacol Exp Ther. 1993;264:496–500. [PubMed] [Google Scholar]
- 55.Huang Y, Erdmann N, Peng H, Zhao Y, Zheng J. The role of TNF related apoptosis-inducing ligand in neurodegenerative diseases. Cell Mol Immunol. 2005;2:113–122. [PubMed] [Google Scholar]
- 56.Hiramoto M, Shimizu N, Sugimoto K, Tang J, Kawakami Y, Ito M, Aizawa S, Tanaka H, Makino I, Handa H. Nuclear targeted suppression of NF-kappa B activity by the novel quinine derivative E3330. J Immunol. 1998;160:810–819. [PubMed] [Google Scholar]