

Abstract
Previous experimental and theoretical work has established that electronic excitation of a guanine cation radical in nucleosides or in DNA itself leads to sugar radical formation by deprotonation from the dexoxyribose sugar. In this work we investigate a ground electronic state pathway for such sugar radical formation in a hydrated one electron oxidized 2′-deoxyguanosine (dG•+ + 7H2O), using density functional theory (DFT) with the B3LYP functional and the 6-31G* basis set. We follow the stretching of the C5′-H bond in dG•+ to gain an understanding of the energy requirements to transfer the hole from the base to sugar ring and then to deprotonate to proton acceptor sites in solution and on the guanine ring. The geometries of reactant (dG•+ + 7H2O), transition state (TS) for deprotonation of C5′ site and product (dG(•C5′, N7-H+) + 7 H2O) were fully optimized. The zero point energy (ZPE) corrected activation energy (TS) for the proton transfer (PT) from C5′ is calculated to be 9.0 kcal/mol and is achieved by stretching the C5′-H bond by 0.13 Å from its equilibrium bond distance (1.099 Å). Remarkably, this small bond stretch is sufficient to transfer the “hole” (positive charge and spin) from guanine to the C5′ site on the deoxyribose group. Beyond the TS, the proton (H+) spontaneously adds to water to form a hydronium ion (H3O+) as an intermediate. The proton subsequently transfers to the N7 site of the guanine (product). The 9 kcal/mol barrier suggests slow thermal conversion of the cation radical to the sugar radical but also suggests that localized vibrational excitations would be sufficient to induce rapid sugar radical formation in DNA base cation radicals.
Keywords: Proton coupled hole transfer (PCHT), Proton coupled electron transfer (PCET), Sugar radical, deoxyguanosine radical cation, oxidation of deoxyguanosine, DFT study
Introduction
Interaction of radiation with DNA results in ionization and leads to the formation of a variety of radicals in DNA.1–7 The specific mechanisms of formation of these intermediates have been of considerable interest. For example, in a recent study,6 it was reported that irradiation of DNA by a high-energy Argon ion-beam (high linear energy transfer (LET) radiation) produced a far greater yield of sugar radicals than was found by -irradiation (a low LET radiation). Since these sugar radicals were formed predominantly along the ion track, where excitations and ionizations are in proximity, it was proposed that excited-state cation radicals could be the direct precursors of the neutral sugar radicals.6,7 A test of this hypothesis was performed in a number of studies,8,9 in which the formation of sugar radicals was observed upon visible photoexcitation of radical cations of guanine (G•+) and adenine (A•+) in model systems of deoxyribonucleotides, ribonucleotides as well as DNA and RNA oligos. Direct conversion of G•+ and A•+ to deoxyribose sugar radicals was found in each case with high yields (50% in DNA and 80–100% in model systems) confirming the excitation hypothesis.8,9a,b Photoexcitation of one electron oxidized deoxyguanosine (dG•+) produces C1′•, C3′• and C5′• sugar radicals9a,b whereas one electron oxidized adenine in 2′-deoxynucleosides resulted in near complete conversion to sugar radicals, predominantly C5′• with a small contribution of C3′•.9b,f The underlying mechanism of sugar radical formation proposed by us was that photoexcitation induces hole transfer from one electron oxidized DNA base radical to the sugar ring and this is followed by rapid deprotonation (-H+) at specific carbon sites on the sugar ring.7, 8 Note that hole transfer from base to sugar corresponds to an electron transfer from sugar to the one electron oxidized base.
Theoretical treatments of the excited states of one electron oxidized deoxyribonucleosides and several dinucleosides, using time-dependent density functional theory (TD-DFT) calculations,9 also supported the proposed hypothesis. It was reported that excited states which were formed by light exposure in the near-UV-visible range all originate from filled inner shell (core) molecular orbitals (MOs) and involve hole transfer from the base to the sugar ring. This supports the first step in the proposed mechanism, i.e., excitation induced transfer of the hole from base to sugar. It is the subsequent deprotonation from specific carbon sites of the one electron oxidized sugar ring (see Scheme 1) that prevents the back transfer of the hole to the base via de-excitation. The overall sugar radical formation process is clearly a proton coupled hole transfer (PCHT) (or proton coupled electron transfer (PCET)). Proton coupled electron transfer (PCET) reactions in DNA are quite important processes that play crucial role in controlling the hole and electron transfer in DNA.10–12 The intrabase proton transfer between one electron oxidized base pairs in DNA can either slow down or stop the electron and hole transfer in DNA.13 – 15 The seminal work of Steenken16 showed that in comparison to their neutral state, one electron ionized bases become quite acidic (low pKa) and deprotonate quickly while one electron adducts to DNA bases become basic (high pKa) and protonate. Shafirovich et al.17 showed the solvent kinetic isotope effects on the oxidation of guanine in 2′-deoxyguanosine 5′-monophosphate (dGMP), single- and double- stranded DNA. The electron transfer rate associated with the oxidation of guanine was found to be ca. 2 times smaller in D2O than in H2O.17 This suggested that electron transfer from neutral guanine was accompanied with deprotonation of guanine radical cation. The reductive repair of DNA guanyl radical by aniline derivatives was also suggested to involve a PCET mechanism.18 In addition to experiments, theoretical studies were also done to show the involvement of PCET reactions, e.g., in DNA-acrylamide complexes,19,20 and the mechanism of repair of guanyl radical by aromatic amino acids.21
Scheme 1.

Formation of the neutral C5′• sugar radical (C5′•) through proton coupled electron transfer (PCET) mechanism in 2′-deoxyguanosine radical cation (dG•+). The proton transfers from C5′ to N7 of guanine through waters and results in electron transfer from C5′ to guanine yielding the product, (dG(•C5′, N7-H+) + 7 H2O).
In this work we employ density functional theory (DFT) to investigate sugar radical formation in a hydrated deoxyguanosine cation radical (dG•+) via a ground state PCHT mechanism. Simple extension of the C5′-H bond induces hole transfer from guanine to the sugar ring as deprotonation to water takes place. In these calculations, sufficient water molecules are included to span from C5′-site to the O6 atom of the guanine ring and allow for proton transfer shuttle between C5′-site to N7 site of guanine (see Scheme 1 and Figures 1 – 3). The present work differs from our earlier works14,15 on the deprotonation of N1-H site in guanine radical cation as the sugar ring is the focus of deprotonation. In addition this work differs from our work on the deprotonation from sugar after photoexcitation of guanine cation in DNA and nucleosides described above. In the present work, we investigate hole transfer and the deprotonation process along the ground state potential energy surface. The barrier along this surface is found to be remarkably low and deprotonation occurs with only a small extension of the bond. This work suggests an alternative pathway to sugar radical formation from the guanine cation in DNA via ground state electronic surfaces.
Figure 1.
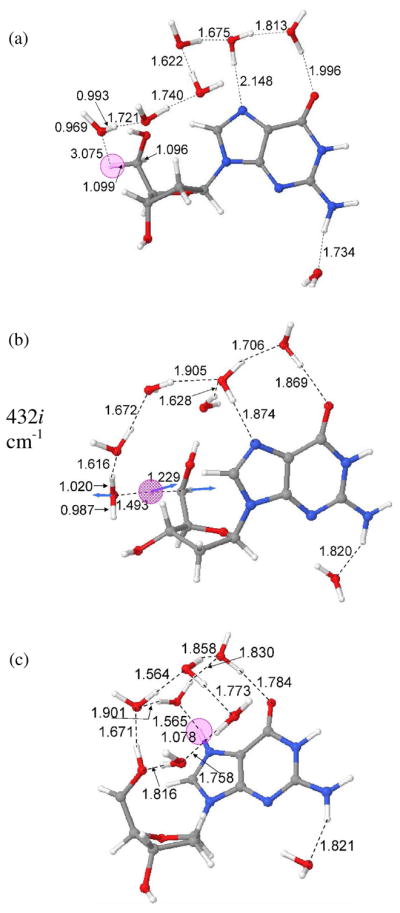
B3LYP/6-31G* optimized geometries of (a) dG•++7 H2O (reactant), (b) transition state (TS) and (c) dG(•C5′, N7-H+) + 7 H2O (product).
Figure 3.

BHandHLYP/6-31G*//B3LYP/6-31G* calculated spin density distribution during proton transfer from C5′ on the deoxyribose group to N7 site on guanine in dG•+ + 7 H2O. The stretching of the C5′-H bond from its equilibrium bond length (1.099) to 1.23 Å results in the transfer of the hole from guanine to the C5′ site which is equivalent to electron transfer from the C5′ site to guanine. The pink circle highlights the transferring proton when not obscured by the spin distribution.
Methods of calculation
The molecular geometries of guanosine radical cation (dG•+) in the presence of seven waters (dG•+ + 7H2O) (reactant), the transition state (TS) for deprotonation (-H+) from C5′-site to water and the final product after proton transfer is complete, dG(•C5′, N7-H+) + 7 H2O are shown in Figure 1. Each structure was fully optimized using the B3LYP functional and 6-31G* basis set as implemented in Gaussian 03 suite of programs.22 Further, frequency analyses, using the same method and basis set, were performed to ensure the existence of bothe reactant and product as local minimum structures and the characterization of the TS as a saddle point of 1st order. For reactant (dG•+ + 7H2O) and product (dG(•C5′, N7-H+) + 7 H2O) positive frequencies were obtained while for the TS a negative frequency was obtained. The molecular structures were drawn using the JMOL molecular modeling program.23 Figures 1 – 3 are presented in the text while Figures S1 – S14 are presented in the supporting information.
The placement and choice of the number of waters was based in part on the previous work of Schneider et al.24 These investigators studied the water binding sites around the DNA bases in different DNA conformations, in their study they calculated the hydration densities (defining the location of water) around the bases. In guanine (B-DNA conformation) three water binding sites near N7, O6 and N3 were found.24 Based on these observations the initial structure was generated as follows: (a) We placed three water molecules near N7, O6 and N3 atoms of the guanine in appropriate hydrogen binding arrangements. (b) In addition to these three water molecules, we placed four more water molecules with appropriate hydrogen bonding arrangements spanning from C5′ towards the N7 site of the guanine ring. The initial structure of dG•+ + 7H2O thus generated was used for full geometry optimization. This partial solvation reduces the CPU time as well as makes the calculations feasible. This structure also provides sufficient waters to provide a minimal solvation for the hydronium ion formed as an intermediate.
Results and Discussion
The atom numbering scheme and the PCHT/PCET mechanism for the formation of the neutral C5′ sugar radical (C5′•) by deprotonation from the C5′ site of dG•+ is shown in Scheme 1. In this scheme, the hole is localized on the guanine base while the sugar moiety remains neutral. Deprotonation from the C5′ site of the sugar ring to water occurs in a concerted fashion as the hole transfers from guanine to the C5′ site of the sugar ring. This proton transfer to water leads to the formation of a neutral sugar radical (C5′•) and a hydronium ion. In Table 1, we present the ZPE-corrected changes in energy (ΔE) at 0 K (without thermal correction), enthalpy (ΔH) and free energy (ΔG) (in kcal/mol) thermodynamically corrected to 298 K, employing the B3LYP/6-31G* method, for several points along the potential energy surface shown in Figure 2 for the proton transfer reaction from the C5′ site to the N7 of guanine in dG•+ + 7 H2O. The values in the potential energy surface (PES), shown in Figure 2, are the ZPE-corrected energies (ΔE) without thermal correction.
Table 1.
The zero point energy (ZPE) corrected energies (ΔE), enthalpies (ΔH) and free energies (ΔG) in kcal/mol calculated using the B3LYP/6-31G* method for the proton transfer from C5′ to N7 site of guanine.a,b
| Systema | Without ZPE-correction | ZPE-corrected Energetics | ||||
|---|---|---|---|---|---|---|
| Total Energyc | Relative energyd,e (ΔE) | Total Energyc | (ΔE)d,e | (ΔH)b,d | (ΔG)b,d | |
| 1 (reactant) | −1498.30213 | 0.0 | −1497.87540 | 0.0 | 0.0 | 0.0 |
| 2 (TS) | −1498.28718 | 9.38 | −1497.86111 | 8.97 | 6.52 | 14.87 |
| 3 | −1498.29022 | 7.47 | −1497.86437 | 6.92 | 4.05 | 12.76 |
| 4 | −1498.29460 | 4.73 | −1497.86811 | 4.58 | 1.49 | 10.82 |
| 5 | −1498.30200 | 0.08 | −1497.87280 | 1.63 | −1.46 | 8.99 |
| 6 (product) | −1498.32544 | −14.63 | −1497.89585 | −12.83 | −14.45 | −7.47 |
For structures see Figures (1, 2 and S3 – S8 in the supporting information).
Enthalpy (ΔH) and free energy (ΔG) were calculated at 298 K while ΔE was calculated at 0 K.
Total energy in atomic units (A.U.).
Energies in kcal/mol and calculated with respect to system 1, see Figure 2.
The energy (ΔE) values presented here refer to the activation energy for the TS and the relative energy for systems 3–5. For system 6 (product), ΔE represents the thermodynamic reaction energy with respect to the reactant.
Figure 2.

B3LYP/6-31G* calculated potential energy surface (PES) for proton transfer from C5′ on the deoxyribose group to N7 on guanine in dG•+ + 7 H2O. The zero point energy corrected energies (ΔE) at 0 K and C5′-H bond distances are given in kcal/mol and angstroms (Å), respectively. The pink circle highlights the transferring proton. For atom numbering, see scheme 1.
The B3LYP/6-31G* optimized geometries for the reactant (dG•+ + 7 H2O), TS, i.e., the incipient proton transfer from C5′ to water, and the product (dG(•C5′, N7-H+) + 7 H2O), are shown in Figure 1. From the optimized geometry of dG•+ + 7 H2O (Figure 1a), it is evident that six water molecules span from the C5′ site of sugar ring to O6 atom of guanine in a serially hydrogen bonded fashion. The hydrogen bond distances between the six water molecules vary from 1.622 to 1.813 Å (see Figures S3 – S8 in the supporting information). The water molecules hydrogen bonded to N7 and O6 atoms of guanine have hydrogen bond distances 2.148 and 1.996 Å, respectively. During geometry optimization, the seventh water molecule shifted from N3 towards N2 to make a hydrogen bond with the N2-H with a hydrogen bond distance of 1.734 Å. The transition state for deprotonation of C5′ to water, shown in Figure 1b, occurs at 1.299 Å with a single imaginary frequency of 432i cm−1. The complete deprotonation of C5′ leads to the formation of N7 protonated guanine (dG(•C5′, N7-H+) + 7 H2O) as shown in Figure 1c.
To elucidate the mechanism of C5′ sugar radical formation, we scanned the potential energy surface (PES), shown in Figure 2, for the proton transfer from C5′ to N7 site of guanine through the intervening hydrogen bonded water network using B3LYP/6-31G* level of calculation. The different structures present during proton transfer from reactant (dG•+ + 7 H2O) to product (dG(•C5′, N7-H+) + 7 H2O), shown in Figure 2, are numbered from 1 – 6. The ZPE-corrected energy (ΔE), presented in Figure 2, is given in kcal/mol. The energies (ΔE) at each step of the proton transfer reaction were calculated with respect to reactant (dG•+ + 7 H2O). The TS for the deprotonation of C5′ site to a water is found by stretching the C5′-H bond from 1.099 to 1.229 Å (see structure 2 in Figure 2 and Figure 1b). The activation energy (ΔE) is found to be 9.0 kcal/mol. This transition state is characterized by an imaginary frequency 432i cm−1 (see Figures 1b and S11). The visual inspection of this vibrational frequency (432i cm−1) clearly shows the incipient formation of hydronium ion (H3O+) as an intermediate. For complete proton transfer from C5′-site (proton donor site) to proton acceptor sites (N7 and O6) of the guanine, we further scanned the PES by increasing the C5′-H bond distance from its transition state distance (1.229 Å) to 1.70 Å in the step size of 0.05 Å. The structure of dG•+ + 7 H2O was fully optimized with frequency calculation holding the C5′-H bond distance fixed at each chosen distance on the PES. Interestingly, we found that the detachment of proton from C5′-site and formation of the N7 ring protonated product (dG(N7 – H+) + 7 H2O) is an exothermic process by −12.8 kcal/mol. At C5′-H bond distance of 1.35 Å (see structure number 3 in Figure 2 and Figure S5) the ZPE-corrected relative energy at 1.35 Å is 6.9 kcal/mol. The frequency calculation shows this structure has an imaginary frequency 167i cm−1 (see Figure S12). In our calculation, we find a single negative frequency for all the optimized structures at each step of the proton transfer on the PES beyond the TS at 1.229 Å. The frequencies decrease beyond the TS from 432i cm−1 to as low as 70i cm−1 then rise to lower maximum of 167i cm−1 at 1.35 Å. Also, from the vibrational analyses we noted that the structure at 1.229 Å involves the rotation of the H-C5′-H group to align one of its hydrogen atoms with the oxygen atom of the neighboring water molecule while at 1.35 Å the structure involves a pure stretching of the C5′-H bond, see structure numbers 2 and 3 in Figure 3 and vibrational spectra (Figures S11 and S12 in the supporting information). Further stretching the C5′-H bond up to 1.50 Å, we found the initial formation of the H3O+ with ZPE-corrected relative energy of 4.6 kcal/mol, see structure number 4 in Figure 2 and Figure S6. The increase of the C5′-H bond up to 1.70 Å transfers the proton into the water network with the formation of the H3O+ which is stabilized by three neighboring water molecules having hydrogen bond distances 1.552, 1.824 and 1.430 Å, respectively, see structure number 5 in Figure 2 and Figure S7. In addition to the proton’s high mobility, the stabilization of a proton in an aqueous medium on formation of H3O+ strongly hydrogen bonded with three water molecules is a well known phenomenon.25 Thus the short hydrogen bond distances to the hydronium ion we find are expected. A further increase in the C5′-H bond beyond 1.70 Å leads to the complete transfer of the proton from C5′ through the intervening waters to the N7 atom of the guanine resulting into the formation of dG(•C5′, N7-H+) + 7 H2O complex, see Figures 1, 2 and Figure S8 and S13. These results are in accord with a report employing fast atom bombardment tandem mass spectrometry,26a suggests the N7 site of guanine is the most favorable site for the proton attachment in the gas phase with a proton affinity of 227 ± 0.1 kcal/mol. In Table 1 it is evident that conversion of dG•+ + 7 H2O (reactant) to dG(•C5′, N7-H+) + 7 H2O (product) is exergonic with a calculated ZPE-corrected free energy changes (ΔGo) at 298 K of −7.5 kcal/mol. The free energy of activation (ΔG‡) is ca. 15 kcal/mol (Table 1) which involves a significant entropy cost (ΔS‡ = −28.0 cal K−1 mol−1). Clearly the reorganization of water around the proton creates a more organized state. The solvation of the proton in water is an extremely exergonic process (ΔGo = −264 kcal/mol) but is not favored entropically (ΔS‡ = −37.0 cal K−1 mol−1).26b At the transition state a similar entropic cost is found.
Since B3LYP functional is known to overestimate delocalization of spin density owing to the self interaction error,27 we plotted the spin density distribution using the BHandHLYP functional (which has 50% Hartree-Fock exchange contribution) and the 6-31G* basis set with the optimized structures shown in Figure 2 from the B3LYP/6-31G* method. The spin densities calculated using the B3LYP/6-31G* method are shown in Figure S2 in the supporting information. The spin densities, shown in Figures 3 and S2, are plotted at 0.002 spin contour level using Molekel program28 at each step of the proton transfer reaction indicated in the PES in Figure 2. From Figure 3, we see that in dG•+ + 7 H2O (reactant), the spin is totally delocalized into the p molecular orbital of guanine, which is in agreement with the experimental observation.1 – 4, 9,14,15 At the TS, a large part of the spin density is found on the C5′ while a small portion is found on a water molecule adjacent to the C5′ site, see Figure 3. After complete proton transfer from C5′ to guanine (dG(•C5′, N7-H+) + 7 H2O (product)), the spin density is localized mainly on C5′ site as shown in Figure 3. A comparison of Figures 3 and S2 shows that both the methods give similar spin density distributions except at the TS where the B3LYP/6-31G* level of calculation shows the presence of a small amount of spin density on the guanine ring, see structure number 2 in Figures 3 and S2, respectively. The spin density distribution, shown in Figure 3, shows that a small stretch in the C5′-H bond of only ~ 0.13 Å from its equilibrium distance (1.099 Å, see Figure S3) transfers the “hole” (positive charge) from guanine to the C5′ site or in other words transfers electron from C5′ site to the guanine ring making guanine ring neutral as proposed in Scheme 1. From the present calculation, it is obvious that the first vibrational excitation of C5′-H bond would overcome the 9 kcal/mol barrier and result in the transfer of the “hole” from guanine to the C5′ site. The hole transfer creates a very acidic C5′ site which deprotonates into the aqueous environment. The B3LYP/6-31G* calculated stretching frequency of C5′-H bond is 3040 cm−1 which suggests a proton transfer time of some multiple of the ~11 fs vibrational time for a vibrationally excited species.
To show the influence of the water environment on the proton transfer process we considered a system without the water network. We employed the B3LYP/6-31G* optimized TS structure (structure number 2 in Figure 2) and then increased the C5′-H bond up to 4 Å and calculated the spin density distribution using B3LYP/6-31G* method. The calculated spin density distribution is totally localized on the detached hydrogen atom, see Figure 4. A hydrogen atom has an IP of 13.6 eV, thus the proton easily acquires an electron from the dG system leaving a singlet cation at the C5′ site. In the aqueous environment a proton transfers leaving behind the full spin at the C5′ site (see structure 5 in Figures 3 and S2). These results clearly show that in a polar solvent such as water, proton transfer takes place while in gas phase, or by extension nonpolar systems, hydrogen atom transfer could be operative. We note that in nonpolar systems, such as for one electron ionized hydrocarbons, hydrogen atom formation is commonly invoked as a mechanism in competition with deprotonation reactions.29 We further note that, since DNA has a variety of proton acceptor sites, the direct proton transfer to the other effective proton acceptors sites such as N7, O6 and N3 of guanine could occur even without water.
Figure 4.

B3LYP/6-31G* calculated spin density distribution at a C5′-H bond distance of 4 Å without waters. In the calculation, the surrounding waters were removed from the B3LYP/6-31G* optimized TS (structure number 2 in Figure 2) and the C5′-H bond distance increased to 4 Å. A single point B3LYP/6-31G* calculation was then performed. The full spin density is localized on the detached hydrogen atom and positive charge is maintained on the remaining structure mainly at C5′.
Conclusions
The spin density for the hydrated deoxyguanosine cation radical calculated in this study, dG•+ + 7H2O, is fully localized on the guanine base, as expected, since one electron oxidation of 2′-deoxyguanosine (2′-dG) is known to initially result in a hole on the guanine.1,9 However, a small stretch in the C5′-H bond of only 0.13 Å from the equilibrium value of 1.099 Å to 1.229 Å costs only 9 kcal/mol to reach the TS toward deprotonation into water. This bond stretch is sufficient to transfer the hole from guanine to C5′ site which makes the C5′ site highly acidic and induces deprotonation to water. The critical role of the proton acceptor, i.e., the waters of hydration, is made clear by Figure 4 which shows that, without a protonation site on water, hydrogen atom loss could occur instead of proton loss. The role of oxidation of the guanine base on the C5′-H bond strength is also of interest. To illustrate this, a qualitative comparison of the potential in its neutral and cation radical states are energy surfaces of C5′-H bond scission in 2′-dG presented in Figure 5. The C5′-H bond in the neutral 2′-dG has a bond strength toward homolytic cleavage of 97 kcal/mol30 which would not be fully cleaved until extension of several angstroms. Comparison of this with the 9 kcal/mole and 0.13 Å found in this work for the cation radical, dG•+ + 7H2O, shows the dramatic effect of oxidation of the guanine base on the reactivity of the sugar group.
Figure 5.

Qualitative plots of dG + 7 H2O in neutral and cation radical states as a function of C5′-H bond length. Not to scale. The neutral C5′-H bond energy is 97 kcal/mol (Ref. 30) where as the cation radical of dG + 7 H2O requires only 9 kcal/mol to overcome the barrier and is energetically downhill thereafter. Note that the cation radical is ca. 7 eV higher in energy (ionization potential, Ref. 3) than the neutral. The curves are shown together to compare the barriers to bond rupture.
The 9 kcal/mol activation energy (ΔE) for proton transfer from C5′ site to the water would suggest a significant rate of conversion of the guanine cation radical in DNA to sugar radicals at ambient temperatures, e.g., the activation energy for the proton transfer decreases by to 2 – 3 kcal/mol at 298 K (see Table 1). However, the free energy of activation (ΔG‡ =15 kcal/mol, see Table 1) is substantially greater than the activation energy (see Table 1) owing to a significant entropic penalty on forming the transition state. This entropic cost would clearly lower the rate of reaction. Experimentally it is found that on the time scale of the lifetime of G•+ in DNA no such reaction occurs and a faster reaction of addition of water directly to the C8 on guanine to form the 8-hydroxyl guanine radical occurs.31–34 Subsequently, after a second one electron oxidation 8-oxoguanine is formed.31 However, our work suggests that simple vibrational excitation of the C5′-H bond in dG•+ would provide the bond stretching needed to cause the PCHT and sugar radical formation.
The proton transfer from C5′ site to the N7 of guanine is an overall exothermic process with a free energy change (ΔGo) of −7.5 kcal/mol at 298 K. This study clearly shows that, in aqueous environment, proton coupled hole transfer (PCHT) can be an effective route to C5′ sugar radical (C5′•) formation. In the present study, we model the proton transfer reaction through water network considering the simple model of a nucleoside, however, in the actual DNA system the phosphate (PO4) groups are present and will be a favored site for proton addition. The pKa of the phosphate group and its proximity to the C5′ site would make it an effective competitor with the guanine N7 site. Experimentally it is found that the thermal reaction modeled in this work is slow and electronic excitation was necessary to induce deprotonation from the sugar of the guanine cation radical in DNA or model systems.8,9 The low barrier found in this work suggests vibrational excitation would be sufficient to induce deprotonation as well. The apparent stability of the guanine cation in DNA systems toward the sugar radical formation is explained by the activation barrier, and the more rapid reaction of the guanine base cation with water.
Supplementary Material
Acknowledgments
This Work was supported by the NIH NCI Grant No. R01CA045424. The authors thank the Arctic Region Supercomputing Center (ARSC) for a generous grant of CPU time and facilities. These computational studies were also supported by a computational facilities grant, NSF CHE-072689.
Footnotes
Supporting Information Available: Scheme showing the mechanism of sugar radical formation, figures showing potential energy surface (PES), optimized geometries, spin density distributions and vibrational spectra. Video clips showing the proton transfer process (PT.avi), rotational and stretching nature of the transition states (TS1.avi and TS2.avi) are provided as media files. This material is available free of charge via the internet at http://pubs.acs.org.
References
- 1.(a) von Sonntag C. The Chemical Basis of Radiation Biology. Taylor & Francis; New York: 1987. [Google Scholar]; (b) Pogozelski WK, Tullius TD. Chem Rev. 1998;98:1089. doi: 10.1021/cr960437i. [DOI] [PubMed] [Google Scholar]
- 2.(a) Sevilla MD, Becker D. Electron Paramagn Reson. 2004;19:243. [Google Scholar]; (b) Swarts SG, Sevilla MD, Becker D, Tokar CJ, Wheeler KT. Radiat Res. 1992;129:333. [PubMed] [Google Scholar]; (c) Sevilla MD, Becker D, Yan M, Summerfield SR. J Phys Chem. 1991;95:3409. [Google Scholar]; (d) Yan M, Becker D, Summerfield SR, Renke P, Sevilla MD. J Phys Chem. 1992;96:1983. [Google Scholar]; (e) Wang W, Sevilla MD. Radiat Res. 1994;9:17. [PubMed] [Google Scholar]
- 3.Kumar A, Sevilla MD. In: Radiation Induced Molecular Phenomena in Nucleic Acid: A Comprehensie Theoretical and Experimental Analysis. Shukla MK, Leszczynski J, editors. Springer-Verlag; Berlin, Heidelberg, New York: 2008. p. 577. [Google Scholar]
- 4.Becker D, Adhikary A, Sevilla MD. In: The Role of Charge and Spin Migration in DNA Radiation Damage In Charge Migration in DNA Physics, Chemistry and Biology Perspectives. Chakraborty T, editor. Springer-Verlag; Berlin Heidelberg: 2007. p. 139. [Google Scholar]
- 5.(a) Spaletta RA, Bernhard WA. Radiat Res. 1993:143. [PubMed] [Google Scholar]; (b) Razskazovskiy Y, Debije MG, William A, Bernhard WA. Radiat Res. 2003;159:663. doi: 10.1667/0033-7587(2003)159[0663:sbpixc]2.0.co;2. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Steenken S, Goldbergerova L. J Am Chem Soc. 1998;120:3928. [Google Scholar]
- 6.Becker D, Bryant-Friedrich A, Trzasko C, Sevilla MD. Radiat Res. 2003;160:174. doi: 10.1667/rr3037. [DOI] [PubMed] [Google Scholar]
- 7.Becker D, Razskazovskii Y, Callaghan MU, Sevilla MD. Radiat Res. 1996;146:361. [PubMed] [Google Scholar]
- 8.(a) Shukla LI, Pazdro R, Huang J, DeVreugd C, Becker D, Sevilla MD. Radiat Res. 2004;161:582. doi: 10.1667/rr3167. [DOI] [PubMed] [Google Scholar]; (b) Shukla LI, Pazdro R, Becker D, Sevilla MD. Radiat Res. 2005;163:591. doi: 10.1667/rr3347. [DOI] [PubMed] [Google Scholar]
- 9.(a) Adhikary A, Malkhasian AYS, Collins S, Koppen J, Becker D, Sevilla MD. Nucleic Acids Res. 2005;33:5553. doi: 10.1093/nar/gki857. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Adhikary A, Collins S, Koppen J, Becker D, Sevilla MD. Nucleic Acid Res. 2006;34:1501. doi: 10.1093/nar/gkl026. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Adhikary A, Kumar A, Sevilla MD. Radiat Res. 2006;165:479. doi: 10.1667/rr3563.1. [DOI] [PubMed] [Google Scholar]; (d) Adhikary A, Collins S, Khanduri D, Sevilla MD. J Phys Chem B. 2007;111:7415. doi: 10.1021/jp071107c. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Khanduri D, Collins S, Kumar A, Adhikary A, Sevilla MD. J Phys Chem B. 2008;112:2168. doi: 10.1021/jp077429y. [DOI] [PubMed] [Google Scholar]; (f) Adhikary A, Khanduri D, Kumar A, Sevilla MD. J Phys Chem B. 2008;112:15844. doi: 10.1021/jp808139e. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Kumar A, Sevilla MD. J Phys Chem B. 2006;110:24181. doi: 10.1021/jp064524i. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Stubbe J, Nocera DG, Yee CS, Chang MCY. Chem Rev. 2003;103:2167. doi: 10.1021/cr020421u. [DOI] [PubMed] [Google Scholar]
- 11.Costentin C. Chem Rev. 2008;108:2145. doi: 10.1021/cr068065t. [DOI] [PubMed] [Google Scholar]
- 12.Boussicault F, Robert M. Chem Rev. 2008;108:2622. doi: 10.1021/cr0680787. [DOI] [PubMed] [Google Scholar]
- 13.Kawai K, Osakada Y, Majima T. Chem Phys Chem. 2009 doi: 10.1002/cphc.200900148. [DOI] [PubMed] [Google Scholar]
- 14.Adhikary A, Khanduri D, Sevilla MD. J Am Chem Soc. 2009;131:8614. doi: 10.1021/ja9014869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.(a) Kumar A, Sevilla MD. J Phys Chem B. 2009 doi: 10.1021/jp903403d. [DOI] [Google Scholar]; (b) Adhikary A, Kumar A, Becker D, Sevilla MD. J Phys Chem B. 2006;110:24171. doi: 10.1021/jp064361y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Steenken S. Chem Rev. 1989;89:503. [Google Scholar]
- 17.(a) Kuzmin VA, Dourandin A, Shafirovich V, Geacintov NE. Phys Chem Chem Phys. 2000;2:1531. [Google Scholar]; (b) Shafirovich V, Dourandin A, Luneva NP, Geacintov NE. J Phys Chem B. 2000;104:137. [Google Scholar]; (c) Shafirovich V, Dourandin A, Luneva NP, Geacintov NE. J Phys Chem B. 2001;105:8431. [Google Scholar]
- 18.Ly A, Tran NQ, Sullivan K, Bandong SL, Milligan JR. Org Biomol Chem. 2005;3:917. doi: 10.1039/b418681h. [DOI] [PubMed] [Google Scholar]
- 19.Taylor J, Eliezer I, Sevilla MD. J Phys Chem B. 2001;105:1614. [Google Scholar]
- 20.Carra C, Iordanova N, Hammes-Schiffer S. J Phys Chem B. 2002;106:8415. [Google Scholar]
- 21.Jena NR, Mishra PC. J Phys Chem B. 2009 doi: 10.1021/jp810468m. [DOI] [Google Scholar]
- 22.Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheesemean JR, Montogomery JJA, Vreven T, Kudin KN, Burant JC, Millam JM, Iyengar SS, Tomasi J, Barone V, Mennucci B, Cossi M, Scalmani G, Rega N, Petersson GA, Nakatsuji H, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Klene M, Li X, Knox JE, Hratchian HP, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochtersk JW, Ayala PY, Morokuma K, Voth GA, Salvador P, Dannenberg JJ, Zakrzewski VG, Dapprich S, Daniels AD, Strain MC, Farkas O, Malick DK, Rabuck AD, Raghavachari K, Foresman JB, Oritz JV, Cui Q, Baboul AG, Clifford S, Cioslowski J, Stefanov BB, Liu G, Liashenko A, Piskorz P, Komaromi I, Martin RL, Fox DJ, Keith T, Al-Laham MA, Peng CY, Nanayakkara A, Challacombe M, Gill PMW, Johnson B, Chen W, Wong MW, Gonzalez C, Pople JA. GAUSSIAN 03 (Rev. C.02) Gaussian Inc; Wallingford, CT: 2004. [Google Scholar]
- 23.http://jmol.sourceforge.net, Jmol development team, An Open-Science Project © 2004
- 24.Schneider B, Cohen DM, Schleifer L, Srinivasan AR, Olson WK, Berman HM. Biophys J. 1993;65:2291. doi: 10.1016/S0006-3495(93)81306-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Marx D, Tuckerman ME, Hutter J, Parrinello M. Nature. 1999;397:601. [Google Scholar]
- 26.(a) Greco F, Liguori A, Sindona G, Uccella N. J Am Chem Soc. 1990;11(2):9092. [Google Scholar]; (b) Tissandier MD, Cowen KA, Feng WY, Gundlach E, Cohen MH, Earhart AD, Coe JV, Tuttle TR. J Phys Chem A. 1998;102:7787. [Google Scholar]
- 27.Mantz YA, Gervasio FL, Laino T, Parrinello M. J Phys Chem A. 2007;111:105. doi: 10.1021/jp063080n. [DOI] [PubMed] [Google Scholar]
- 28.Molekel 43. www.cscs.ch/molekel/
- 29.(a) Sakurai H, Shiotani M, Ichikawa T. Radiat Phys Chem. 1999;54:235. [Google Scholar]; (b) Nielsen ML, Budnik BA, Haselmann KF, Olsen JV, Zubarev RA. Chem Phys Lett. 2000;330:558. [Google Scholar]
- 30.Li MJ, Liu L, Wei K, Fu Y, Guo QX. J Phys Chem B. 2006;110:13582. doi: 10.1021/jp060331j. [DOI] [PubMed] [Google Scholar]
- 31.Angelov D, Spassky A, Berger M, Cadet J. J Am Chem Soc. 1997;119:11373. [Google Scholar]
- 32.Reynisson J, Steenken S. Phys Chem Chem Phys. 2001;4:527. [Google Scholar]
- 33.Gervasio FL, Laio A, Iannuzzi M, Parrinello M. Chem Eur J. 2004;10:4846. doi: 10.1002/chem.200400171. [DOI] [PubMed] [Google Scholar]
- 34.Shukla LI, Adhikary A, Pazdro R, Becker D, Sevilla MD. Nucleic Acids Res. 2004:6565. doi: 10.1093/nar/gkh989. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.