

Abstract
Expression of B7-family costimulatory molecules CD80 (B7–1) and CD86 (B7–2) on tumor cells enhances host immunity. However, the role of the two B7 receptors, CD28 and CTLA4 (CD152), on T cells in antitumor immune response has not been clearly elucidated. Based on the effects of anti-CD28 and anti-CTLA4 mAbs on T cell response, it was proposed that CD28-B7 interaction promotes antitumor immunity, whereas B7-CTLA4 interaction down-regulates it. A critical test for the hypothesis is whether selective engagement of CTLA4 receptors by their natural ligands CD80 and CD86 enhances or reduces antitumor immunity. Here we used tumors expressing wild-type and mutant CD80, as well as mice with targeted mutation of CD28, to address this issue. We report that in syngeneic wild-type mice, B7W (W88>A), a CD80 mutant that has lost binding to CD28 but retained binding to CTLA4, can enhance the induction of antitumor cytotoxic T lymphocytes (CTL); B7Y (Y201>A), which binds neither CD28 nor CTLA4, fails to do so. Consistent with these observations, B7W-transfected J558 plasmocytoma and EL4 thymoma grow significantly more slowly than those transfected with either vector alone or with B7Y. Optimal tumor rejection requires wild-type CD80. Moreover, expression of a high level of CD80 on thymoma EL4 cells conveys immunity in mice with a targeted mutation of CD28 gene. Taken together, our results demonstrate that B7-CTLA4 interaction enhances production of antitumor CTL and resistance to tumor challenge and that optimal enhancement of antitumor immunity by CD80 requires its engagement of both CD28 and CTLA4.
The importance of costimulatory molecules CD80 and CD86 in the induction of optimal immune responses, including antitumor immunity, is well established (1–5). CD80 and CD86 interact with both CD28 and CTLA4, which are produced in different kinetics and locate at different compartments in T cells (6–11). The CD28 is expressed constitutively at a significant level on the T cell surface, and its expression is enhanced after T cell activation (12, 13). In contrast, CTLA4 is expressed at low levels in resting T cells and resides primarily within the cytoplasm (13–15). Activation of T cells leads to both increased CTLA4 gene expression and trafficking of CTLA4 protein to the cell surface (13–15). Although it is generally agreed that engagement of CD28 enhances T cell activation, the function of CTLA4 is still unclear. Two hypotheses have been proposed. First, optimal costimulatory activity of B7 requires its interaction with both CD28 and CTLA4 (13, 16, 17). Alternatively, it was suggested that CD28 and CTLA4 play opposite roles in T cell activation; CD28-B7 interaction promotes T cell activation, whereas B7-CTLA4 inhibits it (14, 18).
Several groups used anti-CTLA4 mAbs to probe the function of CTLA4 engagement during immune responses (13, 14, 18–23). Although anti-CTLA4 mAbs do not costimulate T cell activation in vitro, they enhance T cell activation in the presence of anti-CD28 (13, 14). These results initially were interpreted as evidence that CD28 and CTLA4 act synergistically in inducing T cell activation (13). It was later noted, however, that both intact and Fab fragments of anti-CTLA4 mAbs enhance T cell response in a number of in vivo and in vitro models, whereas cross-linked anti-CTLA4 mAbs appear to inhibit T cell activation (14, 18–23). In other models, anti-CTLA4 mAb inhibits T cell proliferation (18, 19). If one assumes that Fab fragment of the anti-CTLA4 mAb is not an agonist, it is possible that anti-CTLA4 mAbs enhance T cell activation by blocking a negative signal from CTLA4 engagement. Subsequently, it was reported that mice with a targeted mutation of CTLA4 develop fatal lymphoproliferative disease (24, 25).
The sources for most of the controversies are how one interprets the effects of anti-CTLA4 mAbs and whether the fatal lymphoproliferative diseases in CTLA4-deficient mice are due to alteration in the repertoire of T cells selected in the absence of CTLA4, or to lack of an inhibitory signal during the activation of T cells. Both issues remain to be addressed experimentally. To avoid some of these caveats, we recently have taken a genetic approach, namely, utilization of either a mutant B7 molecule that binds CTLA4 but not CD28, or T cells lacking CD28 because of a targeted mutation of the gene to address the biological consequences of B7-CTLA4 interaction (16). Here we report that expression of mutant CD80 that binds CTLA4 but not CD28 on thymoma and plasmocytoma enhances their immunogenicity while reducing their tumorigenicity. In contrast, expression of mutant CD80 that binds neither CD28 nor CTLA4 on the same tumors fails to do so. Moreover, wild-type CD80 reduces tumorigenicity of thymoma in CD28-deficient mice. These results demonstrated that B7-CTLA4 interaction enhances production of antitumor CTL and increases resistance to tumor challenge. Thus, B7-CTLA4 interaction enhances, rather than inhibits, T cell responses in vivo.
MATERIALS AND METHODS
Experimental Animals and Tumor Cell Lines.
Male BALB/c, C57BL/6j mice were purchased from the National Cancer Institute (Rockville, MD). Mice with a targeted mutation of CD28 (CD28-KO) (26) were kindly provided by Tak Mak (University of Toronto, Canada). The CD28-KO mice were backcrossed to C57BL/6j mice for 8 generations. CD28(+/−) littermates were used as controls.
Two tumor lines were used: J558 is a plasmocytoma derived from BALB/c (H-2d) mice, and EL4 is a thymoma derived from C57BL/6j (H-2b) mice. Both tumor lines were obtained from American Type Culture Collection.
Transfection of Tumor Cell Lines with Either Wild-Type or Mutant CD80 Molecules.
J558-B7 and EL4 cells were transfected with either wild-type or mutant murine CD80 by electroporation, as described (4). Two CD80 mutants, generated by site-directed mutagenesis, were used: B7W has a mutation at position 88 from W>A, which leads to selective loss of binding to CD28Ig but not CTLA4Ig (16), and B7Y carries a mutation at position 201 from Y>A, which leads to loss of binding to both CD28Ig and CTLA4Ig (27). J558 clones surviving G418 selection (0.4 mg/ml) were screened for cell surface CD80 expression by flow cytometry, and the clone with the highest cell surface CD80 was selected from each group for the study. EL4 tumors were electroporated by the same method. After selection with G418 (0.6 mg/ml), CD80+ tumor cells were sorted using a fluorescence-activated cell sorter.
Cell Adhesion Assay.
The assay process was adopted from Linsley et al. (28). Briefly, varying numbers of CHO cells transfected with either CD28 or CD80 genes were cultured in a 96-well plate for 2 h to allow firm adhesion. After removing the medium, the CHO cell monolayers were fixed with 1% paraformaldehyde at room temperature for 20 min. After three washings with tissue culture medium containing 2.5% fetal calf serum and 2.5 mM EDTA, 51Cr-labeled tumor cells (105/well) were added to the monolayers of fixed cells and incubated at 37°C for 2 h. Unbound cells were washed away by four gentle washes, and labeled cells adhering to the CHO cell monolayers were lysed by 1% Triton X-100. The amount of radioactivity in the lysates was quantified by an LKB beta plate counter. The percentage of cells bound was calculated based on the amount of radioactivity retained by the CHO cell monolayer as a percentage of radioactivity input.
Monoclonal Antibodies, Fusion Proteins, and Flow Cytometry.
mAbs and fusion proteins used for the study are as follows: mAbs 3A12 (ref. 8) and 10.16A.1 (ref. 29) are specific for murine CD80 and are used for cell sorting and blocking studies; 4G6 is specific for PC.1 and is used to deplete J558 tumor cells (30); anti-CD28 mAbs were used to sort CD28-transfected CHO cells. Fusion protein CTLA4Ig was prepared as described (27) and used to determine the receptor specificity of wild-type and mutant CD80 molecules. The data presented are mean fluorescences (MF) of CTLA4Ig binding, after normalization of CD80 expression, according to the following formula:
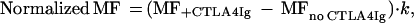 |
where k = MFanti-CD80 binding of WT/MFanti-CD80 binding of the mutants.
Cytotoxicity of Tumor-Infiltrating Lymphocytes (TIL).
As has been described (4), the cytotoxicity of TIL was measured in a 6-h 51Cr-release assay, using tumor cell lines J558-B7 and macrophage cell line P388D1 pulsed with 10 μg/ml of either P1A antigenic peptide (LPYLGWLVF, ref. 31) or a control Db-binding peptide from influenza virus NP366–374 (ASNENMDTM), both produced by Research Genetics (Huntsville, AL).
Kinetics of Tumor Growth.
BALB/c mice were injected with 5 × 106/mouse of J558 plasmocytoma, whereas C57BL/6j or H-2b CD28-deficient mice were injected with 2–5 × 105/mouse of EL4-thymoma. The injections were subcutaneous and were in the left inguen. Both the tumor incidences and tumor diameters were determined by physical examination. The tumor diameters presented are mean diameters of each group with eight mice per group. Mice with no tumor also are included in the calculation. This treatment increases the standard derivations when tumor rejection is underway, but it gives a more comprehensive description of tumorigenicity.
RESULTS
Characterization of Mutant CD80-Transfected J558 Tumor Cell Lines.
Our previous studies (16, 27) have identified mutant CD80 molecules that lack binding activity to either CD28 alone (B7W) or both CD28 and CTLA4 (B7Y). To study the role of B7-CTLA4 interaction in antitumor immunity, we transfected these CD80 mutants into plasmocytoma J558 and produced J558-B7 (WT-CD80), J558-B7W (W88>A), J558-B7Y (Y201>A), and J558-Neo (vector alone). We have used the clones that express the highest levels of wild-type (WT) and mutant CD80 for the study to reveal potentially weak CD28-independent function of CD80. As shown in Fig. 1a, all clones used in the study express significant levels of CD80. The levels of B7W and B7Y are comparable, although they are approximately 2- to 3-fold lower than that of WT CD80. After normalization based on cell surface amount of CD80, it is clear that B7W binds CTLA4Ig at high levels, whereas B7Y does not bind CTLA4Ig (Fig. 1b).
Figure 1.

CTLA4Ig binding to wild-type and mutant CD80 as revealed by flow cytometry. (a) CD80 expression. J558 cells were transfected with either vector alone (J558-Neo), wild-type CD80 (J558-B7), or CD80 mutants (W88>A, J558-B7W; Y201>1, J558-B7Y). J558 cells were stained with a saturating amount of biotinylated anti-CD80 mAb 10.16A, and the amount of mAb bound was determined by phycoerythorin-avidin. (b) Binding of J558-B7, J558-B7W, J558-B7Y, and J558-Neo to varying concentrations of CTLA4Ig. Data presented are mean fluorescence after normalization of cell surface CD80 expression as described in Materials and Methods. The experiments have been repeated three times with similar results.
Using the COS cell overexpression system, we have demonstrated that B7W and B7Y do not bind to CD28Ig (16, 27). Because the levels of CD80 expressed on the J558 cell lines allow only moderate binding to CD28Ig (data not shown), we used a more sensitive CHO cell adhesion assay to measure CD28-B7 interaction. As shown in Fig. 2a, J558-B7, but not J558-Neo, binds CD28-transfected CHO cells (CHO.CD28) in a dose-dependent manner. Neither J558-B7 nor J558-Neo bound significantly to control CD80-transfected CHO cells (CHO.B7). In addition, this binding is blocked by anti-CD80 mAb 3A12, but not by control hamster mAb 20C9 (ref. 32) (Fig. 2b). Therefore, we used this assay to measure binding of CD80 and its mutant to CD28. As shown in Fig. 2c, J558-B7W, J558-B7Y, and J558-Neo did not adhere to CD28-transfected CHO cells. Because the CD28-transfected CHO cells express 10- to 20-fold higher CD28 than normal T cells (data not shown), these results indicate that it is unlikely for J558-B7W and J558-B7Y to engage CD28 on T cells in vivo. Moreover, these results, together with our previous findings in transiently transfected COS cells using CD28Ig fusion protein (16, 27), demonstrate that both B7W and B7Y have lost their binding to CD28. The utilization of cell adhesion assay here also ruled out a possibility that B7W and B7Y expressed at these levels may bind to membrane-bound CD28.
Figure 2.

Comparison of CD28-binding activity of J558-B7 and J558-B7W by using cell adhesion assay. (a) Specificity of the binding. 51Cr-labeled J558-B7 were added to fixed CD28-transfected CHO cells (CHO.CD28) or CD80-transfectants (CHO.B7). The amount of cells added was quantified by the radioactivity retained on the CHO cell monolayer. (b) Blocking by of J558-B7 binding by anti-CD80 mAb 3A12. As in a, except that anti-CD80 mAb (anti-B7) or control hamster mAb 20C9 (anti-HSA) were added at the beginning of the incubation. (c) Specificity of mutant CD80 molecules. As in a, except that 51Cr-labeled J558-B7, J558-B7W, J558-B7Y, and J558-Neo were used. Data presented in c are specific binding after subtracting the binding to CHO-B7 monolayer. The results have been repeated three times.
B7-CTLA4 Interaction Is Sufficient to Costimulate Antitumor CTL Responses.
Our previous studies have shown that the TIL in J558-B7 tumor contain a substantial number of CD8 T cells with ex vivo antitumor cytotoxicity (4). We used this experimental model to test whether costimulation by B7-CTLA4 interaction is sufficient to induce antitumor CTL responses. We compared the TIL from different tumors for cytotoxicity to J558-B7, because our previous study indicated that CD8 T cells are responsible for cytotoxicity in this model (33). As shown in Fig. 3a, TIL from J558-B7 and J558-B7W have strong cytotoxicity toward J558-B7 target. In contrast, TIL from J558-Neo tumors are not cytotoxic. Because B7W has lost binding to CD28 while retaining binding to CTLA4 (ref. 16 and this study), this result demonstrates that B7-CD28 interaction is not essential for induction of antitumor CTL in vivo. In addition, because the only known receptor for B7W is CTLA4, it is likely that B7-CTLA4 interaction is responsible for the induction of CTL within tumor. To test whether B7-CTLA4 interaction is necessary in vivo, we compared CTL within TIL isolated from J558-B7, J558-B7W, and J558-B7Y. As shown in Fig. 3b, although potent CTL were detected in TIL isolated from J558-B7 and J558-B7W, antitumor CTL is not detectable within J558-B7Y.
Figure 3.

Antitumor CTL activity in TIL isolated from wild-type or mutant CD80-transfected tumors at 2–3 weeks after tumor injection. (a) CD28-B7 interaction is not essential for induction of antitumor CTL: comparison of antitumor CTL in J558-B7, J558-B7W, and J558-Neo. (b) In the absence of CD28-binding activity, B7-CTLA4 interaction is required for induction of antitumor CTL in vivo: comparison of CTL activity in TIL isolated from J558-B7W or J558-B7Y. TIL were isolated between 2 and 3 weeks after syngeneic mice were challenged with 5 × 106 tumor cells subcutaneously. The J558-B7 is used as target, whereas the TIL were used as effector cells. Data shown are representative of two independent experiments.
Our recent study revealed that a major tumor antigen in the experimental model is P1A (AA35–43) presented by H-2Ld (34). To test whether B7-CTLA4 interaction is sufficient to costimulate anti-P1A CTL, we tested TIL for the anti-P1A CTL. As shown in Fig. 4, TIL from J558-B7 and J558-B7W tumors contains a comparable level of anti-P1A CTL, whereas TIL from J558-B7Y are poorly cytotoxic to P1A-peptide pulsed targets, with maximal CTL activity less than 10%. Because B7Y and B7W differ in their ability to bind CTLA4, it is concluded that B7-CTLA4 interaction is sufficient to costimulate production of antitumor CTL, and, in the absence of CD28-B7 interaction, B7-CTLA4 interaction is necessary for the induction of antitumor CTL. The lack of mutant B7 that binds CD28 but not CTLA4 at this stage has prevented us from addressing whether B7-CD28 interaction alone is sufficient to costimulate the induction of antitumor CTL.
Figure 4.

Anti-P1A CTL within the TIL isolated from different types of tumors. As in Fig. 3 except that target cells used were P388D1 cells pulsed with either a P1A peptide or a control peptide from influenza virus (NP).
B7-CTLA4 Interaction Increases Host Resistance to Tumor Challenge.
We compared tumorigenicity of cell lines expressing wild-type and mutant B7 molecules in syngeneic mice. We used two tumor models, J558 (H-2d, derived from BALB/c mice) and EL4 (thymoma derived from C57BL/6j mice) for this purpose. As shown in Fig. 5, J558-B7 tumors were rejected rapidly in the majority of the mice, whereas J558-Neo and J558-B7Y tumors grew progressively. J558-B7W gave an intermediate phenotype. All mice inoculated with J558-B7W tumor cells developed tumors at a rate similar to those inoculated with J558-Neo and J558-B7Y (Fig. 5a). However, the J558-B7W tumors grew substantially slower than J558-B7Y and J558-Neo (Fig. 5b). Although it is necessary to sacrifice mice inoculated with J558-Neo and J558-B7Y between 3 and 4 weeks, mice bearing J558-B7W tumors can be kept for 6–7 weeks before euthanasia becomes necessary (data not shown). Moreover, the difference in tumorigenicity is a result of T cell-mediated immune rejection, because these tumor lines have similar tumorigenicity in BALB/c nu/nu mice (data not shown).
Figure 5.

Growth kinetics of J558 tumors transfected with WT or mutant CD80. Syngeneic mice were challenged with either J558-B7, J558-B7W, J558-B7-Neo, or J558-B7Y. The data presented are tumor incidence (a) and mean tumor diameters (b) with eight mice per group. Data shown are representative of two experiments.
Previous studies indicated that in immunocompetent mice, CD80-transfected thymoma EL4 is rejected in a CD8 T cell-dependent fashion (34). Therefore, we also transfected wild-type and mutant CD80 into thymoma EL4 and compared tumorigenicity of EL4-transfectant in immune competent mice (Fig. 6). Regardless of CD80 expression, all EL4-transfectants cause palpable tumor within 7–12 days. EL4-B7 tumors are rejected in all mice, whereas all EL4-B7Y and EL4 tumors grow progressively. A small proportion of mice (approximately 25%) rejected EL4-B7W. The remaining mice in the group did not reject the tumor, although the growth of the EL4-B7W tumors is substantially slower than the EL4-Neo and EL4-B7Y tumors.
Figure 6.

Growth kinetics of thymoma EL4 cells expressing either B7, B7W, B7Y, or vector alone (Neo). (a) Expression of wild-type and mutant CD80 molecules. Cell surface expression of wild-type or mutant CD80 was measured by saturating amount of anti-CD80 mAb 10.16A (10 μg/ml). (b) Tumor incidences: percentage of mice with palpable tumors. (c) Tumor growth kinetics as measured by the diameters of tumors at different time points after inoculation. Syngeneic mice were inoculated subcutaneously with 5 × 105 tumor cells, and the tumor incidences and sizes were recorded every 3 days. Representative of two independent experiments with five to eight mice per group.
To confirm the CD28-independent costimulatory activity of CD80, we compared tumorigenicity of the EL4-B7 and EL4-Neo tumors in H-2bCD28(+/−) and CD28(−/−) mice after 8 generations of backcross to C57BL/6j. As shown in Fig. 7, CD28(+/−) mice rejected CD80-transfected (EL4-B7) but not vector-transfected (EL4-Neo) EL4 tumors, as expected. Interestingly, CD28-deficient mice also rapidly reject EL4-B7. The overwhelming difference in growth rate and tumor incidence between EL4-Neo and EL4-B7 in CD28-deficient mice strongly indicates that CTLA4-B7 interaction promotes antitumor immunity.
Figure 7.

CD28-independent induction of tumor rejection by CD80. CD28(−/−) mice were backcrossed to C57BL/6j mice for 8 generations. CD28(+/−) and CD28(−/−) littermates were challenged with 5 × 105 of either CD80- (EL4-B7) or vector-transfected (EL4-Neo) EL4 cells. The enhancement of tumor immunogenicity by CD80 in CD28(−/−) mice was repeated in five independent experiments using 1–5 × 105 tumor cells and mice backcrossed for 6 or 8 generations.
DISCUSSION
Costimulatory molecule B7 interacts with both CD28 and CTLA4 (6–11). Although it is generally agreed that B7-CD28 interaction promotes T cell activation (35–39), the role of B7-CTLA4 interaction remains controversial. We have demonstrated previously that B7-CTLA4 interaction is sufficient to induce T cell clonal expansion in vitro (16). Here we have extended the study to antitumor immunity in vivo. We have utilized mutant CD80, which binds to CTLA4 but not CD28, as well as mice with a targeted mutation of CD28 to address the function of B7-CTLA4 interaction in antitumor immunity. Our results demonstrated that B7-CTLA4 interaction promotes antitumor immunity, although optimal immunity appears to be induced only when both CD28 and CTLA4 are engaged by CD80.
Of critical importance for the current study is the specificity of the mutant CD80 molecules B7W (W88>A) and B7Y (Y201>A). Extensive characterizations in COS-cell-transient transfection systems (16, 27) have revealed that B7W binds CTLA4Ig but not CD28Ig, whereas B7Y binds neither fusion protein. We confirm this specificity in the stable cell line expressing either mutant. We also have used CHO cell adhesion assay to show that B7W and B7Y do not bind significantly to membrane-bound CD28. Moreover, using T cell proliferation assay as the basic readout, we have demonstrated previously that all costimulatory activity of B7W can be attributed to its binding to CTLA4, and that essentially all reduction of the costimulatory activity as a result of mutation in B7W is a result of its lack of CD28-binding activity (16). Thus, the mutant B7W and B7Y offer a valuable tool to evaluate the contribution of CD28 and CTLA4. Moreover, in CD28-deficient mice, all CD28-independent costimulatory activity of CD80 is mediated by CTLA4 (16). CD28-deficient mice therefore can be used as an alternative tool to evaluate the contribution of B7-CTLA4 interaction in antitumor immunity. Our observations reported here indicate that B7-CTLA4 interaction is sufficient to promote antitumor CTL responses. Moreover, in the absence of CD28-B7 interaction, B7-CTLA4 interaction is essential for induction of antitumor CTL.
Corresponding to the enhanced antitumor CTL response, mutant B7W reduces tumorigenicity of the two tumor cell lines tested here, plasmocytoma J558 and thymoma EL4. Surprisingly, although TIL from J558-B7W contain substantial amounts of antitumor CTL, wild-type B7 transfectants are rejected much more efficiently than B7W transfectants. In fact, B7W does not appear to provide sufficient costimulation for tumor rejection in most experiments. Because B7W binds CTLA4 somewhat less well than WT-B7 (Fig. 1 and ref. 16), and because the binding assay used in the current study is less quantitative than that reported by others recently (7), it is possible that the reduced binding to CTLA4 and undetectable binding to CD28 both may contribute to the lower potency of B7W. In addition, B7W appears less stable than WT-B7 in vitro, and analysis of B7W transfectants recovered from tumor-bearing mice revealed significant reduction of B7W levels (data not shown). The lower B7W expression and instability of B7W in vivo also can explain the apparent contradiction between lack of B7W rejection and complete rejection of EL4-B7 in CD28-deficient mice. Thus, although it is likely that B7-CD28 interaction is required for optimal antitumor immunity, B7-CTLA4 interaction is sufficient to enhance antitumor immunity.
We have backcrossed CD28(−/−) locus into C57BL/6j for various generations and tested whether B7 expression on EL4 thymoma leads to tumor rejection. The CD28-independent function of CD80 is most clearly demonstrated by the reduced tumorigenicity of B7-transfected tumor cells in CD28-deficient mice. Our conclusion differs from a recent report that B7-transfected EL4 cells are not rejected in CD28-deficient mice (40). The difference can be a result of different levels of B7 expression and/or tumorigenicity of EL4 sublines used in different laboratories. Nevertheless, it is important to note that although the previous study has demonstrated an important role for CD28 in B7-induced antitumor immunity, it has not attempted to test whether B7-CTLA4 interaction enhances or down-regulates antitumor immunity, because tumorigenicity of B7-transfected EL4 cells was not compared with untransfected thymoma. Our study reveals a clear reduction in tumorigenicity of EL4-B7 as compared with EL4-Neo in CD28-deficient mice. Because we have demonstrated in vitro that all CD28-independent function of CD80 is mediated by CTLA4 (16), the results in this report demonstrate that B7-CTLA4 interaction promotes antitumor immunity, although we have not formally ruled out a possibility that B7 may have a third functional receptor other than CD28 and CTLA4.
Taken together, our results demonstrate that B7-CTLA4 interaction promotes antitumor immunity. Our results are not compatible with the hypothesis that CTLA4-B7 interaction down-regulates T cell responses (14, 18–25). Two important points must be considered to reconcile these different interpretations. First, the hypothesis that CTLA4 delivers a negative signal for T cell activation is only one of the potential interpretations of the effect of anti-CTLA4 mAb (13, 14, 17, 18). To interpret these earlier results based on the function of anti-CTLA4 mAb, one has to make an assumption as to whether Fab fragment of the mAb is an agonist or an antagonist. Our study, however, involves measurement of the functional outcome of B7-CTLA4 interaction and therefore is devoid of such caveat. Second, because CTLA4 and B7 are expressed in normal thymus (41), T cells in CTLA4-deficient mice mature and live in the absence of B7-CTLA4 interaction. The lymphoproliferative diseases in CTLA4-deficient mice are not necessarily attributable to a lack of negative regulation during T cell activation. A recent study revealed that major T cell subsets are produced normally in CTLA4-deficient thymi (42). In addition, CTLA4 is not essential for clonal deletion of H-Y-specific T cells in male mice (43). However, the critical issue, whether T cells that cause the fatal lymphoproliferative disease in CTLA4-deficient mice after activation exist normally in wild-type mice in a resting state, remains to be determined. In this regard, it is worth noting that the lymphoproliferative diseases in CTLA4-deficient mice are cured by expressing lymphocytic choriomeningitis virus-specific TCR on all T cells (43). It is possible that the lymphoproliferative diseases can be attributed at least in part to a failure of immune tolerance, either in the thymus or in the periphery, as has been suggested recently (17, 43–45).
Expression of costimulatory molecules CD80 and CD86 on a variety of tumor cells substantially increases tumor immunogenicity (1–5). However, a major obstacle to the application of the CD80 molecule is the difficulty in expressing costimulatory molecules on tumor cells in vivo. Because B7 interacts with CD28 and CTLA4, understanding the function of each receptor may provide insights for the next generation of immunotherapy targeted at this costimulatory pathway.
Acknowledgments
We thank Dr. Tak Mak for providing us with CD28-deficient mice, Dr. John Hirst for flow cytometry, and Fran Hitchcock for critical reading of the manuscript. This study was supported by National Institutes of Health Grant CA69016 and a grant from the Council for Tobacco Research, Inc. Y.W. was supported in part by National Institutes of Health Training Grant CA09161.
ABBREVIATIONS
- CTL
cytotoxic T lymphocytes
- TIL
tumor-infiltrating lymphocytes
- MF
mean fluorescences
References
- 1.Chen L P, Ashe S, Brady W A, Hellstrom I, Hellstrom K E, Ledbetter J A, McGowan P, Linsley P S. Cell. 1992;71:1093–1102. doi: 10.1016/s0092-8674(05)80059-5. [DOI] [PubMed] [Google Scholar]
- 2.Townsend S E, Allison J P. Science. 1993;259:368–370. doi: 10.1126/science.7678351. [DOI] [PubMed] [Google Scholar]
- 3.Baskar S, Ostrand-Rosenberg S, Nabavi N, Glimcher L. Proc Natl Acad Sci USA. 1993;90:7015–7019. doi: 10.1073/pnas.90.12.5687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ramarathinam L, Castle M, Wu Y, Liu Y. J Exp Med. 1994;179:1205–1214. doi: 10.1084/jem.179.4.1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yang G, Hellstrom K E, Hellstrom I, Chen L. J Immunol. 1995;154:2794–2800. [PubMed] [Google Scholar]
- 6.Linsley P S, Brady W, Grosmaire L, Damle N K, Ledbetter J A. J Exp Med. 1991;174:561–569. doi: 10.1084/jem.174.3.561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Linsley P S, Green J L, Brady W, Bajorath J, Ledbetter J A, Peach R. Immunity. 1994;1:793–801. doi: 10.1016/s1074-7613(94)80021-9. [DOI] [PubMed] [Google Scholar]
- 8.Wu Y, Guo Y, Liu Y. J Exp Med. 1993;178:1789–1793. doi: 10.1084/jem.178.5.1789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Azuma M, Ito D, Yagita H, Okumura K, Philips J H, Lanier L L, Somora C. Nature (London) 1993;366:76–78. doi: 10.1038/366076a0. [DOI] [PubMed] [Google Scholar]
- 10.Hathcock K S, Laszlo G, Dickler H B, Bradshaw J, Linsley P S, Hodes R J. Science. 1993;262:905–907. doi: 10.1126/science.7694361. [DOI] [PubMed] [Google Scholar]
- 11.Freeman G J, Borriello F, Hodes R J, Reiser H, Gribben J G, Ng J W, Kim J, Goldberg J W, Hathcock K, Laszlo G, et al. J Exp Med. 1993;178:2185–2192. doi: 10.1084/jem.178.6.2185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gross J A, Callas E, Allison J P. J Immunol. 1992;149:2380–2388. [PubMed] [Google Scholar]
- 13.Linsley P S, Green J A, Tan P, Bradshaw J, Ledbetter J A, Anasetti C, Damle N K. J Exp Med. 1992;176:1595–1604. doi: 10.1084/jem.176.6.1595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Walunas T L, Lenchow D J, Bakker C, Linsly P S, Freeman G J, Green J M, Thompson C B, Bluestone J A. Immunity. 1995;1:405–414. doi: 10.1016/1074-7613(94)90071-x. [DOI] [PubMed] [Google Scholar]
- 15.Linsley P S, Bradshaw J, Greene J, Peach R, Bennett K L, Mittler R S. Immunity. 1996;4:535–543. doi: 10.1016/s1074-7613(00)80480-x. [DOI] [PubMed] [Google Scholar]
- 16.Wu Y, Guo Y, Huang A, Zheng P, Liu Y. J Exp Med. 1997;185:1327–1335. doi: 10.1084/jem.185.7.1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liu Y. Immunol Today. 1997;18:569–572. doi: 10.1016/s0167-5699(97)01170-5. [DOI] [PubMed] [Google Scholar]
- 18.Krummel M F, Allison J P. J Exp Med. 1995;182:459–466. doi: 10.1084/jem.182.2.459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Krummel M F, Allison J P. J Exp Med. 1996;183:2533–2540. doi: 10.1084/jem.183.6.2533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Walnunas T L, Bakker C Y, Bluestone J A. J Exp Med. 1996;183:2541–2550. doi: 10.1084/jem.183.6.2541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kearney E R, Walunas T L, Karr R W, Morton R A, Loh D Y, Bluestone J A, Jenkins M K. J Immunol. 1995;155:1032–1036. [PubMed] [Google Scholar]
- 22.Leach D R, Krummel M F, Allison J P. Science. 1996;271:1734–1736. doi: 10.1126/science.271.5256.1734. [DOI] [PubMed] [Google Scholar]
- 23.Karandikar N J, Vanderlugt C L, Walunas T L, Miller S D, Bluestone J A. J Exp Med. 1996;184:783–788. doi: 10.1084/jem.184.2.783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Waterhouse P, Penninger J M, Yimms E, Wakeham A, Shahinian A, Lee K P, Thompson C B, Griesser H, Mak T W. Science. 1995;270:985–987. doi: 10.1126/science.270.5238.985. [DOI] [PubMed] [Google Scholar]
- 25.Tivol E A, Borriello F, Schweitzer A N, Lynch W P, Bluestone J A, Sharpe A H. Immunity. 1995;3:541–547. doi: 10.1016/1074-7613(95)90125-6. [DOI] [PubMed] [Google Scholar]
- 26.Shahinian A, Pfeffer K, Lee K P, Kundig K P, Kishihara T M, Wakeham A, Kawai K, Ohashi P S, Thompson C B, Mak T W. Science. 1993;261:609–612. doi: 10.1126/science.7688139. [DOI] [PubMed] [Google Scholar]
- 27.Guo Y, Wu Y, Zhao M, Kong X P, Liu Y. J Exp Med. 1995;181:1345–1355. doi: 10.1084/jem.181.4.1345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Linsley P S, Clark E A, Ledbetter J A. Proc Natl Acad Sci USA. 1990;87:5031–5035. [Google Scholar]
- 29.Razi-Wolf Z, Freeman G J, Galvin F, Benacerraf B, Nadler L M, Reiser H. Proc Natl Acad Sci USA. 1992;89:4210–4214. doi: 10.1073/pnas.89.9.4210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dumont F J, Habbrtsett R C, Coker L C, Nichols E A, Treffinger E A. Cell Immunol. 1985;96:327–337. doi: 10.1016/0008-8749(85)90364-8. [DOI] [PubMed] [Google Scholar]
- 31.Lethe B, Van der Eynde B, Van Pel A, Corradin G, Boon T. Eur J Immunol. 1992;22:2283–2288. doi: 10.1002/eji.1830220916. [DOI] [PubMed] [Google Scholar]
- 32.Liu Y, Jones B, Sullivan K, Aruffo A, Linsley P S, Janeway C A., Jr J Exp Med. 1992;175:437–445. doi: 10.1084/jem.175.2.437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ramarathinam R, Sarma S, Maric M, Zhao M, Yang G, Chen L, Liu Y. J Immunol. 1995;155:5323–5329. [PubMed] [Google Scholar]
- 34.Johnson J V, Malacko A R, Mizumo M T, McCowan P, Hellstrom I, Hellstrom K, Marquardt H, Chen L P. J Exp Med. 1996;183:791–800. doi: 10.1084/jem.183.3.791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Harding F, McAuther J, Gross J A, Allison J P. Nature (London) 1992;356:607–609. doi: 10.1038/356607a0. [DOI] [PubMed] [Google Scholar]
- 36.Green J M, Noel P J, Sperling A I, Walunas T L, Gray G S, Bluestone J A, Thompson C B. Immunity. 1994;1:501–508. doi: 10.1016/1074-7613(94)90092-2. [DOI] [PubMed] [Google Scholar]
- 37.June C H, Ledbetter J A, Gillispie M M, Linsten T, Thompson C B. Mol Cell Biol. 1987;7:4472–4481. doi: 10.1128/mcb.7.12.4472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hara T, Fu S M, Hansen J A. J Exp Med. 1985;161:1513–1524. doi: 10.1084/jem.161.6.1513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Moretta A, Pantaleo G, Lopez-Botet M, Moratta L. J Exp Med. 1985;162:823–838. doi: 10.1084/jem.162.3.823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wen T, Kono K, Shahinian A, Kiessling R, Mak T W, Klein G. Eur J Immunol. 1997;27:1988–1993. doi: 10.1002/eji.1830270824. [DOI] [PubMed] [Google Scholar]
- 41.Wagner D H, Jr, Hagman J, Linsley P S, Hodsdon W, Freed J H, Newell M K. J Exp Med. 1996;184:1631–1638. doi: 10.1084/jem.184.5.1631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chamber C A, Cado D, Troung T, Allison J P. Proc Natl Acad Sci USA. 1997;94:9296–9301. doi: 10.1073/pnas.94.17.9296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Waterhouse P, Bachmann M, Penninger J M, Ohashi P S, Mak T W. Eur J Immunol. 1997;27:1887–1892. doi: 10.1002/eji.1830270811. [DOI] [PubMed] [Google Scholar]
- 44.Bluestone J A. J Immunol. 1997;158:1989–1993. [PubMed] [Google Scholar]
- 45.Perez V L, Van Parijs L, Biuckians A, Zheng X-X, Strom T B, Abbas A K. Immunity. 1997;6:411–417. doi: 10.1016/s1074-7613(00)80284-8. [DOI] [PubMed] [Google Scholar]