

Abstract
Pertussis toxin (PTx) has been shown to exert a variety of effects on immune cells independent of its ability to ADP-ribosylate G proteins. Of these effects, the binding subunit of PTx (PTxB) has been shown to block signaling via the chemokine receptor CCR5, but the mechanism involved in this process is unknown. Here, we show that PTxB causes desensitization of a related chemokine receptor, CXCR4, and explore the mechanism by which this occurs. CXCR4 is the receptor for the chemokine stromal cell-derived factor 1α (SDF-1α) and elicits a number of biological effects, including stimulation of T cell migration. PTxB treatment causes a decrease in CXCR4 surface expression, inhibits G protein-associated signaling, and blocks SDF-1α-mediated chemotaxis. We show that PTxB mediates these effects by activating the TCR signaling network, as the effects are dependent on TCR and ZAP70 expression. Additionally, the activation of the TCR with anti-CD3 mAb elicits a similar set of effects on CXCR4 activity, supporting the idea that TCR signaling leads to cross-desensitization of CXCR4. The inhibition of CXCR4 by PTxB is rapid and transient; however, the catalytic activity of PTx prevents CXCR4 signaling in the long term. Thus, the effects of PTx holotoxin on CXCR4 signaling can be divided into two phases: short term by the B subunit, and long term by the catalytic subunit. These data suggest that TCR crosstalk with CXCR4 is likely a normal cellular process that leads to cross-desensitization, which is exploited by the B subunit of PTx.
CXCR4 is a chemokine receptor abundantly expressed on immune cells, including T cells, B cells, and monocytes (1–4). Its ligand, SDF-1α3 (stromal cell-derived factor 1α, CXCL12), has been shown to induce CXCR4-mediated signaling activities that culminate in chemotaxis (5–7). Typically, CXCR4 undergoes homologous desensitization following activation by SDF-1α, but it has also been shown to be cross- or heterologously desensitized by other signaling receptors such as CD4, the BCR, and the κ opioid receptor (8–11). Mechanistically, evidence suggests that homologous and heterologous desensitization are mediated by signaling pathways that activate protein kinases, which in turn phosphorylate the intracellular regions of receptors such as CXCR4 (12–14). These phosphorylation events prevent receptor signaling by blocking access to G proteins and promoting internalization, ultimately resulting in receptor down-regulation from the cell surface (13).
The TCR is an important immunological receptor expressed on T cells. It is stimulated by Ag-presenting MHC and results in T cell activation characterized by clonal expansion and cytokine production (15, 16). Recent reports indicate that the TCR and several chemokine receptors, including CXCR4, are intimately coupled (17). Studies show that many proteins predominantly thought to be required only for TCR signaling (CD45, Zap70, Lck, and the TCR) are also activated in response to various chemokines and needed for optimal chemotaxis (18–24). Additionally, many chemokine receptor-associated proteins, such as G protein-coupled receptor kinase 2 (GRK2) and Gαq/11, also interact with the TCR (25, 26). Furthermore, reports show that the TCR and CXCR4 physically associate upon SDF-1α treatment and have been shown to affect each other's function and expression (22, 27).
Pertussis toxin (PTx) is a complex AB5 toxin made and secreted by the pathogenic bacterium Bordetella pertussis. It has been shown to have profound effects on the immune system, including the ability to induce lymphocyte proliferation and modify cellular signaling pathways (28–31). The A subunit of PTx (S1) has enzymatic activity in which it ADP-ribosylates cellular G proteins in the cytoplasm, thus preventing their signaling functions (32, 33). The B subunit (PTxB) is a pentamer (comprised of S2, S3, S4, and S5) responsible for the binding and delivery of the A subunit to target cells (34–36). Several studies have revealed that PTxB alone is capable of activating a variety of signals in lymphocytes (28, 29, 37–40). More specifically, PTxB promotes activation of many proteins associated with TCR signaling, resulting in a phenotype similar to an activated T cell (28, 41). In addition to activating TCR signals, studies also show that pretreatment of primary lymphocytes with PTxB results in a rapid block of Ca2+ response from the chemokine receptor CCR5 (31). This block occurs without interfering with ligand binding and appears to be reversed by protein kinase C (PKC) inhibitors (31). Although the detailed mechanism by which PTxB blocks CCR5 is currently unclear, collectively these studies suggest a mechanism of receptor crosstalk between an unknown PTxB receptor and chemokine receptors. In several scenarios, crosstalk between two cell surface receptors has been demonstrated to result in a ligand-activated receptor inducing cross-desensitization of an inactive receptor by uncoupling it from its cognate signaling molecules and/or promoting its internalization (8, 13).
In this report we show that PTx, via the B subunit and independent of its enzymatic A subunit, regulates CXCR4 signaling and function by utilization of a TCR signaling pathway. Using primary human T cells and T cell lines with well-characterized TCR deficiencies, we show that expression and signaling from the TCR pathway is required for the effects of PTxB activity on CXCR4. This phenomenon is additionally corroborated by mimicking TCR engagement with an anti-CD3 mAb. These studies elucidate a novel pathogenic mechanism of blocking chemotaxis through utilization of a previously undefined cellular process by which TCR signaling can cross-regulate CXCR4.
Materials and Methods
Cell lines and reagents
The human Jurkat T cell lymphoma line (clones E6.1, P116, and RT3) was obtained from the American Type Culture Collection and maintained in RPMI 1640 supplemented with 10% FBS, penicillin (100 U/ml), and streptomycin (100 μg/ml). PTx and PTxB were purchased from List Biological Laboratories. The PTx9K/129G mutant was prepared at Chiron Bioscience and kindly provided by Rino Rappuoli (64, 65). The presence of contaminating PTx holotoxin in PTxB preparations was assessed using the Chinese hamster ovary cell-clustering assay (66). The concentration of residual PTx holotoxin in PTxB preparations was found to be <2%. No effect was observed in experiments using the PTx holotoxin at these concentrations in short-term assays; therefore, we conclude that the observed effects are due to PTxB and not contaminating catalytic subunit activity. Anti-CXCR4 mAb clones 12G5 and 4G10 were purchased from BD Pharmigen and Santa Cruz Biotechnology, respectively. Anti-CD3 mAb clone HIT3a was purchased from Millipore. PMA and staurosporine were purchased from Sigma-Aldrich. Recombinant SDF-1α was purchased from PeproTech.
Primary T cell isolation
PBMCs were obtained from buffy coats of healthy donor whole blood by Ficoll separation. T cells were enriched according to the manufacturer's protocol using the Pan T Cell Isolation Kit II from Miltenyi Biotec. Following isolation, cells were maintained in RPMI 1640 supplemented with 10% FBS, penicillin (100 U/ml), and streptomycin (100 μg/ml) for up to 24 h as indicated. CD3 expression was measured with the anti-CD3 Ab (clone SK7) from BD Biosciences, and all samples were found to be >75% CD3-positive.
CXCR4 internalization
Internalization was assessed by washing (106 cells/sample) in serum-free RPMI 1640 twice before treatment. Cells were warmed to 37°C and incubated with 0.25–10 μg/ml PTxB, 2 μg/ml PTx, 25 ng/ml PMA, or 10 nM SDF-1α for the indicated amount of time at 4°C or 37°C. Where indicated, samples were also exposed to 1 or 5 μM staurosporin for 1 h before treatment with any of the above ligands. Following stimulation, cells were put on ice for 10 min before addition of mAb and remained on ice there after. Cells were stained with anti-CXCR4 (12G5 or 4G10) at (1/100) for 1 h and then with anti-mouse PE (1/100) for 1 h. Cells were washed twice and FACS analyzed using a FACSCalibur (BD Biosciences). Internalization was calculated by determining the percentage mean fluorescence of treated cells compared with untreated cells or cells stained following each treatment at 4°C to control for ligand/Ab receptor competition.
CXCR4 phosphorylation
Cells (1.5 × 107 cells/sample) were washed twice with phosphate-free DMEM and labeled with 150 μCi of [32P]orthophosphate (PerkinElmer) for 3 h. After washing the cells in PBS, they were suspended in a 0.5-ml volume and stimulated with 10 μg/ml PTxB, 10 μg/ml anti-CD3 mAb, or 20 nM SDF-1α for 15 min and placed on ice. Cells were pelleted and lysed with 0.5 ml of lysis buffer (150 mM NaCl, 1% Nonidet P-40, 0.1% SDS, 0.1% deoxycholate, 50 mM Tris, Complete protease inhibitor (Roche), 10 mM sodium fluoride, 20 mM sodium vanidate) for 15 min. Lysates were clarified by centrifugation and precleared with protein A- and G-Sepharose (Calbiochem) for 1 h at 4°C before the addition of 3 μg of anti-CXCR4 mAb (clone 4G10) and fresh protein A- and G-Sepharose. The mixture was incubated for 2 h at 4°C with constant agitation followed by extensive washing and elution in 60 μl of 3× Laemmli sample buffer. The immunoprecipitates were subjected to SDS-PAGE and autoradiography. Samples were quantified using ImmageQuant software and graphically represented using GraphPad Prism.
cAMP assays
Cells (106/sample) were washed with serum-free RPMI 1640 and suspended in 0.1 ml. After cells were warmed to 37°C, 2 μg/ml PTxB or anti-CD3 mAb were added and incubated at 37°C for 15 min. Cells were then assayed for cAMP inhibition by the simultaneous addition of 10 nM SDF-1α and 10 μM forskolin for 30 additional minutes. Cells were placed on ice and 0.1 ml of Tris/EDTA buffer containing 0.1% BSA was added, followed by heating at 95°C for 10 min. Lysates were clarified by centrifugation and analyzed for cAMP levels by a PKA competition assay. Briefly, 50 μl of the lysate was added to 50 μl of a known concentration of [3H]cAMP and mixed well before 100 μl of 0.6 mg/ml PKA was added. Equilibrium was allowed to occur as the mixture was incubated at 4°C for 2 h. All unbound cAMP was absorbed with activated charcoal in Tris/EDTA buffer containing 2% BSA. The clarified lysate was counted with a scintillation counter and compared with a standard curve of cAMP concentrations ranging from 0.125 to 128 pmol. Data are analyzed by percentage remaining activity when SDF-1α-induced inhibition of forskolin activity is set at 100%.
Ca2+ flux assays
Cells (2 × 105/sample) were washed with serum-free RPMI 1640 and plated. Cells were loaded with the Fluo-4 NW (no wash) calcium assay kit (Invitrogen) according to the manufacturer's protocol and treated with PBS (without Ca2+/Mg2+), 2 μg/ml PTxB, or 2 μg/ml anti-CD3 mAb for 30 min before the addition of 10 nM SDF-1α. Ca2+ flux was analyzed by the increase in fluorescence reported by the Flex Station II (Molecular Probes) using SoftMax Pro software. Data were imported into GraphPad Prism software for analysis and production of graphs.
Chemotaxis assays
For migration of Jurkat T cells, 2 × 106 cells/sample were washed twice with serum-free RPMI 1640, warmed to 37°C, and plated in the top chamber of an 8-μm transwell in 0.2 ml of serum-free RPMI 1640. Cells were left untreated or were pretreated with 2 μg/ml PTxB, PTxWT, PTx9K/129G, anti-CD3 mAb, or 25 ng/ml PMA for 15 min. Cells pretreated for 18 h were treated in serum-free media for 2 h before serum-containing RPMI 1640 was added for the remaining time before the migration assay. These samples were then processed as above. The enzymatically inactive mutant of PTx was used to asses the function of the B subunit in these longer time frame assays, as the PTxB preparations were found to contain low levels of contaminating holotoxin that could interfere with longer time frame assays (data not shown). Primary T cells were assayed as described above for Jurkat cells except that 5-μm pore inserts were used in place of 8-μm pores. Also, to keep the cell concentration consistent with Jurkat cells during PTxB treatments, 1 × 106 primary T cells were treated with 4 μg/ml PTxB for 2 h in 0.1 ml before the volume was doubled to make samples with 5 × 105 cells/sample in 0.1 ml with 2 μg/ml PTxB. Each transwell insert was then placed into a bottom chamber containing 0.6 ml serum-free media or serum-free media containing 10 nM (Jurkat) or 50 nM (primary T cells) SDF-1α. Cells were allowed to migrate for 4 h and cells in the bottom chamber were counted for 30 s at a rate of ∼75 μl/min using a FACSCalibur (BD Biosciences).
Results
PTx induces CXCR4 internalization
To investigate whether the B subunit of PTx may cross-regulate chemokine receptors, we chose to analyze the effects of PTxB on cell surface expression of the chemokine receptor CXCR4, which is endogenously expressed in Jurkat T cells. Jurkat cells were treated with various concentrations of PTxB (42), and CXCR4 surface expression was then analyzed by FACS using an anti-CXCR4 mAb (clone 12G5). As shown in Fig. 1A, CXCR4 surface expression decreases in a dose-dependent manner following PTxB treatment, with a maximal response of a 45% decrease in cells treated with 2 μg/ml PTxB for 45 min. The effect of the B subunit on CXCR4 cell surface expression was confirmed using PTx holotoxin (Fig. 1B) and an enzymatically inactive mutant, PTx9K/129G (data not shown), both of which promoted a loss in CXCR4 cell surface expression. Fig. 1B also shows that cells treated with PTx at 37°C, but not at 4°C for the same length of time, have decreased CXCR4 surface expression, indicating that PTx is not competing with the Ab by directly binding to CXCR4.
Figure 1.
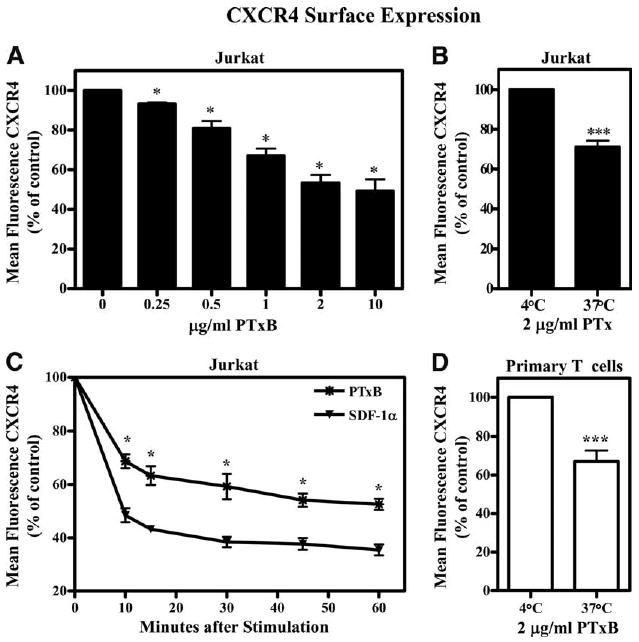
PTxB promotes CXCR4 internalization. A, Jurkat cells were treated with 0, 0.25, 0.5, 1, 2, or 10 μg/ml PTxB at 37°C for 45 min, placed on ice, and stained with the 12G5 anti-CXCR4 mAb. Mean fluorescence for each sample was collected by FACS, analyzed, and graphed as percentage of mean fluorescence of untreated cells. *, p < 0.05 for all values of the dose response when each concentration is compared with no treatment (n = 4). B, Jurkat cells were treated with 2 μg/ml PTx at 4°C or 37°C for 60 min, placed on ice, and stained with the 12G5 anti-CXCR4 mAb. Mean fluorescence for each sample was collected by FACS, analyzed, and graphed as percentage of mean fluorescence of cells treated at 4°C. ***, p < 0.001 for PTxB treatments at 37°C when compared with the same treatment at 4°C (n = 6). C, Jurkat cells were treated with 10 nM SDF-1α or 2 μg/ml PTxB for 10, 15, 30, 45, or 60 min and analyzed as in A. *, p < 0.05 for all values of PTxB treatments when compared with untreated and SDF-1α treatments at each corresponding time point (n = 6). D, Primary human T cells were treated with 2 μg/ml PTxB at 4°C or 37°C for 60 min, placed on ice, and stained with the 4G10 anti-CXCR4 mAb. Mean fluorescence for each sample was collected by FACS, analyzed, and graphed as percentage of mean fluorescence of cells treated at 4°C. ***, p < 0.001 for PTxB treatments at 37°C when compared with the same treatment at 4°C (n = 8).
Since PTx induces a robust internalization of CXCR4, we next wished to determine the kinetics of this process and compare the level of PTxB-induced internalization to SDF-1α-induced internalization. Data in Fig. 1C show that 2 μg/ml PTxB decreases CXCR4 surface expression 25% within 10 min of treatment. This reduction reaches a maximum of a 45% decrease within 45 min. When compared with ligand-induced internalization by SDF-1α, PTxB-induced internalization is equally as rapid, but not as robust as SDF-1α, which causes a maximum of 65% internalization.
Although the Jurkat T cell line is very useful for studying many aspects of T cell signaling and function, we sought to validate the effects of PTx on CXCR4 internalization in primary human T cells. Similar to what we observed in Jurkat T cells, Fig. 1D shows that primary human T cells from healthy donors demonstrate a significant decrease in CXCR4 surface expression following treatment with PTxB. The data indicate that PTx similarly affects CXCR4 in both primary human T cells and Jurkat T cells, suggesting that the Jurkat cell line would serve as a good model to explore the mechanism by which PTx regulates CXCR4.
The TCR and its associated signaling pathways are required for the effects of PTx on CXCR4
The speed and strength of the PTxB-induced CXCR4 internalization (Fig. 1C) suggest that PTx might be activating the signaling of an unidentified receptor resulting in cross-regulation of CXCR4. To further define the mechanism by which PTxB induces CXCR4 internalization, we sought to identify potential PTxB receptors responsible for initiating signals that result in CXCR4 internalization. Interestingly, the TCR signaling pathway is both intimately coupled to CXCR4 and is activated by PTxB (20, 22–24, 27–29, 43, 44). To determine whether PTxB utilizes a TCR signaling pathway to induce CXCR4 internalization, we examined CXCR4 internalization in TCR-deficient (RT3) Jurkat T cells (45). Fig. 2A shows that PTxB-induced CXCR4 internalization is significantly reduced in TCR− Jurkat cells compared with wild-type (WT) Jurkat cells. We confirmed this result using a second independently derived TCR− cell line termed OKT3.3 and observed similar effects regarding PTxB regulation of CXCR4 internalization (data not shown). To determine whether signaling molecules downstream of the TCR were responsible for the effects of PTx, we also analyzed the effect of PTxB on CXCR4 surface expression in ZAP70− (P116) Jurkat cells. Fig. 2B shows that in comparison to WT Jurkat cells, ZAP70− Jurkat cells treated with PTxB demonstrate a significant reduction in CXCR4 internalization. For control purposes, we analyzed the ability of SDF-1α and PMA to stimulate CXCR4 internalization in TCR− and ZAP70− cells (13, 14). In contrast to PTxB, SDF-1α and PMA both stimulated CXCR4 internalization in TCR− and ZAP70− cells equal to that observed in WT cells (Fig. 2, A and B), indicating that both homologous desensitization induced by SDF-1α and heterologous desensitization mediated by PKC activity remain completely intact in the mutant cell lines. Collectively, these data suggest that PTx depends on the expression and the signaling functions of the TCR to induce CXCR4 down-regulation.
Figure 2.
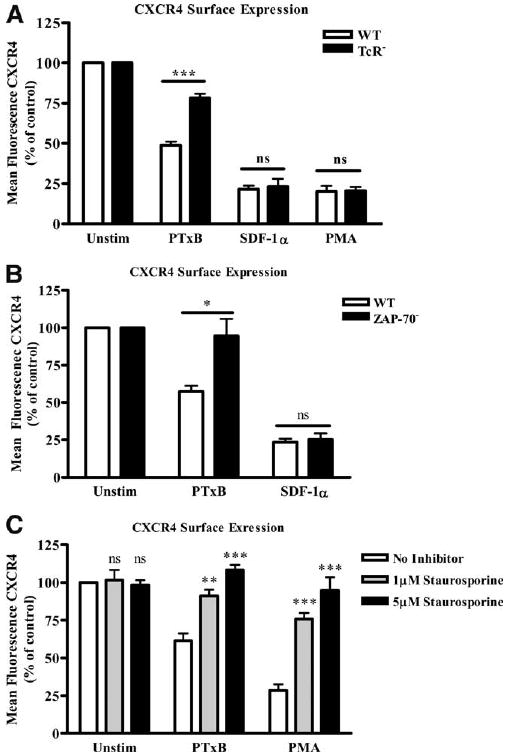
PTxB utilizes the TCR to induce CXCR4 internalization. A, WT and TCR− Jurkat cells or (B) WT and ZAP70− Jurkat cells were treated with 2 μg/ml PTxB, 10 nM SDF-1α, or 25 ng/ml PMA (WT and TCR− only) for 45 min. Cells were placed on ice and stained with anti-CXCR4 mAb. Mean fluorescence was collected by FACS, analyzed, and graphed as percentage of mean fluorescence of untreated cells. ***, p < 0.001 in WT vs TCR− (n = 6) and *, p < 0.05 in WT vs ZAP70− in PTxB-treated cells (n = 4). C, WT Jurkat cells were treated with 0, 1, or 5 μM staurosporine for 60 min before treating with 2 μg/ml PTxB or 25 ng/ml PMA for 45 min. All cells were placed on ice and stained with 12G5 anti-CXCR4 mAb. Mean fluorescence was collected by FACS, analyzed, and graphed as percentage of mean fluorescence of untreated cells. **, p < 0.01 and ***, p < 0.001 when 1 or 5 μM staurosporine pretreated cells were compared with cells that received no inhibitor in each group (n = 3). ns, not significant.
Since the effects of PTx require the presence of the TCR and its associated signaling molecule ZAP70, we speculated that PTxB stimulates CXCR4 internalization by activating protein kinases downstream of ZAP70, such as those in the PKC family. Members of the PKC family have been shown play active roles in many downstream TCR signaling pathways (35, 46). To investigate this hypothesis, we treated WT Jurkat cells with several concentrations of the PKC inhibitor staurosporine before the cells were treated with PTxB (47). Fig. 2C shows that there is a significant decrease in the ability of PTxB to induce CXCR4 internalization in cells pretreated with 1 or 5 μM staurosporine. To control for the effect of staurosporine pretreatment on PKC activity, we also analyzed CXCR4 surface expression in cells treated with the PKC activator PMA. The data show that the same concentrations of staurosporine that block PTxB-induced CXCR4 internalization also block PMA-induced CXCR4 internalization. Collectively, Fig. 2 suggests that PTxB utilizes the TCR to activate a TCR signaling pathway that drives the internalization of CXCR4 through the activity of PKC.
PTx/TCR signaling induces CXCR4 phosphorylation
Since chemokine receptor desensitization and internalization can occur as a result of receptor phosphorylation, we investigated if CXCR4 was phosphorylated after PTxB or anti-CD3 mAb treatments. Using SDF-1α as a control, we stimulated cells with PTxB or anti-CD3 and immunoprecipitated CXCR4 from [32P]orthophosphate-labeled cells followed by SDS-PAGE and autoradiographic analysis. As shown in Fig. 3A (upper panel), after 15 min of treatment with SDF-1α, PTxB, or anti-CD3 mAb, a phosphorylated protein species that migrates at ∼46 kDa was specifically immunoprecipitated from Jurkat lysates using an anti-CXCR4 Ab. The identity of this protein was confirmed to be CXCR4, as an isotype-matched control Ab failed to immunoprecipitate this protein. When the blots were quantified (Fig. 3A, lower panel), it was determined that SDF-1α caused a 5-fold increase in CXCR4 phosphorylation over basal levels. PTxB also significantly increased CXCR4 phosphorylation, as there was a 2.1-fold increase when compared with basal levels. A relatively equal increase in phosphorylation was also detected by directly stimulating the TCR with anti-CD3 mAb. These data show for the first time that TCR signaling causes a direct change in the phosphorylation state of CXCR4. In comparison to SDF-1α-induced phosphorylation of CXCR4, PTxB- or anti-CD3-dependent induction of CXCR4 phosphorylation is not as robust, only approaching 50% of that induced by SDF-1α. This suggests that SDF-1α causes phosphorylation of a stoichiometrically higher number of the 18 putative CXCR4 phosphorylation sites or stimulates phosphorylation of a higher percentage of receptors per cell than does TCR signaling. Taken together, these data show that PTxB and TCR signaling both rapidly induce CXCR4 phosphorylation.
Figure 3.
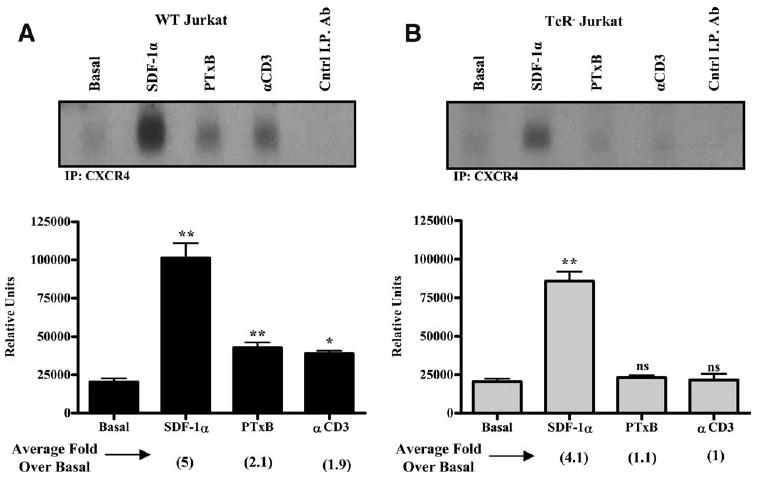
PTxB Utilizes the TCR to promote CXCR4 phosphorylation. A, WT and (B) TCR− Jurkat cells were radiolabled with [32P]orthophosphate and treated with 20 nM SDF-1α, 10 μg/ml PTxB, or 10 μg/ml anti-CD3 (αCD3) mAb for 15 min. CXCR4 was immunoprecipitated (I.P.) with the 4G10 anti-CXCR4 mAb, and an anti-CD28 mAb was utilized as a negative control for the immunoprecipitation reactions. Immunoprecipitations were subjected to SDS-PAGE followed by autoradiographic analysis. Representative autoradiographs are shown and five independent experiments were quantified in the lower panels and are graphically depicted. *, p < 0.05 and **, p < 0.01 when treated cells are compared with basal levels. ns, not significant.
To determine whether PTxB utilizes the TCR to facilitate CXCR4 phosphorylation, we analyzed CXCR4 phosphorylation in TCR− cells. Fig. 3B shows that similar to WT Jurkat cells, CXCR4 phosphorylation increases 4.1-fold over basal levels in SDF-1α-treated TCR− cells. In contrast to WT Jurkat cells, TCR− Jurkat cells show no change in CXCR4 phosphorylation when treated with PTxB or the negative control anti-CD3 mAb. Collectively, Fig. 3 reveals that TCR signaling is required to promote CXCR4 phosphorylation in response to either PTxB or αCD3.
PTx/TCR signaling blocks CXCR4-mediated G protein activation
CXCR4, like most other chemokine receptors, signals through Gαi proteins in response to SDF-1α stimulation (48–50). Gαi activation results in a variety of signaling events, including the inhibition of cAMP accumulation (49). During desensitization, receptor phosphorylation is thought to alter the receptor such that there is an inhibition of G protein coupling (51). Since TCR signaling activated by anti-CD3 mAb or PTxB results in phosphorylation and internalization of CXCR4, we next sought to determine whether PTxB or anti-CD3 mAb was able to inhibit CXCR4 signaling through Gαi proteins. Gαi activity was assessed by measuring inhibition of forskolin-stimulated adenylate cyclase. When WT Jurkat cells are treated with PTxB or anti-CD3 before the addition of SDF-1α, >90% of the SDF-1α-induced Gαi activity is blocked at 45 min (Fig. 4A). Additionally, there is no cAMP increase with PTxB or anti-CD3 mAb alone (data not shown), indicating that the loss of cAMP inhibition is due to blocked CXCR4 Gαi function, not increased basal cAMP. In contrast to WT cells, TCR− cells show no decrease in SDF-1α-induced Gαi activity seen when cells are pretreated with PTxB or anti-CD3 mAb (Fig. 4B). Taken together, these data show that activating TCR signaling by either PTxB or anti-CD3 mAb rapidly blocks CXCR4 signaling through Gαi.
Figure 4.
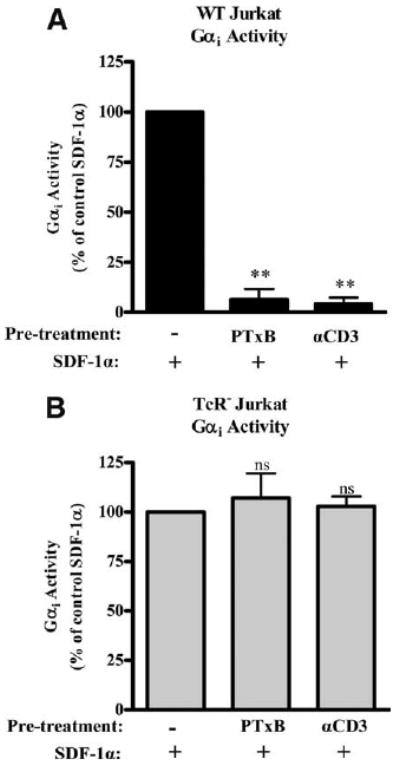
PTxB/TCR signaling prevents CXCR4-stimulated Gαi activity. A, WT and (B) TCR− cells were untreated or treated with 2 μg/ml PTxB or 2 μg/ml anti-CD3 (αCD3) mAb for 15 min before simultaneous treatment with 10 μM forskolin and 10 nM SDF-1α for 30 min. Cells were harvested and cAMP was quantified by competition assay. SDF-1α inhibition of forskolin was set to 100% in each cell type and data are shown as percentage Gαi activity remaining. **, p < 0.01 compared with control (n = 12). ns, not significant.
PTx/TCR signaling blocks CXCR4-mediated Ca2+ release
In addition to Gαi activity, intracellular release of Ca2+ induced by Gβγ subunits is indicative of CXCR4 signaling (49, 52). To analyze how PTxB signaling through the TCR affects CXCR4 signaling mediated by Gβγ, we tested the ability of CXCR4 to induce Ca2+ flux after 30 min of pretreatment with PBS, PTxB, or anti-CD3 mAb. Ca2+ traces in Fig. 5A (upper panels) show that WT cells pretreated with 2 μg/ml PTxB or anti-CD3 mAb display dramatic inhibition of the SDF-1α response compared with cells pretreated with PBS. Quantification of the peak responses to 10 and 50 nM SDF-1α demonstrates that the observed inhibition by PTxB is >80% (Fig. 5B, left panel). In contrast to WT cells, when TCR-negative cells were pretreated with 2 μg/ml PTxB or anti-CD3 mAb, there was no significant inhibition of CXCR4 Ca2+ signaling in response to 10 or 50 nM SDF-1α (Fig. 5). This further supports our hypothesis that PTxB utilizes TCR signaling to rapidly block CXCR4 signaling.
Figure 5.
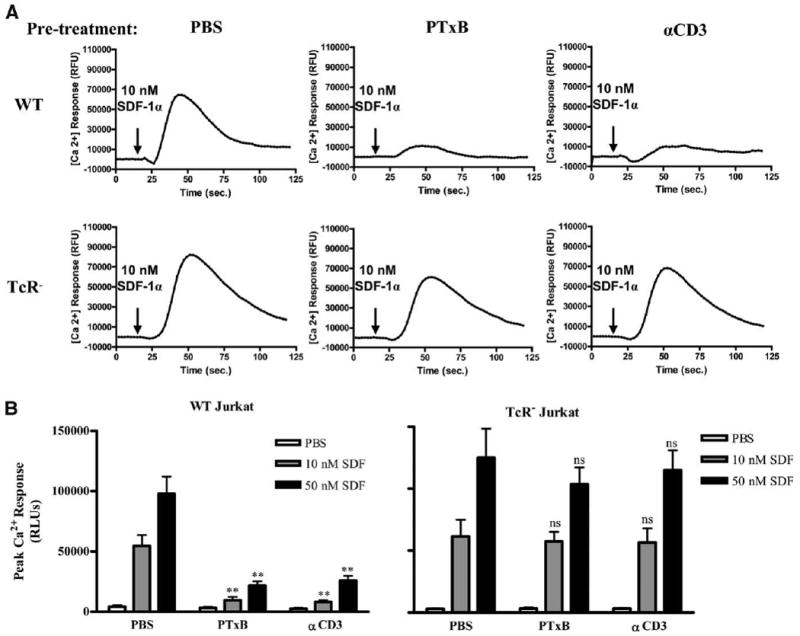
PTxB/TCR signaling blocks CXCR4-stimulated Gβγ activity. WT and TCR− Jurkat cells were loaded with the Fluo4 Ca2+ sensing dye (Invitrogen) and treated with PBS, 2 μg/ml PTxB, or 2 μg/ml anti-CD3 (αCD3) mAb for 30 min before stimulation with PBS or 10 or 50 nM SDF-1α. Data were collected for 2 min using a FlexStation II (Molecular Devices). Arrows indicate the time at which ligand was added (20 s). A, Representative time courses displaying relative Ca2+ fluorescence with 10 nM SDF-1 treatment. B, Average peak response of three independent experiments done in triplicate are graphed. **, p < 0.01 compared with their PBS pretreated match.
PTx/TCR signaling inhibits CXCR4-mediated chemotaxis
After demonstrating that PTxB induces CXCR4 internalization and phosphorylation and blocks signaling initiated by G proteins, we next sought to determine whether PTxB could block CXCR4 function at a biological level. We predicted that the decrease in surface CXCR4 expression and subsequent loss of signaling after PTxB or anti-CD3 mAb treatment would cause a block in CXCR4-mediated cell migration to SDF-1α. To test this hypothesis, PTxB or anti-CD3 mAb was added to WT and TCR− Jurkat cells 15 min before they were analyzed for their ability to migrate from the top chamber of an 8-μm transwell to the bottom chamber containing 10 nM SDF-1α. Since PKC phosphorylation of CXCR4 results in desensitization and internalization of CXCR4, we also assessed the effects of PMA pretreatment on migration to SDF-1α. Data in Fig. 6A show that WT cells pretreated with PTxB display a 75% decrease in migration to SDF-1α, while cells treated with anti-CD3 mAb display a 40% decrease in migration.
Figure 6.
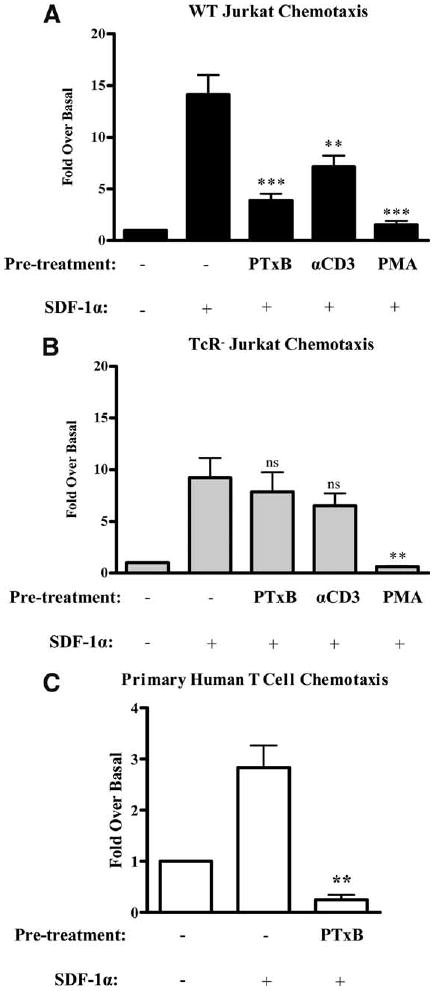
PTxB activates a TCR-dependent signaling pathway to prevent CXCR4-mediated chemotaxis. A, WT and (B) TCR− Jurkat cells were left untreated or treated with 2 μg/ml PTxB, 2 μg/ml anti-CD3 (αCD3) mAb, or 25 ng/ml PMA for 15 min before being placed into the upper well of 8-μm-pore transwell chambers. The transwell chambers were assembled such that the bottom wells contained either media alone or media supplemented with 10 nM SDF-1α. Cells were then allowed to migrate for 4 h, removed from the bottom of the transwell chamber, and counted using a flow cytometer. ***, p < 0.001 or **, p < 0.01 when pretreated cells were compared with untreated cells migrating to SDF-1α (n = 15 for WT Jurkat cells and n = 12 for TCR− Jurkat cells). C, Human primary T cells were treated with 4 μg/ml PTxB for 2 h, diluted, and placed into the upper well of 5-μm-pore transwell chambers. The transwell chambers were assembled such that the bottom wells contained either media alone or media supplemented with 50 nM SDF-1α. Cells were then allowed to migrate for 4 h, removed from the bottom of the transwell chamber, and counted using a flow cytometer. ***, p < 0.001 or **, p < 0.01 when pretreated cells were compared with untreated cells migrating to SDF-1α (n = 3).
In contrast to WT cells, TCR− cells showed no significant decrease in CXCR4-mediated migration with either PTxB or anti-CD3 mAb pretreatments (Fig. 6B). Note that TCR− Jurkat cells exhibit a slightly reduced overall capacity to chemotax to SDF-1α as previously reported (19, 20, 22, 27). Additionally, cells pretreated with PMA show a complete block on CXCR4-mediated migration in both WT and TCR− cells. This indicates that TCR− cells have a fully functional PKC pathway that is able to induce CXCR4 internalization (Fig. 2) and block migration.
To verify that the effects of PTx on T cell chemotaxis also occur in normal T cells, we examined CXCR4-mediated chemotaxis in primary human T cells pretreated with PTxB. T cells were isolated from healthy donors and cultured for 24 h. Cells were then pretreated PTxB for 2 h before they were assayed for their ability to chemotax to 50 nM SDF-1α. Similar to WT Jurkat cells, PTxB blocks the ability of primary T cells to chemotax to SDF-1α, as shown in Fig. 6C. Collectively, Fig. 6 reveals that PTx, through its B subunit, uses a TCR signaling pathway to efficiently block T cell chemotaxis to SDF-1α.
Lastly, we wanted to further characterize the timing and duration of PTxB-mediated cross-desensitization of CXCR4. Since it is known that the A subunit of PTx inactivates chemokine receptor signaling several hours after toxin exposure, it is important to establish the time frame in which the effects of PTxB are observed relative to that of the A subunit (32, 33). To test this, WT or TCR− cells were pretreated with PTx holotoxin (PTxWT) or a PTx enzymatically inactive mutant (PTx9K/129G) for various times before analyzing their ability ability to chemotax to SDF-1α. In Fig. 7A, WT cells were pretreated with PTxWT and PTx9K/129G for 15 min before assaying their migration to SDF-1α. The data show that WT cells pretreated for 15 min with PTxWT and PTx9K/129G have a substantially decreased chemotactic capability, similar to results for 15-min PTxB pretreatments shown in Fig. 6. Additionally, PTxWT pretreated cells in Fig. 7A show a statistically significant, but slightly increased, inhibition of chemotactic capability compared with PTx9K/129G pretreated cells, demonstrating that the A subunit effects can be detected at this early time point, but that the short-term block on migration is predominantly mediated by PTxB. In Fig. 7B, WT cells were pretreated for 18 h with either toxin before analyzing their ability to migrate to SDF-1α. Fig. 7B indicates that after 18 h of pretreatment with PTx9K/129G, WT cells are able to migrate to SDF-1α as effectively as untreated cells. In contrast, WT cells pretreated for the same time with PTxWT completely lack the ability to migrate. Of note, the magnitude of SDF-1α-mediated chemotaxis in control untreated cells throughout Fig. 7 is slightly higher than in earlier experiments (∼30-fold vs ∼18 fold), as the experimental conditions to allow for analyses of PTx effects at the 18 h time point dictated that the cell density be higher during the time frame preceding the start of these experiments. These data illustrate that TCR-mediated CXCR4 desensitization is transient when compared with G protein inactivation by the PTx holotoxin.
Figure 7.
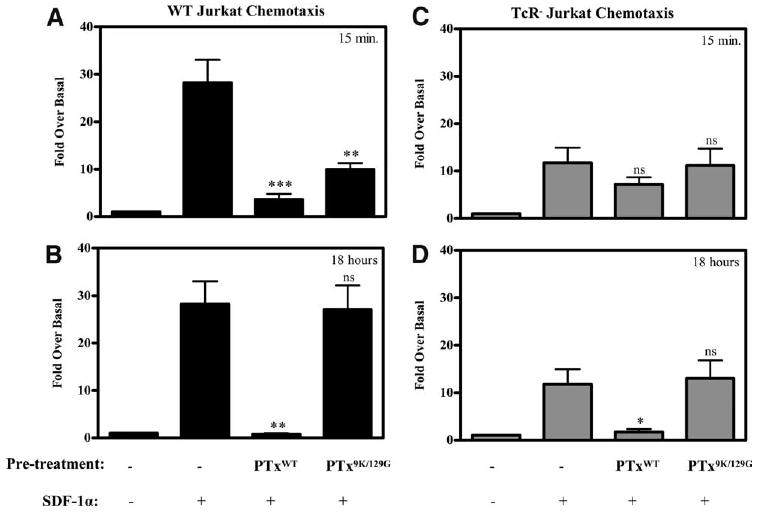
The effects of PTxB are transient in comparison to PTx holotoxin. A and B, WT and (C and D) TCR− Jurkat cells were left untreated or treated with 2 μg/ml PTxWT or 2 μg/ml PTx9K/129G (enzymatic dead mutant) for 15 min or 18 h before placing them in the top chamber of an 8-μm transwell. The top chamber was then inserted into a bottom chamber containing media alone or supplemented with 10 nM SDF-1α. Cells were allowed to migrate for 4 h before they were removed from the bottom and counted using a flow cytometer. ***, p < 0.001; **, p < 0.01; and *, p < 0.05 when pretreated samples were compared with untreated cells migrating to SDF-1α (n = 4).
These same parameters were conducted with TCR− cells, and the data demonstrate that TCR− cells are again insensitive to the B subunit (PTx9K/129G) alone after 15-min and 18-h pretreatments (Fig. 7, C and D). In contrast to PTx9K/129G, pretreatment with PTxWT significantly blocks migration after 18 h, showing that the effects of the A subunit are not TCR-specific and that the TCR is not required for toxin access to the cytoplasm. These data show that while TCR− cells are not susceptible to the signaling effects initiated by the B subunit, the A subunit of PTx still inactivates G proteins regardless of TCR expression.
Discussion
In these studies we found that TCR signaling, initiated by PTx or anti-CD3 mAb, causes cross- or heterologous desensitization of CXCR4 (summarized in Fig. 8). Specifically, the data indicate that treatment of T cells with PTx decreases CXCR4 surface expression, blocks SDF-1α-induced G protein signaling, and prevents cellular migration. These observations are mediated by TCR signaling events, as the effects of PTx were defective in cells lacking a functional TCR signaling pathway. Furthermore, all PTx effects could be mimicked by stimulating cells with an anti-CD3 mAb. Although TCR signaling rapidly blocks CXCR4 activity, the data show that this block is transient and likely mediated by a decrease in CXCR4 cell surface expression and/or uncoupling from G proteins.
Figure 8.
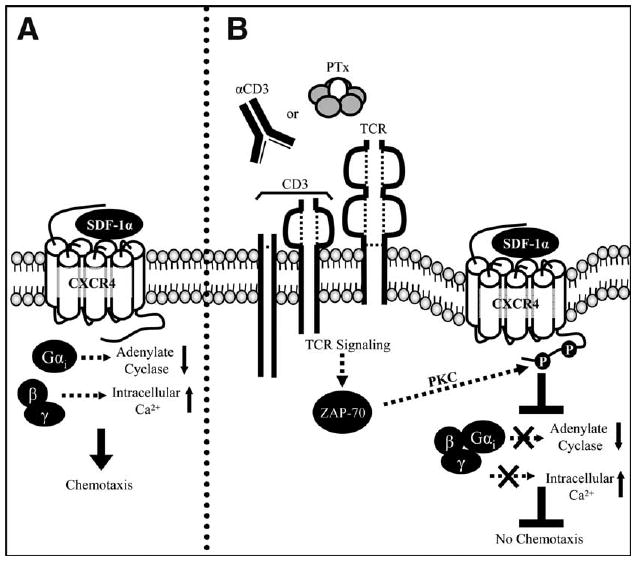
Schematic diagram depicting the mechanism of PTxB-mediated desensitization of CXCR4. A, SDF-1α binding to the extracellular domains of CXCR4 causes a conformational change in CXCR4, enabling activation of associated G proteins. Gαi proteins inhibit adenylate cyclase while the βγ proteins trigger an intracellular Ca2+ release. These events, in addition to others, result in cellular chemotaxis to SDF-1α. B, Treatment with the B subunit of PTx or an anti-CD3 (αCD3) mAb initiates the TCR signaling pathway resulting in the activation of many proteins, including a variety of kinases. We propose that kinases (PKCs or GRKs) activated by the TCR can phosphorylate CXCR4 to promote its internalization and uncoupling from G proteins. Subsequently, the decreased availability of CXCR4 on the cell surface and/or is inability to couple to G proteins results in CXCR4 that cannot respond to SDF-1α. Thus, TCR signaling results in a rapid and transient block of chemotaxis to SDF-1α.
While PTx has been known to prevent chemokine receptor signaling through the enzymatic activity of the A subunit, our present studies reveal a novel mechanism in which PTx modulates the immune system through the function of the B subunit (32, 33). One mechanism is localized to the PTx A subunit and is mediated by enzymatic inactivation of G proteins, while the other is localized to the PTx B subunit and is mediated by the interaction with the TCR. This indicates that Bordetella pertussis has developed two distinct mechanisms to block chemotactic responses. These two mechanisms vary in speed, duration, and toxin dose. The B subunit interacts with extracellular receptors, such as the TCR, to produce rapid effects on CXCR4 that can be detected within 10 min of treatment (30, 53, 54). These effects fade and CXCR4 function is regained within 18 h. The transient nature of this PTx effect could be due to the depletion of toxin from the extracellular space or to desensitization of the PTxB receptor (i.e., TCR) itself. Thus far, the block of CXCR4 activity is only attributed to T cells, but other cell types may ultimately also be found to be susceptible to PTxB. In contrast to the rapid and transient effect of PTxB, the A subunit must gain access to the cell's cytoplasm to inactivate G proteins, a process that can take hours, but permanently modifies G proteins (33). The activity of the A subunit is not cell type specific and requires lower concentrations of toxin. The multifaceted approach utilized by the A and B subunits of PTx to inhibit chemotaxis suggests that altering immune cell signaling is critical for a successful and productive B. pertussis infection (55).
Throughout these studies we have used PTx to study receptor crosstalk between the TCR and CXCR4. By exploiting PTx as a reagent to study TCR signaling, we have uncovered a novel cellular mechanism to inhibit chemotaxis. Although several studies have shown a general link between TCR signaling and chemotaxis, this report is the first to show that TCR signaling, activated by anti-CD3 mAb, directly regulates chemokine receptor signaling by physically altering CXCR4. The ability of the TCR to control CXCR4 is a mechanism that likely evolved as a means to prevent newly activated T cells, engaged by MHC presenting Ag, from inappropriately migrating away from the site of an immune challenge. A similar mechanism has been reported in B cells, as BCR engagement also results in a block of CXCR4 signaling (10). Although in the present report we focused on interactions between the TCR and CXCR4, it is possible that the TCR can cross-regulate a variety of chemokine receptors.
Another chemokine receptor that is likely regulated by PTx and TCR signaling is CCR5 (31). CCR5, like CXCR4, is an HIV coreceptor (31). Previous studies have shown that primary lymphocyte populations treated with PTxB are unable to respond to the CCR5 ligand, MIP-1β, and cannot support R5 coreceptor-mediated HIV entry (31, 56–58). Whether there are extensive similarities in the mechanisms by which PTxB affects CXCR4 and CCR5 remains to be explored. Interestingly, in these earlier studies CXCR4 signaling or CXCR4 coreceptor utilization was not affected by PTxB pretreatment (31). Such results could differ from the results presented herein because the earlier study used a mixed lymphocyte population, and in this report we use isolated primary T cells and clonal T cell lines. Since other cells, such as B cells and monocytes, can also express CXCR4, the non-T cells present in the mixed lymphocyte population could mask any effects mediated by the TCR in the T cell subpopulation.
Mechanistically, the TCR appears to promote CXCR4 phosphorylation and subsequent desensitization by activating a protein kinase cascade involving ZAP70 and PKC. The protein kinases responsible for directly phosphorylating CXCR4 remain unclear, but our studies point to the PKC family as a likely candidate. There are 18 possible phosphorylation sites in the cytoplasmic tail of CXCR4, many of which have been shown to be important for CXCR4 internalization in response to PKC activators (10, 51). These phosphorylation sites make CXCR4 a potential substrate for PKCs activated by the TCR. Both PKCθ and PKCα are activated by TCR signaling and are shown to phosphorylate CD3, thereby exposing a special dileucine motif critical for TCR internalization (59–61). Interestingly, CXCR4 also contains a dileucine motif similar to that found in the intracellular domains of CD3 (14). Since CXCR4 is physically associated with the TCR, PKCs activated by the TCR could also phosphorylate CXCR4, thus exposing the dileucine motif and promoting internalization. In support of this idea, studies show that PKC activation and the CXCR4 dileucine motif are both required for the cross-desensitization of CXCR4 by the BCR (10). An additional candidate that could mediate TCR and CXCR4 crosstalk is GRK2. GRK2 is known to be a major contributor to homologous desensitization of many chemokine receptors, including CXCR4, by inducing phosphorylation and internalization (51). Intriguingly, GRK2 has also recently been found to be associated with the TCR complex (26). If the function of GRK2 is modulated following activation of the TCR signaling pathway, it could be one of the kinases responsible for mediating CXCR4 internalization. Additionally, it seems likely that PTxB through the activity of PKC and/or GRK2 could regulate a wide range of chemokine receptors in addition to CXCR4. It will be interesting to explore the effects of PTxB on other GPCRs and to determine what effect this has on T cell biology.
Throughout this report, our data show that the decrease of CXCR4 signaling is of a greater magnitude than the decrease in cell surface CXCR4 at comparable treatments and time points. For example, while PTxB treatments cause a 75% decease in chemotaxis and 90% decrease in Gαi signaling, CXCR4 surface expression decreases only ∼40–50%. There are two hypotheses to explain how a 50% loss in receptor from the cell surface could result in a 75–90% loss in function. Since CXCR4 is thought to act as dimer when signaling, one hypothesis could be that removing half of the total CXCR4 from the surface could drop the receptor concentration below a critical threshold required for signaling (62, 63). As a result, the 50% CXCR4 reduction would lead to a more severe phenotypic effect on CXCR4 signaling in which each activated receptor can no longer find a partner receptor to effectively transmit signals. An alternative hypothesis is that although 50% of the CXCR4 remains on the cell surface, it is not able to respond to SDF-1α because it is phosphorylated and/or uncoupled from G proteins. This CXCR4 would be detected by FACS analysis because it has not yet been internalized, but no signaling would be detected because it is locked in an inactive conformation.
Future studies are required to further refine the mechanisms and intracellular signaling pathways utilized by PTxB and the TCR to modify CXCR4 activity. A better understanding of the mechanism by which PTx and TCR-mediated CXCR4 cross-desensitization occurs is also needed to clarify the differences, if any, between traditional TCR signaling and that initiated by PTx. Finally, it will be interesting to determine how PTxB-mediated cross-regulation of CXCR4 participates in B. pertussis pathogenesis and to determine whether other pathogens similarly regulate chemotactic proteins such as CXCR4 during infection.
Acknowledgments
We thank Arthur Weiss for providing cell lines and reagents, Dennis McGraw for providing us with access to the Molecular Dynamics FlexStation, and John Monaco, David Freidmann, and Joseph Sherrill for critical review of the manuscript. We also thank all friends, family, and members of the department that served as our healthy blood donors and Scott Millen for his frightening, but accurate ability to draw blood.
Footnotes
This work was supported by National Institutes of Health Grant R56 AI023695 (to A.A.W. and W.E.M.) and by start-up funds from the University of Cincinnati (to W.E.M.). A.A.W. acknowledges support from the Epidemiology and Surveillance Division in the National Immunization Program at the Centers for Disease Control and Prevention. O.D.S. was supported by a National Institutes of Health Training Grant T32 AI055406.
Abbreviations used in this paper: SDF-1α, stromal cell-derived factor 1α; GRK2, G protein-coupled receptor kinase 2; PKC, protein kinase C; PTx, pertussis toxin; WT, wild type.
Disclosures: The authors have no financial conflicts of interest.
References
- 1.Hesselgesser J, Liang M, Hoxie J, Greenberg M, Brass LF, Orsini MJ, Taub D, Horuk R. Identification and characterization of the CXCR4 chemokine receptor in human T cell lines: ligand binding, biological activity, and HIV-1 infectivity. J Immunol. 1998;160:877–883. [PubMed] [Google Scholar]
- 2.Bleul CC, Wu L, Hoxie JA, Springer TA, Mackay CR. The HIV coreceptors CXCR4 and CCR5 are differentially expressed and regulated on human T lymphocytes. Proc Natl Acad Sci USA. 1997;94:1925–1930. doi: 10.1073/pnas.94.5.1925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhang L, He T, Talal A, Wang G, Frankel SS, Ho DD. In vivo distribution of the human immunodeficiency virus/simian immunodeficiency virus coreceptors: CXCR4, CCR3, and CCR5. J Virol. 1998;72:5035–5045. doi: 10.1128/jvi.72.6.5035-5045.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Durig J, Schmucker U, Duhrsen U. Differential expression of chemokine receptors in B cell malignancies. Leukemia. 2001;15:752–756. doi: 10.1038/sj.leu.2402107. [DOI] [PubMed] [Google Scholar]
- 5.D'Apuzzo M, Rolink A, Loetscher M, Hoxie JA, Clark-Lewis I, Melchers F, Baggiolini M, Moser B. The chemokine SDF-1, stromal cell-derived factor 1, attracts early stage B cell precursors via the chemokine receptor CXCR4. Eur J Immunol. 1997;27:1788–1793. doi: 10.1002/eji.1830270729. [DOI] [PubMed] [Google Scholar]
- 6.Okabe S, Fukuda S, Kim YJ, Niki M, Pelus LM, Ohyashiki K, Pandolfi PP, Broxmeyer HE. Stromal cell-derived factor-1α/CXCL12-induced chemotaxis of T cells involves activation of the RasGAP-associated docking protein p62Dok-1. Blood. 2005;105:474–480. doi: 10.1182/blood-2004-03-0843. [DOI] [PubMed] [Google Scholar]
- 7.Blades MC, Manzo A, Ingegnoli F, Taylor PR, Panayi GS, Irjala H, Jalkanen S, Haskard DO, Perretti M, Pitzalis C. Stromal cell-derived factor 1 (CXCL12) induces human cell migration into human lymph nodes transplanted into SCID mice. J Immunol. 2002;168:4308–4317. doi: 10.4049/jimmunol.168.9.4308. [DOI] [PubMed] [Google Scholar]
- 8.Van Drenth C, Jenkins A, Ledwich L, Ryan TC, Mashikian MV, Brazer W, Center DM, Cruikshank WW. Desensitization of CXC chemokine receptor 4, mediated by IL-16/CD4, is independent of p56lck enzymatic activity. J Immunol. 2000;165:6356–6363. doi: 10.4049/jimmunol.165.11.6356. [DOI] [PubMed] [Google Scholar]
- 9.Geay JF, Buet D, Zhang Y, Foudi A, Jarrier P, Berthebaud M, Turhan AG, Vainchenker W, Louache F. p210BCR-ABL inhibits SDF-1 chemotactic response via alteration of CXCR4 signaling and down-regulation of CXCR4 expression. Cancer Res. 2005;65:2676–2683. doi: 10.1158/0008-5472.CAN-04-2152. [DOI] [PubMed] [Google Scholar]
- 10.Guinamard R, Signoret N, Ishiai M, Marsh M, Kurosaki T, Ravetch JV. B cell antigen receptor engagement inhibits stromal cell-derived factor (SDF)-1α chemotaxis and promotes protein kinase C (PKC)-induced internalization of CXCR4. J Exp Med. 1999;189:1461–1466. doi: 10.1084/jem.189.9.1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Finley MJ, Chen X, Bardi G, Davey P, Geller EB, Zhang L, Adler MW, Rogers TJ. Bi-directional heterologous desensitization between the major HIV-1 co-receptor CXCR4 and the κ-opioid receptor. J Neuroimmunol. 2008;197:114–123. doi: 10.1016/j.jneuroim.2008.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ali H, Richardson RM, Haribabu B, Snyderman R. Chemoattractant receptor cross-desensitization. J Biol Chem. 1999;274:6027–6030. doi: 10.1074/jbc.274.10.6027. [DOI] [PubMed] [Google Scholar]
- 13.Haribabu B, Richardson RM, Fisher I, Sozzani S, Peiper SC, Horuk R, Ali H, Snyderman R. Regulation of human chemokine receptors CXCR4: role of phosphorylation in desensitization and internalization. J Biol Chem. 1997;272:28726–28731. doi: 10.1074/jbc.272.45.28726. [DOI] [PubMed] [Google Scholar]
- 14.Signoret N, Oldridge J, Pelchen-Matthews A, Klasse PJ, Tran T, Brass LF, Rosenkilde MM, Schwartz TW, Holmes W, Dallas W, et al. Phorbol esters and SDF-1 induce rapid endocytosis and down modulation of the chemokine receptor CXCR4. J Cell Biol. 1997;139:651–664. doi: 10.1083/jcb.139.3.651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Weiss A, Imboden J, Wiskocil R, Stobo J. The role of T3 in the activation of human T cells. J Clin Immunol. 1984;4:165–173. doi: 10.1007/BF00914963. [DOI] [PubMed] [Google Scholar]
- 16.Weiss A, Irving BA, Tan LK, Koretzky GA. Signal transduction by the T cell antigen receptor. Semin Immunol. 1991;3:313–324. [PubMed] [Google Scholar]
- 17.Patrussi L, Baldari CT. Intracellular mediators of CXCR4-dependent signaling in T cells. Immunol Lett. 2008;115:75–82. doi: 10.1016/j.imlet.2007.10.012. [DOI] [PubMed] [Google Scholar]
- 18.Dar WA, Knechtle SJ. CXCR3-mediated T-cell chemotaxis involves ZAP-70 and is regulated by signalling through the T-cell receptor. Immunology. 2007;120:467–485. doi: 10.1111/j.1365-2567.2006.02534.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fernandis AZ, Cherla RP, Ganju RK. Differential regulation of CXCR4-mediated T-cell chemotaxis and mitogen-activated protein kinase activation by the membrane tyrosine phosphatase, CD45. J Biol Chem. 2003;278:9536–9543. doi: 10.1074/jbc.M211803200. [DOI] [PubMed] [Google Scholar]
- 20.Inngjerdingen M, Torgersen KM, Maghazachi AA. Lck is required for stromal cell-derived factor 1α (CXCL12)-induced lymphoid cell chemotaxis. Blood. 2002;99:4318–4325. doi: 10.1182/blood.v99.12.4318. [DOI] [PubMed] [Google Scholar]
- 21.Patrussi L, Ulivieri C, Lucherini OM, Paccani SR, Gamberucci A, Lanfrancone L, Pelicci PG, Baldari CT. p52Shc is required for CXCR4-dependent signaling and chemotaxis in T cells. Blood. 2007;110:1730–1738. doi: 10.1182/blood-2007-01-068411. [DOI] [PubMed] [Google Scholar]
- 22.Peacock JW, Jirik FR. TCR activation inhibits chemotaxis toward stromal cell-derived factor-1: evidence for reciprocal regulation between CXCR4 and the TCR. J Immunol. 1999;162:215–223. [PubMed] [Google Scholar]
- 23.Ticchioni M, Charvet C, Noraz N, Lamy L, Steinberg M, Bernard A, Deckert M. Signaling through ZAP-70 is required for CXCL12-mediated T-cell transendothelial migration. Blood. 2002;99:3111–3118. doi: 10.1182/blood.v99.9.3111. [DOI] [PubMed] [Google Scholar]
- 24.Ottoson NC, Pribila JT, Chan AS, Shimizu Y. Cutting edge: T cell migration regulated by CXCR4 chemokine receptor signaling to ZAP-70 tyrosine kinase. J Immunol. 2001;167:1857–1861. doi: 10.4049/jimmunol.167.4.1857. [DOI] [PubMed] [Google Scholar]
- 25.Stanners J, Kabouridis PS, McGuire KL, Tsoukas CD. Interaction between G proteins and tyrosine kinases upon T cell receptor.CD3-mediated signaling. J Biol Chem. 1995;270:30635–30642. doi: 10.1074/jbc.270.51.30635. [DOI] [PubMed] [Google Scholar]
- 26.DeFord-Watts LM, Young JA, Pitcher LA, van Oers NS. The membrane-proximal portion of CD3 epsilon associates with the serine/threonine kinase GRK2. J Biol Chem. 2007;282:16126–16134. doi: 10.1074/jbc.M609418200. [DOI] [PubMed] [Google Scholar]
- 27.Kumar A, Humphreys TD, Kremer KN, Bramati PS, Bradfield L, Edgar CE, Hedin KE. CXCR4 physically associates with the T cell receptor to signal in T cells. Immunity. 2006;25:213–224. doi: 10.1016/j.immuni.2006.06.015. [DOI] [PubMed] [Google Scholar]
- 28.Schneider OD, Weiss AA, Miller WE. Pertussis toxin utilizes proximal components of the T-cell receptor complex to initiate signal transduction events in T cells. Infect Immun. 2007;75:4040–4049. doi: 10.1128/IAI.00414-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Witvliet MH, Vogel ML, Wiertz EJ, Poolman JT. Interaction of pertussis toxin with human T lymphocytes. Infect Immun. 1992;60:5085–5090. doi: 10.1128/iai.60.12.5085-5090.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wong WS, Luk JM. Signaling mechanisms of pertussis toxin-induced myelomonocytic cell adhesion: role of tyrosine phosphorylation. Biochem Biophys Res Commun. 1997;236:479–482. doi: 10.1006/bbrc.1997.6986. [DOI] [PubMed] [Google Scholar]
- 31.Alfano M, Schmidtmayerova H, Amella CA, Pushkarsky T, Bukrinsky M. The B-oligomer of pertussis toxin deactivates CC chemokine receptor 5 and blocks entry of M-tropic HIV-1 strains. J Exp Med. 1999;190:597–605. doi: 10.1084/jem.190.5.597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pizza M, Bartoloni A, Prugnola A, Silvestri S, Rappuoli R. Subunit S1 of pertussis toxin: mapping of the regions essential for ADP-ribosyltransferase activity. Proc Natl Acad Sci USA. 1988;85:7521–7525. doi: 10.1073/pnas.85.20.7521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Moss J, Bruni P, Hsia JA, Tsai SC, Watkins PA, Halpern JL, Burns DL, Kanaho Y, Chang PP, Hewlett EL, et al. Pertussis toxin-catalyzed ADP-ribosylation: effects on the coupling of inhibitory receptors to the adenylate cyclase system. J Recept Res. 1984;4:459–474. doi: 10.3109/10799898409042567. [DOI] [PubMed] [Google Scholar]
- 34.Nencioni L, Pizza MG, Volpini G, De Magistris MT, Giovannoni F, Rappuoli R. Properties of the B oligomer of pertussis toxin. Infect Immun. 1991;59:4732–4734. doi: 10.1128/iai.59.12.4732-4734.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hayashi K, Altman A. Protein kinase Cθ (PKCθ): a key player in T cell life and death. Pharmacol Res. 2007;55:537–544. doi: 10.1016/j.phrs.2007.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ehrmann IE, Weiss AA, Goodwin MS, Gray MC, Barry E, Hewlett EL. Enzymatic activity of adenylate cyclase toxin from Bordetella pertussis is not required for hemolysis. FEBS Lett. 1992;304:51–56. doi: 10.1016/0014-5793(92)80587-7. [DOI] [PubMed] [Google Scholar]
- 37.Tonon S, Badran B, Benghiat FS, Goriely S, Flamand V, Willard-Gallo K, Willems F, Goldman M, De Wit D. Pertussis toxin activates adult and neonatal naive human CD4+ T lymphocytes. Eur J Immunol. 2006;36:1794–1804. doi: 10.1002/eji.200535697. [DOI] [PubMed] [Google Scholar]
- 38.Denkinger CM, Denkinger MD, Forsthuber TG. Pertussis toxin-induced cytokine differentiation and clonal expansion of T cells is mediated predominantly via costimulation. Cell Immunol. 2007;246:46–54. doi: 10.1016/j.cellimm.2007.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Latif R, Kerlero de Rosbo N, Amarant T, Rappuoli R, Sappler G, Ben-Nun A. Reversal of the CD4+/CD8+ T-cell ratio in lymph node cells upon in vitro mitogenic stimulation by highly purified, water-soluble S3–S4 dimer of pertussis toxin. Infect Immun. 2001;69:3073–3081. doi: 10.1128/IAI.69.5.3073-3081.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Thom RE, Casnellie JE. Pertussis toxin activates protein kinase C and a tyrosine protein kinase in the human T cell line Jurkat. FEBS Lett. 1989;244:181–184. doi: 10.1016/0014-5793(89)81188-3. [DOI] [PubMed] [Google Scholar]
- 41.Gray LS, Huber KS, Gray MC, Hewlett EL, Engelhard VH. Pertussis toxin effects on T lymphocytes are mediated through CD3 and not by pertussis toxin catalyzed modification of a G protein. J Immunol. 1989;142:1631–1638. [PubMed] [Google Scholar]
- 42.Rambow-Larsen AA, Weiss AA. Temporal expression of pertussis toxin and Ptl secretion proteins by Bordetella pertussis. J Bacteriol. 2004;186:43–50. doi: 10.1128/JB.186.1.43-50.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Molon B, Gri G, Bettella M, Gomez-Mouton C, Lanzavecchia A, Martinez AC, Manes S, Viola A. T cell costimulation by chemokine receptors. Nat Immunol. 2005;6:465–471. doi: 10.1038/ni1191. [DOI] [PubMed] [Google Scholar]
- 44.Nanki T, Lipsky PE. Cutting edge: stromal cell-derived factor-1 is a costimulator for CD4+ T cell activation. J Immunol. 2000;164:5010–5014. doi: 10.4049/jimmunol.164.10.5010. [DOI] [PubMed] [Google Scholar]
- 45.Abraham RT, Weiss A. Jurkat T cells and development of the T-cell receptor signalling paradigm. Nat Rev Immunol. 2004;4:301–308. doi: 10.1038/nri1330. [DOI] [PubMed] [Google Scholar]
- 46.Springael C, Thomas S, Rahmouni S, Vandamme A, Goldman M, Willems F, Vosters O. Rottlerin inhibits human T cell responses. Biochem Pharmacol. 2007;73:515–525. doi: 10.1016/j.bcp.2006.10.034. [DOI] [PubMed] [Google Scholar]
- 47.Yamashita Y, Hasegawa-Sasaki H, Sasaki T. Suppression by staurosporine of Ca2+-mobilization triggered by ligation of antigen-specific receptors on T and B lymphocytes: an essential role of protein tyrosine kinase in the signal transduction. FEBS Lett. 1991;288:46–50. doi: 10.1016/0014-5793(91)81000-x. [DOI] [PubMed] [Google Scholar]
- 48.Chen WJ, Jayawickreme C, Watson C, Wolfe L, Holmes W, Ferris R, Armour S, Dallas W, Chen G, Boone L, et al. Recombinant human CXC-chemokine receptor-4 in melanophores are linked to Gi protein: seven transmembrane coreceptors for human immunodeficiency virus entry into cells. Mol Pharmacol. 1998;53:177–181. doi: 10.1124/mol.53.2.177. [DOI] [PubMed] [Google Scholar]
- 49.Ganju RK, Brubaker SA, Meyer J, Dutt P, Yang Y, Qin S, Newman W, Groopman JE. The α-chemokine, stromal cell-derived factor-1α, binds to the transmembrane G-protein-coupled CXCR-4 receptor and activates multiple signal transduction pathways. J Biol Chem. 1998;273:23169–23175. doi: 10.1074/jbc.273.36.23169. [DOI] [PubMed] [Google Scholar]
- 50.Haribabu B, Zhelev DV, Pridgen BC, Richardson RM, Ali H, Snyderman R. Chemoattractant receptors activate distinct pathways for chemotaxis and secretion: role of G-protein usage. J Biol Chem. 1999;274:37087–37092. doi: 10.1074/jbc.274.52.37087. [DOI] [PubMed] [Google Scholar]
- 51.Orsini MJ, Parent JL, Mundell SJ, Marchese A, Benovic JL. Trafficking of the HIV coreceptor CXCR4: role of arrestins and identification of residues in the C-terminal tail that mediate receptor internalization. J Biol Chem. 1999;274:31076–31086. doi: 10.1074/jbc.274.43.31076. [DOI] [PubMed] [Google Scholar]
- 52.Oberlin E, Amara A, Bachelerie F, Bessia C, Virelizier JL, Arenzana-Seisdedos F, Schwartz O, Heard JM, Clark-Lewis I, Legler DF, et al. The CXC chemokine SDF-1 is the ligand for LESTR/fusin and prevents infection by T-cell-line-adapted HIV-1. Nature. 1996;382:833–835. doi: 10.1038/382833a0. [DOI] [PubMed] [Google Scholar]
- 53.Thomazzi SM, Souza MH, Melo-Filho AA, Hewlett EL, Lima AA, Ribeiro RA. Pertussis toxin from Bordetella pertussis blocks neutrophil migration and neutrophil-dependent edema in response to inflammation. Braz J Med Biol Res. 1995;28:120–124. [PubMed] [Google Scholar]
- 54.Alfano M, Grivel JC, Ghezzi S, Corti D, Trimarchi M, Poli G, Margolis L. Pertussis toxin B-oligomer dissociates T cell activation and HIV replication in CD4 T cells released from infected lymphoid tissue. AIDS. 2005;19:1007–1014. doi: 10.1097/01.aids.0000174446.40379.3b. [DOI] [PubMed] [Google Scholar]
- 55.Paccani SR, Dal Molin F, Benagiano M, Ladant D, D'Elios MM, Montecucco C, Baldari CT. Suppression of T-lymphocyte activation and chemotaxis by the adenylate cyclase toxin of Bordetella pertussis. Infect Immun. 2008;76:2822–2832. doi: 10.1128/IAI.00200-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Alfano M, Pushkarsky T, Poli G, Bukrinsky M. The B-oligomer of pertussis toxin inhibits human immunodeficiency virus type 1 replication at multiple stages. J Virol. 2000;74:8767–8770. doi: 10.1128/jvi.74.18.8767-8770.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lapenta C, Spada M, Santini SM, Racca S, Dorigatti F, Poli G, Belardelli F, Alfano M. Pertussis toxin B-oligomer inhibits HIV infection and replication in hu-PBL-SCID mice. Int Immunol. 2005;17:469–475. doi: 10.1093/intimm/dxh226. [DOI] [PubMed] [Google Scholar]
- 58.Alfano M, Vallanti G, Biswas P, Bovolenta C, Vicenzi E, Mantelli B, Pushkarsky T, Rappuoli R, Lazzarin A, Bukrinsky M, Poli G. The binding subunit of pertussis toxin inhibits HIV replication in human macrophages and virus expression in chronically infected promonocytic U1 cells. J Immunol. 2001;166:1863–1870. doi: 10.4049/jimmunol.166.3.1863. [DOI] [PubMed] [Google Scholar]
- 59.von Essen M, Menne C, Nielsen BL, Lauritsen JP, Dietrich J, Andersen PS, Karjalainen K, Odum N, Geisler C. The CD3γ leucine-based receptor-sorting motif is required for efficient ligand-mediated TCR down-regulation. J Immunol. 2002;168:4519–4523. doi: 10.4049/jimmunol.168.9.4519. [DOI] [PubMed] [Google Scholar]
- 60.Geisler C, Dietrich J, Nielsen BL, Kastrup J, Lauritsen JP, Odum N, Christensen MD. Leucine-based receptor sorting motifs are dependent on the spacing relative to the plasma membrane. J Biol Chem. 1998;273:21316–21323. doi: 10.1074/jbc.273.33.21316. [DOI] [PubMed] [Google Scholar]
- 61.von Essen M, Nielsen MW, Bonefeld CM, Boding L, Larsen JM, Leitges M, Baier G, Odum N, Geisler C. Protein kinase C (PKC)α and PKCθ are the major PKC isotypes involved in TCR down-regulation. J Immunol. 2006;176:7502–7510. doi: 10.4049/jimmunol.176.12.7502. [DOI] [PubMed] [Google Scholar]
- 62.Lagane B, Chow KY, Balabanian K, Levoye A, Harriague J, Planchenault T, Baleux F, Gunera-Saad N, Arenzana-Seisdedos F, Bachelerie F. CXCR4 dimerization and β-arrestin-mediated signaling account for the enhanced chemotaxis to CXCL12 in WHIM syndrome. Blood. 2008;112:34–44. doi: 10.1182/blood-2007-07-102103. [DOI] [PubMed] [Google Scholar]
- 63.Vila-Coro AJ, Rodriguez-Frade JM, Martin De Ana A, Moreno-Ortiz MC, Martinez AC, Mellado M. The chemokine SDF-1α triggers CXCR4 receptor dimerization and activates the JAK/STAT pathway. FASEB J. 1999;13:1699–1710. [PubMed] [Google Scholar]
- 64.Zocchi MR, Contini P, Alfano M, Poggi A. Pertussis toxin (PTX) B subunit and the nontoxic PTX mutant PT9K/129G inhibit Tat-induced TGF-β production by NK cells and TGF-β-mediated NK cell apoptosis. J Immunol. 2005;174:6054–6061. doi: 10.4049/jimmunol.174.10.6054. [DOI] [PubMed] [Google Scholar]
- 65.Pizza M, Covacci A, Bartoloni A, Perugini M, Nencioni L, De Magistris MT, Villa L, Nucci D, Manetti R, Bugnoli M, et al. Mutants of pertussis toxin suitable for vaccine development. Science. 1989;246:497–500. doi: 10.1126/science.2683073. [DOI] [PubMed] [Google Scholar]
- 66.Hewlett EL, Sauer KT, Myers GA, Cowell JL, Guerrant RL. Induction of a novel morphological response in Chinese hamster ovary cells by pertussis toxin. Infect Immun. 1983;40:1198–1203. doi: 10.1128/iai.40.3.1198-1203.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]