

Abstract
Background
Monocyte chemoattractant proteins (MCPs) play an important role in mediating inflammatory processes. Hypertension (HTN) is associated with inflammation as well as impaired cardiac microcirculatory function and structure, but the contribution of MCPs to these alterations remained unclear. This study tested the hypothesis that MCPs regulate cardiac microvascular function and structure in an experimental HTN.
Methods and Results
Pigs (n=6/group) were studied after 10 weeks of normal, renovascular HTN, or renovascular HTN+ bindarit (MCPs inhibitor, 50 mg/kg/day PO). Left ventricular (LV) function, myocardial microvascular permeability, and fractional vascular volume were assessed by fast computed tomography before and after adenosine infusion (400 μg/kg/min). Myocardial fibrosis, inflammation, and microvascular remodeling were determined ex-vivo. Hypertension was not altered by bindarit, but LV hypertrophy and diastolic function were improved. In response to adenosine, myocardial microvascular permeability increased in HTN (from 0.0083±0.0009 to 0.0103±0.0011 AU, p=0.038 vs. baseline) and fractional vascular volume decreased, while both remained unchanged in normal and HTN+bindarit pigs. HTN upregulated endothelin-1 expression, myocardial inflammation and microvascular wall thickening, which were inhibited by bindarit.
Conclusions
MCPs partly mediate myocardial inflammation, fibrosis, vascular remodeling, and impaired vascular integrity induced by hypertension. Inhibition of MCPs could potentially be a therapeutic target in hypertensive cardiomyopathy.
Keywords: MCPs, inflammation, hypertension, microvascular permeability, remodeling
Hypertension (HTN) is a leading cause of congestive heart failure in the United States and impairs left ventricular (LV) function, myocardial perfusion, and microvascular function.1, 2 Myocardial microvascular dysfunction is an important modulator of coronary resistance and myocardial blood flow,3 and often associated with changes in microvascular architecture like rarefaction or thickening.4
Important attributes of microvascular function include microvascular permeability (MP) and fractional vascular volume (FVV), key indices of microvascular structural integrity. MP, the rate of leakage of plasma components to the extravascular tissue, reflects endothelial barrier function.5, 6 FVV, the volume of myocardium occupied by microvessels, represents the number and tone of functional microvessels and myocardial perfusion. 7, 8 MP and FVV are difficult to assess in-vivo, but can be accurately and non-invasively assessed by fast CT.2, 7
The mechanism by which HTN leads to cardiomyopathy may involve inflammation. Perivascular inflammation of intramyocardial arteries is an early response to pressure overload, including induction of monocyte chemoattractant proteins (MCPs) and macrophage infiltration, in particular MCP-1.9, 10 The expression of MCP-1 and its receptor (CCR2) in HTN is regulated by mechanical strain and by release of reactive oxygen species. Furthermore, MCPs mediate vascular inflammation and remodeling by facilitating the secretion of vasoconstrictors like endothelin (ET)-1,11 cytokines and chemokines, and may contribute to vascular endothelial dysfunction.12,13
However, the role of MCPs in HTN-induced alterations of myocardial microvascular structure and function are poorly understood. The current study was designed to test the hypothesis that MCPs contribute to the impairment of the myocardial microcirculation, and their blockade would improve myocardial microvascular structure and function in swine renovascular HTN.
Methods
Animal procedures were approved by the Institutional Animal Care and Use Committee. Female domestic pigs (initially weighing 25-35kg) were randomized into three groups: normal (n=6), renovascular HTN (n=6), and HTN pigs supplemented with bindarit (Angelini Research Center - ACRAF, Italy, 50 mg/kg/day P.O., n=6). Bindarit (2-Methyl-2-[[1-(phenylmethyl)-1H-indazol-3yl]methoxy]propanoic acid) is a specific inhibitor of MCPs 1, 2, and 3 synthesis,14, 15 and at this dose inhibits MCPs' synthesis and inflammation.16 Renovascular HTN was elicited by induction of renal artery stenosis, which increases arterial pressure within 7-10 days.17, 18 Mean arterial pressure (MAP) was measured by a PhysioTel® telemetry system (Data Sciences) implanted at baseline in the left femoral artery.
After 10 weeks, pigs were anesthetized (ketamine 15.7 mg/kg/h and xylazine 2.3 mg/kg/h in saline), intubated, and ventilated. A pigtail catheter in the right atrium served for contrast media injection, and a side-arm for adenosine infusion. Blood samples were collected for measurement of plasma renin activity (PRA), aldosterone, and ET-1 by enzyme immunoassay.19 Fast CT studies were then performed to assess cardiac function and structure in-vivo, MP and FVV (before and after adenosine), LV filling rate, and LV muscle mass (LVMM). A few days later the pigs were euthanized using pentobarbital (100mg/kg), and hearts harvested for in-vitro studies. LV myocardial segments were fresh-frozen or preserved in formalin, and another segment prepared for micro-CT studies. Microvascular architecture was assessed by evaluation of microvascular density and wall thickness, inflammation by the expression of MCP-1, its receptor CCR2, cyclooxygenase (COX)-1, COX-2, IL-6, and macrophage infiltration, and cardiac remodeling by myocardial expression of the angiotensin II receptor type I (AT1R), ET-1, interstitial fibrosis, and myocyte hypertrophy. Vascular integrity was also evaluated by Rho-kinase (ROCK) activity (tested by its downstream product phospho-myosin phosphatase) and tight junction protein expression. Since HTN and inflammation can increase oxidative stress, dihydroethidium (DHE) staining for superoxide production and the expression of the p47 and p67 subunits of NAD(P)H oxidase were assessed20. To evaluate direct effects of MCP-1, cell culture studies were performed using human cardiac fibroblasts (HCF) incubated with MCP-1 (For detail, see the online-only Data Supplement).
In-vivo CT Studies
Pigs were scanned by either electron beam (C-150, Imatron, South San Francisco, CA) or 64-slice multidetector (Somatom Sensation-64, Siemens, Forchheim, Germany) fast CT, which provide very similar assessments of MP and FVV7, and images analyzed with ANALYZE® (Biomedical Imaging Resource, Mayo Clinic, Rochester, MN) (online Data-Supplement).
In-vitro Studies (For detail, see the online-only Data Supplement)
Micro-CT was used to evaluate myocardial microvascular density.21, 22 Myocardial remodeling was evaluated by myocyte cross-sectional areas, and fibrosis with H&E and trichrome staining. Microvascular remodeling was assessed by microvascular wall thickness to lumen ratio using anti-human α-smooth muscle actin (SMA) (DakoCytomation) staining. Immunohistochemistry assessed indices of inflammation and MP, with primary antibodies against MCP-1, macrophage CD163, zonula occludens-1 (ZO-1), and phospho-myosin phosphatase targeting subunit (Thr 696-pMYPT1). The tight junction protein ZO-1 regulates endothelial barrier function and overexpresses in response to strain,23 while phospho-MYPT1 reflects ROCK activity, which also regulates endothelial barrier function by inactivation of Myosin Phosphatase.24 Western Blotting was used for detecting MCP-1, CCR2, IL-6, COX1, COX-2, AT1R, ET-1, endothelial nitric oxide synthase (eNOS), p47 and p67, and ZO-1. GAPDH was used as loading control.
Statistical Analysis
Results are presented as mean±SEM. One-way ANOVA with the Bonferroni correction evaluated differences among the groups followed by an unpaired t-test, and paired Student t tests detected changes within groups; p<0.05 was considered significant.
Results
Cardiac and Microvascular Function
After a 10-week observation MAP was similarly increased in HTN and HTN+bindarit compared to normal (Table 1, ANOVA p=0.01), indicating that bindarit did not influence blood pressure. PRA was not significantly different among the groups, while aldosterone levels were elevated in HTN (ANOVA p=0.03) and unaltered by bindarit (p=0.17 vs. HTN+bindarit), suggesting no significant effect on the systemic renin-angiotensin- aldosterone system. In response to adenosine, MAP significantly decreased in all groups (p<0.05, Table 1), while heart rate remained unchanged. The degree of MAP response was greater in HTN+bindarit compared to normal (p=0.001), but not different from HTN (p=0.68, Table 1). LVMM increased in HTN (p=0.0001 vs. normal), suggesting LV hypertrophy, which was significantly limited (p=0.01 vs. HTN) but not abolished by bindarit. There were no significant differences in stroke volume, ejection fraction, and cardiac output among the groups (p>0.05 for all), suggesting sustained cardiac systolic function. E/A ratio significantly decreased in HTN compared to normal (p=0.0006, Figure 1A), suggesting diastolic dysfunction. Importantly, E/A was increased in HTN+bindarit compared to HTN (Figure 1A, p=0.02), indicating that MCPs inhibition protected cardiac diastolic function, and was not significantly lower than normal (p=0.06).
Table 1.
Systemic and cardiac function and myocardial microvascular density (mean±SEM) assessed by micro-CT in normal, hypertension (HTN), and HTN+bindarit pigs.
| Normal n=6 |
HTN n=6 |
HTN+bindarit n=6 |
|
|---|---|---|---|
| Mean arterial pressure (mmHg) | 101±4 | 120±5* | 124±9* |
| Change post-adenosine (%) | -6±1# | -14±5# | -17±2*# |
| Heart Rate (bpm) | 75±7 | 67±4 | 74±3 |
| Plasma Renin Activity (ng/ml/hour) | 0.17±0.03 | 0.17±0.02 | 0.16±0.06 |
| Systemic Aldosterone (pg/ml) | 684±68 | 1194±156* | 971±139* |
| Endothelin-1 (pg/ml) | 5.6±0.2 | 7.9±0.8* | 5.9±0.3† |
| Stroke Volume (ml) | 54±5 | 46±3 | 47±5 |
| Cardiac Output (Liter/min) | 3.5±0.4 | 2.8±0.1 | 3.4±0.5 |
| E/A Ratio | 1.98±0.17 | 1.04±0.10* | 1.47±0.15† |
| LV Muscle Mass / Body Weight (g/kg) | 2.14±0.16 | 3.43±0.16* | 2.79±0.16*† |
p<0.05 vs. normal;
p<0.05 vs. HTN;
p<0.05 vs. baseline
MAP: mean arterial pressure; LV: left ventricle
Figure 1.
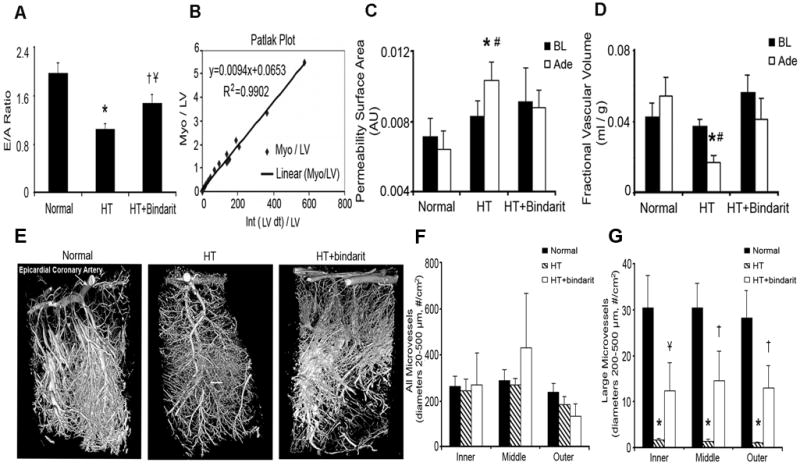
Cardiac diastolic function evaluated by the ratio of left ventricular early (E) to late (A) filling rates, myocardial microvascular permeability (MP) and fractional vascular volume (FVV) assessed by the Patlak graphical analysis method using fast CT, and microvascular density assessed by micro-CT. A. Cardiac diastolic function evaluated by E/A in normal, hypertension (HTN), and HTN+bindarit pigs (n=6 each). E/A ratio significantly decreased in HTN compared to normal and increased in HTN+bindarit compared to HTN. B. Representative Patlak plot from which MP (slope) and FVV (y-axis intercept) are calculated. C, D. MP and FVV at baseline (BL, black bars) and during adenosine (Ade, white bars) infusion. Basal anterior wall myocardial MP and FVV were similar among the groups. Infusion of adenosine induced in HTN a significant increase in MP and decrease in FVV compared to baseline, which were both blunted by bindarit. E. Representative micro-CT images of myocardial microvessels. F, G. Quantitation of microvascular density. There was no difference among the groups in overal microvascular transmural density (from 20 to 500μm in diameter). However, the spatial density of larger microvessels (200-500 μm) was selectively decreased in HTN compared to normal in both the subepicardium and subendocardium, but was significantly preserved by bindarit. * p<0.05 vs. normal, # p<0.05 vs. baseline (BL), † p<0.05 vs. HTN and Ұ p=0.06 vs. normal.
Patlak-derived (Figure 1B) myocardial MP and FVV in the LV anterior wall were similar among the 3 groups at baseline (ANOVA p=0.60 and p=0.14, Figure 1C-D). In response to adenosine, MP significantly increased in HTN (from 0.0083±0.0009 to 0.0103±0.0011 AU, p=0.038 vs. baseline) and achieved levels significantly higher than normal (0.0064±0.0011 AU, p=0.016 vs. HTN, Figure 1C). In contrast, in normal and HTN+bindarit pigs, MP remained unaltered during adenosine. The degree of change in MP in response to adenosine was also significantly greater in HTN compared to normal (+27±10 vs. -20±5%, p=0.003). Furthermore, adenosine induced a significant decrease in FVV in HTN, which bindarit attenuated (Figure 1D). Hence, HTN-induced microvascular dysfunction was blunted by MCPs inhibition.
Microvascular Density
There was no difference among the groups in overall transmural density of microvessels (diameters 20-500μm) (ANOVA p=0.90, Figure 1F). However, subepicardial and subendocardial spatial density of larger microvessels (200-500 μm) was selectively decreased in HTN (all p=0.002 vs. normal, Figure 1G), but preserved by MCPs inhibition (Figure 1G).
Inflammation and Remodeling
In HTN, myocardial MCP-1 expression significantly increased mainly in the intramyocardial arterial wall, but was inhibited by bindarit (p=0.0004, Figure 2), as was perivascular macrophage accumulation (p<0.0001, Figure 2), while IL-6 expression in HTN remained elevated (Figure 2). COX-1 and COX-2 expression was similar among the groups. Myocardial fibrosis in HTN (ANOVA p=0.003) was significantly ameliorated (p=0.04 HTN vs. HTN+bindarit), but not abolished, by bindarit (Figure 3A, D). Myocyte cross-sectional area increased in HTN (p=0.02 vs. normal) and HTN+bindarit (p=0.02 vs. normal) (Figure 3B, E) but was not different between them (p=0.46 vs. HTN), indicating no significant effect of bindarit on myocyte hypertrophy. Contrarily, microvascular media-to-lumen ratio increased from 0.08±0.01 in normal to 0.15±0.01 in HTN (p=0.006) and decreased in HTN+bindarit (to 0.10±0.02, p<0.05 vs. HTN, Figure 3C, F), suggesting inhibition of microvascular wall thickening.
Figure 2.
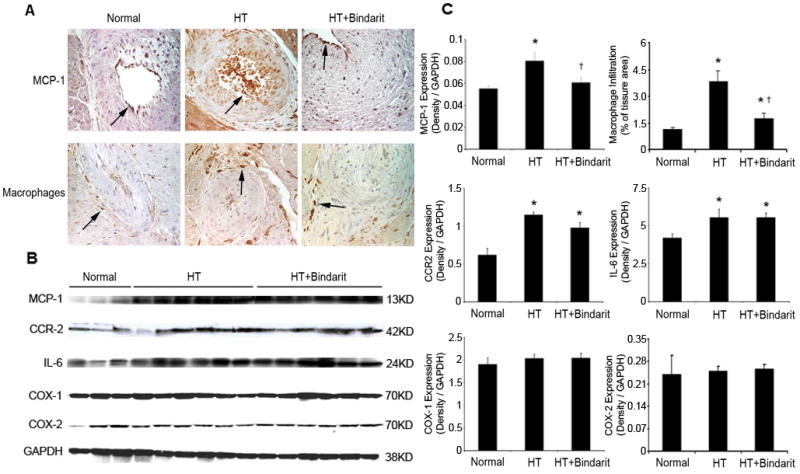
The expression of myocardial MCP-1, its receptor CCR2, cyclooxygenase (COX)-1, COX-2, IL-6, and macrophage infiltration in normal, hypertension (HTN), and HTN+bindarit pigs detected by immunohistochemistry. A. Representative images (magnification ×40) of MCP-1 and macrophage staining (brown, arrows), showing increase in HTN and decrease by bindarit. B. Western blots, showing increased expression of MCP-1, CCR-2 and IL-6 in both HTN and HTN+bindarit. COX-1 and COX-2 expression was similar among the groups. C. Quantitation and densitometry normalized by GAPDH. * p<0.05 vs. normal and † p<0.05 vs. HTN.
Figure 3.
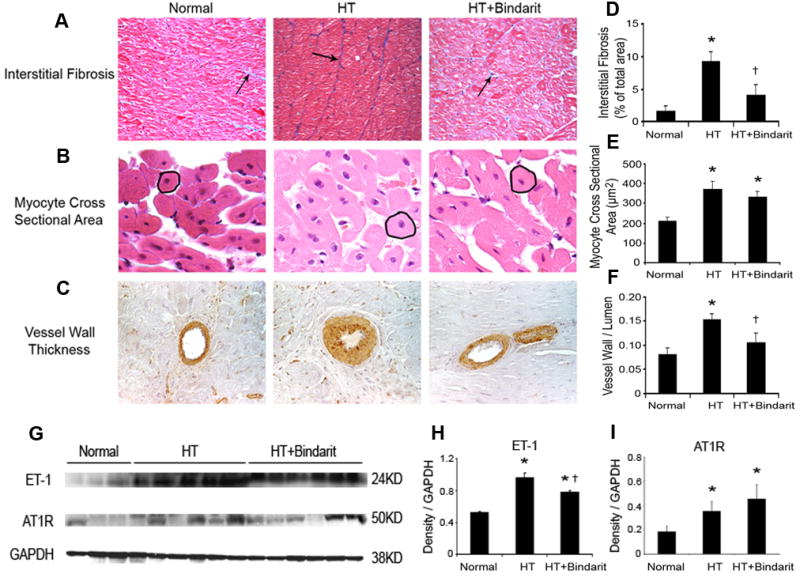
Myocardial fibrosis (A), myocyte cross sectional area (B), and vessel wall to lumen ratio (C) quantified in trichrome, H&E, and SMA stained slides (magnification ×40) and myocardial expression of ET-1 and AT1R detected by western blotting from normal, hypertension (HTN), and HTN+bindarit pigs. Myocardial fibrosis was increased in HTN compared to normal, and significantly ameliorated, but not abolished, by bindarit (D). Myocyte cross sectional area was increased in both HTN and HTN+bindarit compared to normal, but was not different between the two groups (E), indicating that bindarit had no effect on myocyte hypertrophy. Microvascular media-to-lumen ratio increased in HTN compared to normal and decreased in HTN+bindarit compared to HTN, yet remained higher than normal (F). Myocardial tissue ET-1 level significantly increased in HTN, which was significantly preserved by bindarit; AT1R expression was elevated in HTN, but unaffected by bindarit (G, H). * p<0.05 vs. normal and † p<0.05 vs. HTN.
The endothelial and perivascular expression of pMYPT1 increased in both HTN and HTN+bindarit (p=0.02 and p=0.01 vs. normal, respectively), suggesting that bindarit may not influence the ROCK pathway (online supplement, Figure 1SA, D). Endothelial ZO-1 expression was also upregulated in HTN and HTN+bindarit (online supplement, Figure 1SB, E, p=0.04 and p=0.03 vs. normal, respectively), but significantly decreased in bindarit-treated compared to untreated HTN pigs (p=0.04, online supplement, Figure 1SC, F).
In addition, HTN increased both systemic and myocardial ET-1 level, which was preserved by bindarit (Table 1, Figure 3). AT1R expression was also elevated in HTN16, but unaffected by bindarit (p=0.24, Figure 3). On the other hand, myocardial eNOS expression was slightly but significantly decreased in both HTN and HTN+bindarit (p=0.04 and p=0.02, respectively). Superoxide production by DHE was elevated in HTN (p=0.02 vs. normal) and unaltered by bindarit (p=0.7 vs. HTN, online supplement, Figure 2S). NAD(P)H oxidase p67phox expression was increased in both HTN (p=0.01) and HTN+bindarit (p=0.04), while p47phox remained unchanged. Therefore, bindarit did not ameliorate NAD(P)H oxidase-dependent oxidative stress (online supplement, Figure 2S).
Cell culture studies have shown that MCP-1 dose-dependently and significantly increased collagen-I and collagen-III production and TIMP-1 expression in HCF (Online Supplement, Figure 3S).
Discussion
The current study shows that specific inhibition of MCPs in hypertension ameliorated the increase in microvascular permeability and decrease in fractional vascular volume in response to increased cardiac demand, suggesting preserved microvascular integrity and endothelial barrier function. These were accompanied by attenuated microvascular remodeling and myocardial fibrosis, and by improved cardiac diastolic function. These results suggest that the myocardial and microvascular changes induced by hypertension are partly mediated by MCPs and functionally consequential.
HTN is often associated with activation of the renin-angiotensin system, ET-1, oxidative stress, and decreased nitric oxide (NO) bioavailability.25, 26 HTN is additionally characterized by inflammation and overexpression of MCPs, predominantly MCP-127, a critical mediator of macrophage accumulation.26 Activated macrophages, in turn, produce many cytokines, chemokines, growth-factors, and proteases to promote inflammation and facilitate cell proliferation, extracellular-matrix turnover, and angiogenesis. Our previous studies demonstrated that renovascular HTN increased both cardiac and renal MCP-1 protein expression and oxidative stress.17, 25, 28 Furthermore, HTN altered myocardial microvascular function and impaired myocardial perfusion responses to challenge.2, 28, 29 In this study we assessed several inflammatory pathways potentially activated in HTN. COX-1 and COX-2 expression remained unchanged, arguing against their involvement in myocardial alterations in our model. IL-6 increased in HTN but remained elevated in HTN+bindarit, while MCP-1 expression decreased in HTN+bindarit in association with improvement of many aspects of microvascular function and structure, suggesting that it might contribute to these alterations.
Hypertension induces myocyte hypertrophy, interstitial fibrosis, and consequent cardiac stiffness, which are implicated in increased LVMM and impaired diastolic function (e.g. decreased E/A). In this study MCPs synthesis inhibition improved cardiac diastolic function, likely by ameliorating cardiac stiffness and increasing compliance. Interestingly, our HCF study suggests that MCP-1 can directly increase myocardial matrix production and fibrosis, thus its action is not necessarily mediated only by monocyte recruitment. Bindarit decreased interstitial fibrosis, but not myocyte hypertrophy, likely because blood pressure remained elevated, resulting in incomplete improvement of LVMM. Our observations are underscored by previous studies in rodents, in which MCP-1 was inhibited with anti-MCP-1 neutralizing antibody or indirectly reduced using ROCK inhibitor, antioxidants and inactivation of CCR-2.30-33
Importantly, bindarit is able to modulate the levels and attenuate over-production of MCPs in response to inflammation, without entirely blocking their physiological activity or inadvertently increasing MCPs expression, as observed with anti-MCP-1 antibody.34 In line with previous studies showing that bindarit dose-dependently inhibits MCP-1/CCL-2 production and is quite selective among chemokines14, 35, we found that in our model it did not decrease the upregulated IL-6 expression.
Interestingly, we observed that HTN increased MP and decreased FVV in response to adenosine, suggesting vascular dysfunction possibly mediated by inflammation. Hypertension initially evokes coronary endothelial dysfunction and abnormal myocardial perfusion regulation, and subsequently vascular remodeling and dysfunction.4 Endothelial barrier function is important for maintaining vascular integrity and regulation of vascular tone. MCP-1 directly increases blood-brain barrier permeability in inflammation by regulating the expression and rearranging endothelial tight junction proteins36, 37 that maintain endothelial barrier integrity. We found increased expression of the endothelial tight junction component ZO-1, a membrane protein that links the actin cytoskeleton and tight junctions.38 Its endothelial overexpression in HTN may imply tight junction reassembly, a compensatory effect that may reflect disruption of endothelial barrier integrity.39 Notably, subtle changes in barrier function may be disclosed only during challenges like increased cardiac demand.40 ROCK also mediates some of the effects of MCP-1 on brain MP37, but does not seem to be a major modulator of myocardial MP in our model, because bindarit improved MP without changing MYPT (and hence ROCK activity). Nevertheless, other tight-junction proteins might contribute to loss of endothelial integrity in HTN.
Myocardial FVV decreases during cardiac challenge in HTN also indicate microvascular dysfunction, possibly consequent to microvascular wall remodeling and architectural changes4. Indeed, microvascular rarefaction is commonly observed in HTN. We observed a selective decrease in density of larger microvessels (200-500 μm).25 The unchanged total myocardial microvascular density in HTN pigs may reflect an earlier increase in the number of small microvessels needed to support developing LV hypertrophy.2, 4, 25, 28 Prevention of microvascular rarefaction by bindarit in HTN is likely through decreased myocardial and perivascular fibrosis.41 In addition, we observed in HTN increased media-to-lumen ratio, which possibly precedes luminal narrowing.25 The improvement achieved by MCPs inhibition implicates them in microvascular remodeling in HTN. Overall, MCPs inhibition decreased inflammation-induced vascular remodeling, rarefaction, and dysfunction, and blunted the reactive decrease of FVV in response to adenosine.
In addition to microvascular remodeling, myocardial vascular volume (FVV) could be affected by changes in vascular tone. Adenosine normally induces vasodilatation that involves both endothelial-dependent and –independent mechanisms.2 During endothelial dysfunction, a decrease in NO and increased abundance of vasoconstrictors (e.g. ET-1) may lead to paradoxical vasoconstriction.2, 29 By upregulating ET-111 MCPs might contribute to endothelial dysfunction in HTN. This notion is underscored by our observation that MCPs blockade decreased systemic and myocardial ET-1 levels and ameliorated the decrease in FVV in response to cardiac demand. Notably, since FVV responses improved without altering eNOS expression or oxidative stress, a decrease in NO availability was unlikely a critical factor. Adenosine infusion may amplify a decrease in FVV by disclosing functional impairments, a failure in capillary recruitment, or microvascular rarefaction in HTN.42
Systemic PRA increases shortly after induction of experimental renovascular HTN,17, 18 but in the chronic phase returns to basal levels43 and may sustain HTN by activating secondary mechanisms. However, the effects of bindarit in our model were not mediated by inhibition of the renin-angiotensin-aldosterone system, which remained unchanged (PRA, myocardial AT-1R, or circulating aldosterone levels). Because MCP inhibition did not significantly blunt oxidative stress or the renin-angiotensin system, systemic hypertension remained unabated, and the beneficial effects of bindarit on cardiac function were likely derived from direct attenuation of cardiac remodeling (Figure 4). Further studies are needed to dissect the potential interaction of MCPs with the renin-angiotensin system.
Figure 4.
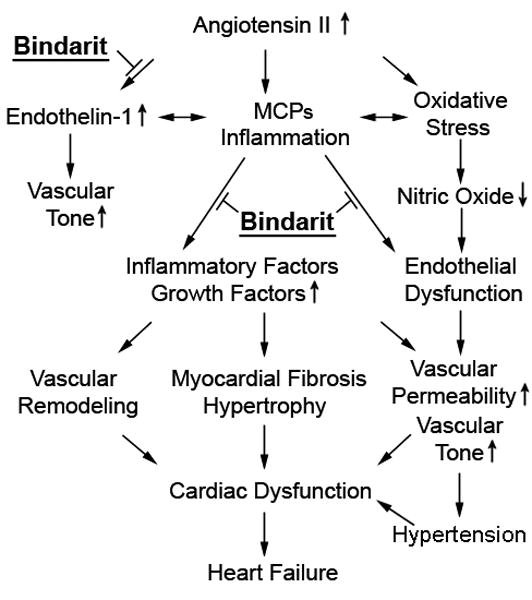
A schematic outlining the pathways blocked by bindarit that are likely responsible for its effects to improve cardiac and myocardial microvascular function. However, MCP inhibition did not substantially blunt oxidative stress or the renin-angiotensin system, which drive systemic hypertension in this model.
Our study was limited by the use of a relatively small number of young pigs. We also used in all groups two fast-CT scanners, which provide comparable assessments of MP and FVV.7 It is possible that some of the tissue studies sampled some unbound circulating proteins, but exanguinated myocardium contains little blood volume, and the preparation procedure was the same for all the groups. Therefore, the fraction of retained unbound cytokines might be negligible and comparable among the groups, and result in minimal interference with our result interpretation. Bindarit inhibits MCPs 1, 2, and 3, but MCP-1 is the key member of the MCP family that has been most commonly and directly linked to cardiovascular disease and hypertension, so that the effects of the drug are likely attributable mainly to blockade of MCP-1. Nevertheless, we cannot exclude the possibility that inhibition of MCPs 2 and 3 contributed to these benefits. Additionally, the dose of bindarit may incompletely inhibit MCPs expression and consequently introduce some variability.
In summary, we observed that hypertension induces myocardial inflammation, fibrosis, and vascular remodeling, and impairs vascular integrity. These effects are partly mediated by the endogenous inflammatory mediator MCPs, and its inhibition attenuated LV hypertrophy and improved cardiac diastolic function (Figure 4). Inhibition of MCPs could therefore be a potential therapeutic target for the downstream end-organ damage induced by hypertension, such as hypertensive cardiomyopathy.
Supplementary Material
Figure 1S. Phospho-myosin phosphatase targeting subunit (pMYPT1, A) and ZO-1 (B and C) expression detected in the myocardium by fluorescent immunostaining (Texas-Red), colorimetric immunostaining and Western blot, respectively, and quantified (D, E and F) at a magnification of ×40. The endothelial and perivascular expression of pMYPT1 was elevated in the HTN and HTN+bindarit. ZO-1 endothelial expression was upregulated in HTN and HTN+bindarit compared to normal. * p<0.05 vs. normal and † p<0.05 vs. HTN.
Figure 2S. Myocardial expression of eNOS and NAD(P)H oxidase (p47 and p67) detected with Western blotting (A) and superoxide production detected by dihydroethidium (DHE) staining (B) in normal, hypertension (HTN), and HTN+bindarit pigs. Densitometry was normalized by GAPDH and DHE staining by nuclei (C). The expression of eNOS was similarly and significantly decreased in both HTN and HTN+bindarit. NAD(P)H oxidase (p67 subunit) was increased, while p47 did not differ among the groups. Superoxide production was elevated in HTN and unaltered by bindarit. *p<0.05 vs. normal and † p<0.05 vs. HTN.
Figure 3S. The effects of MCP-1 on human cardiac fibroblasts (HCFs). A. Western blots, showing the expression of collagen I, collagen III, and TIMP-1 in HCFs treated with different concentration of MCP-1. B. Densitometry normalized by GAPDH. MCP-1 dose-dependently and significantly increased the production of both collagen I and collagen III, as well as the expression of TIMP-1. * p<0.05 vs. control (MCP-1 concentration is 0 ng/ml).
Acknowledgments
Bindarit was kindly provided by Angelini Research Center, Rome, Italy.
This study was partly supported by NIH grants numbers DK73608, HL77131, DK077013, and HL085307.
Footnotes
Disclosures: Dr. Angelo Guglielmotti is an employee of Angelini Research Center – ACRAF. Dr. Lerman provided a one-time consultation to the company regarding this study.
References
- 1.Sironi AM, Pingitore A, Ghione S, De Marchi D, Scattini B, Positano V, Muscelli E, Ciociaro D, Lombardi M, Ferrannini E, Gastaldelli A. Early Hypertension Is Associated With Reduced Regional Cardiac Function, Insulin Resistance, Epicardial, and Visceral Fat. Hypertension. 2008;51:282–288. doi: 10.1161/HYPERTENSIONAHA.107.098640. [DOI] [PubMed] [Google Scholar]
- 2.Rodriguez-Porcel M, Herrman J, Chade AR, Krier JD, Breen JF, Lerman A, Lerman LO. Long-term antioxidant intervention improves myocardial microvascular function in experimental hypertension. Hypertension. 2004;43:493–498. doi: 10.1161/01.HYP.0000111834.03000.e4. [DOI] [PubMed] [Google Scholar]
- 3.Camici PG, Crea F. Coronary microvascular dysfunction. N Engl J Med. 2007;356:830–840. doi: 10.1056/NEJMra061889. [DOI] [PubMed] [Google Scholar]
- 4.Rodriguez-Porcel M, Zhu XY, Chade AR, Amores-Arriaga B, Caplice NM, Ritman EL, Lerman A, Lerman LO. Functional and structural remodeling of the myocardial microvasculature in early experimental hypertension. Am J Physiol Heart Circ Physiol. 2006;290:H978–984. doi: 10.1152/ajpheart.00538.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Birukova AA, Adyshev D, Gorshkov B, Bokoch GM, Birukov KG, Verin AD. GEF-H1 is involved in agonist-induced human pulmonary endothelial barrier dysfunction. Am J Physiol Lung Cell Mol Physiol. 2006;290:L540–548. doi: 10.1152/ajplung.00259.2005. [DOI] [PubMed] [Google Scholar]
- 6.Reddy HK, Campbell SE, Janicki JS, Zhou G, Weber KT. Coronary microvascular fluid flux and permeability: influence of angiotensin II, aldosterone, and acute arterial hypertension. J Lab Clin Med. 1993;121:510–521. [PubMed] [Google Scholar]
- 7.Daghini E, Primak AN, Chade AR, Zhu X, Ritman EL, McCollough CH, Lerman LO. Evaluation of porcine myocardial microvascular permeability and fractional vascular volume using 64-slice helical computed tomography (CT) Invest Radiol. 2007;42:274–282. doi: 10.1097/01.rli.0000258086.78179.90. [DOI] [PubMed] [Google Scholar]
- 8.Mohlenkamp S, Lerman LO, Lerman A, Behrenbeck TR, Katusic ZS, Sheedy PF, 2nd, Ritman EL. Minimally invasive evaluation of coronary microvascular function by electron beam computed tomography. Circulation. 2000;102:2411–2416. doi: 10.1161/01.cir.102.19.2411. [DOI] [PubMed] [Google Scholar]
- 9.Capers Qt, Alexander RW, Lou P, De Leon H, Wilcox JN, Ishizaka N, Howard AB, Taylor WR. Monocyte chemoattractant protein-1 expression in aortic tissues of hypertensive rats. Hypertension. 1997;30:1397–1402. doi: 10.1161/01.hyp.30.6.1397. [DOI] [PubMed] [Google Scholar]
- 10.Hilgers KF, Hartner A, Porst M, Mai M, Wittmann M, Hugo C, Ganten D, Geiger H, Veelken R, Mann JF. Monocyte chemoattractant protein-1 and macrophage infiltration in hypertensive kidney injury. Kidney Int. 2000;58:2408–2419. doi: 10.1046/j.1523-1755.2000.00424.x. [DOI] [PubMed] [Google Scholar]
- 11.Molet S, Furukawa K, Maghazechi A, Hamid Q, Giaid A. Chemokine- and cytokine-induced expression of endothelin 1 and endothelin-converting enzyme 1 in endothelial cells. J Allergy Clin Immunol. 2000;105:333–338. doi: 10.1016/s0091-6749(00)90084-8. [DOI] [PubMed] [Google Scholar]
- 12.Vallbracht KB, Schwimmbeck PL, Seeberg B, Kuhl U, Schultheiss HP. Endothelial dysfunction of peripheral arteries in patients with immunohistologically confirmed myocardial inflammation correlates with endothelial expression of human leukocyte antigens and adhesion molecules in myocardial biopsies. J Am Coll Cardiol. 2002;40:515–520. doi: 10.1016/s0735-1097(02)01990-3. [DOI] [PubMed] [Google Scholar]
- 13.Rittig K, Peter A, Baltz KM, Tschritter O, Weigert C, Andreozzi F, Perticone F, Siegel-Axel DI, Stefan N, Fritsche A, Salih HR, Schleicher E, Machicao F, Sesti G, Haring HU, Balletshofer BM. The CCR2 promoter polymorphism T-960A, but not the serum MCP-1 level, is associated with endothelial function in prediabetic individuals. Atherosclerosis. 2008;198:338–346. doi: 10.1016/j.atherosclerosis.2007.10.028. [DOI] [PubMed] [Google Scholar]
- 14.Sironi M, Guglielmotti A, Polentarutti N, Fioretti F, Milanese C, Romano M, Vigini C, Coletta I, Sozzani S, Bernasconi S, Vecchi A, Pinza M, Mantovani A. A small synthetic molecule capable of preferentially inhibiting the production of the CC chemokine monocyte chemotactic protein-1. Eur Cytokine Netw. 1999;10:437–442. [PubMed] [Google Scholar]
- 15.Guglielmotti A, Aquilini L, D'Onofrio E, Rosignoli MT, Milanese C, Pinza M. Bindarit prolongs survival and reduces renal damage in NZB/W lupus mice. Clin Exp Rheumatol. 1998;16:149–154. [PubMed] [Google Scholar]
- 16.Zoja C, Corna D, Benedetti G, Morigi M, Donadelli R, Guglielmotti A, Pinza M, Bertani T, Remuzzi G. Bindarit retards renal disease and prolongs survival in murine lupus autoimmune disease. Kidney Int. 1998;53:726–734. doi: 10.1046/j.1523-1755.1998.00804.x. [DOI] [PubMed] [Google Scholar]
- 17.Lerman LO, Nath KA, Rodriguez-Porcel M, Krier JD, Schwartz RS, Napoli C, Romero JC. Increased Oxidative Stress in Experimental Renovascular Hypertension. Hypertension. 2001;37:541–546. doi: 10.1161/01.hyp.37.2.541. [DOI] [PubMed] [Google Scholar]
- 18.Lerman LO, Schwartz RS, Grande JP, Sheedy PF, Romero JC. Noninvasive evaluation of a novel swine model of renal artery stenosis. J Am Soc Nephrol. 1999;10:1455–1465. doi: 10.1681/ASN.V1071455. [DOI] [PubMed] [Google Scholar]
- 19.Chade AR, Herrmann J, Zhu X, Krier JD, Lerman A, Lerman LO. Effects of proteasome inhibition on the kidney in experimental hypercholesterolemia. J Am Soc Nephrol. 2005;16:1005–1012. doi: 10.1681/ASN.2004080674. [DOI] [PubMed] [Google Scholar]
- 20.Herrmann J, Saguner AM, Versari D, Peterson TE, Chade A, Olson M, Lerman LO, Lerman A. Chronic proteasome inhibition contributes to coronary atherosclerosis. Circ Res. 2007;101:865–874. doi: 10.1161/CIRCRESAHA.107.152959. [DOI] [PubMed] [Google Scholar]
- 21.Zhu XY, Rodriguez-Porcel M, Bentley MD, Chade AR, Sica V, Napoli C, Caplice N, Ritman EL, Lerman A, Lerman LO. Antioxidant Intervention Attenuates Myocardial Neovascularization in Hypercholesterolemia. Circulation. 2004;109:2109–2115. doi: 10.1161/01.CIR.0000125742.65841.8B. [DOI] [PubMed] [Google Scholar]
- 22.Rodriguez-Porcel M, Lerman A, Ritman EL, Wilson SH, Best PJM, Lerman LO. Altered Myocardial Microvascular 3D Architecture in Experimental Hypercholesterolemia. Circulation. 2000;102:2028–2030. doi: 10.1161/01.cir.102.17.2028. [DOI] [PubMed] [Google Scholar]
- 23.Collins NT, Cummins PM, Colgan OC, Ferguson G, Birney YA, Murphy RP, Meade G, Cahill PA. Cyclic Strain-Mediated Regulation of Vascular Endothelial Occludin and ZO-1: Influence on Intercellular Tight Junction Assembly and Function. Arterioscler Thromb Vasc Biol. 2006;26:62–68. doi: 10.1161/01.ATV.0000194097.92824.b3. [DOI] [PubMed] [Google Scholar]
- 24.van Nieuw Amerongen GP, Beckers CM, Achekar ID, Zeeman S, Musters RJ, van Hinsbergh VW. Involvement of Rho kinase in endothelial barrier maintenance. Arterioscler Thromb Vasc Biol. 2007;27:2332–2339. doi: 10.1161/ATVBAHA.107.152322. [DOI] [PubMed] [Google Scholar]
- 25.Zhu XY, Daghini E, Chade AR, Rodriguez-Porcel M, Napoli C, Lerman A, Lerman LO. Role of oxidative stress in remodeling of the myocardial microcirculation in hypertension. Arterioscler Thromb Vasc Biol. 2006;26:1746–1752. doi: 10.1161/01.ATV.0000227469.40826.01. [DOI] [PubMed] [Google Scholar]
- 26.Taylor WR. Hypertensive vascular disease and inflammation: Mechanical and humoral mechanisms. Current Hypertension Reports. 1999;1:96–101. doi: 10.1007/s11906-999-0079-5. [DOI] [PubMed] [Google Scholar]
- 27.Tucci M, Quatraro C, Frassanito MA, Silvestris F. Deregulated expression of monocyte chemoattractant protein-1 (MCP-1) in arterial hypertension: role in endothelial inflammation and atheromasia. J Hypertens. 2006;24:1307–1318. doi: 10.1097/01.hjh.0000234111.31239.c3. [DOI] [PubMed] [Google Scholar]
- 28.Zhu XY, Daghini E, Chade AR, Napoli C, Ritman EL, Lerman A, Lerman LO. Simvastatin prevents coronary microvascular remodeling in renovascular hypertensive pigs. J Am Soc Nephrol. 2007;18:1209–1217. doi: 10.1681/ASN.2006090976. [DOI] [PubMed] [Google Scholar]
- 29.Rodriguez-Porcel M, Lerman A, Herrmann J, Schwartz RS, Sawamura T, Condorelli M, Napoli C, Lerman LO. Hypertension exacerbates the effect of hypercholesterolemia on the myocardial microvasculature. Cardiovasc Res. 2003;58:213–221. doi: 10.1016/s0008-6363(03)00250-5. [DOI] [PubMed] [Google Scholar]
- 30.Kuwahara F, Kai H, Tokuda K, Takeya M, Takeshita A, Egashira K, Imaizumi T. Hypertensive Myocardial Fibrosis and Diastolic Dysfunction: Another Model of Inflammation? Hypertension. 2004;43:739–745. doi: 10.1161/01.HYP.0000118584.33350.7d. [DOI] [PubMed] [Google Scholar]
- 31.Tokuda K, Kai H, Kuwahara F, Yasukawa H, Tahara N, Kudo H, Takemiya K, Koga M, Yamamoto T, Imaizumi T. Pressure-independent effects of angiotensin II on hypertensive myocardial fibrosis. Hypertension. 2004;43:499–503. doi: 10.1161/01.HYP.0000111831.50834.93. [DOI] [PubMed] [Google Scholar]
- 32.Ishimaru K, Ueno H, Kagitani S, Takabayashi D, Takata M, Inoue H. Fasudil attenuates myocardial fibrosis in association with inhibition of monocyte/macrophage infiltration in the heart of DOCA/salt hypertensive rats. J Cardiovasc Pharmacol. 2007;50:187–194. doi: 10.1097/FJC.0b013e318064f150. [DOI] [PubMed] [Google Scholar]
- 33.Ishibashi M, Hiasa K, Zhao Q, Inoue S, Ohtani K, Kitamoto S, Tsuchihashi M, Sugaya T, Charo IF, Kura S, Tsuzuki T, Ishibashi T, Takeshita A, Egashira K. Critical role of monocyte chemoattractant protein-1 receptor CCR2 on monocytes in hypertension-induced vascular inflammation and remodeling. Circ Res. 2004;94:1203–1210. doi: 10.1161/01.RES.0000126924.23467.A3. [DOI] [PubMed] [Google Scholar]
- 34.Haringman JJ, Gerlag DM, Smeets TJ, Baeten D, van den Bosch F, Bresnihan B, Breedveld FC, Dinant HJ, Legay F, Gram H, Loetscher P, Schmouder R, Woodworth T, Tak PP. A randomized controlled trial with an anti-CCL2 (anti-monocyte chemotactic protein 1) monoclonal antibody in patients with rheumatoid arthritis. Arthritis Rheum. 2006;54:2387–2392. doi: 10.1002/art.21975. [DOI] [PubMed] [Google Scholar]
- 35.Mirolo M, Fabbri M, Sironi M, Vecchi A, Guglielmotti A, Mangano G, Biondi G, Locati M, Mantovani A. Impact of the anti-inflammatory agent bindarit on the chemokinome: selective inhibition of the monocyte chemotactic proteins. Eur Cytokine Netw. 2008;19:119–122. doi: 10.1684/ecn.2008.0133. [DOI] [PubMed] [Google Scholar]
- 36.Song L, Pachter JS. Monocyte chemoattractant protein-1 alters expression of tight junction-associated proteins in brain microvascular endothelial cells. Microvasc Res. 2004;67:78–89. doi: 10.1016/j.mvr.2003.07.001. [DOI] [PubMed] [Google Scholar]
- 37.Stamatovic SM, Keep RF, Kunkel SL, Andjelkovic AV. Potential role of MCP-1 in endothelial cell tight junction ‘opening’: signaling via Rho and Rho kinase. J Cell Sci. 2003;116:4615–4628. doi: 10.1242/jcs.00755. [DOI] [PubMed] [Google Scholar]
- 38.Willott E, Balda MS, Heintzelman M, Jameson B, Anderson JM. Localization and differential expression of two isoforms of the tight junction protein ZO-1. Am J Physiol Cell Physiol. 1992;262:C1119–1124. doi: 10.1152/ajpcell.1992.262.5.C1119. [DOI] [PubMed] [Google Scholar]
- 39.Antonios TF. Microvascular rarefaction in hypertension--reversal or over-correction by treatment? Am J Hypertens. 2006;19:484–485. doi: 10.1016/j.amjhyper.2005.11.010. [DOI] [PubMed] [Google Scholar]
- 40.Rodriguez-Porcel M, Lerman A, Best PJ, Krier JD, Napoli C, Lerman LO. Hypercholesterolemia impairs myocardial perfusion and permeability: role of oxidative stress and endogenous scavenging activity. J Am Coll Cardiol. 2001;37:608–615. doi: 10.1016/s0735-1097(00)01139-6. [DOI] [PubMed] [Google Scholar]
- 41.Amann K, Tornig J, Buzello M, Kuhlmann A, Gross ML, Adamczak M, Ritz E. Effect of antioxidant therapy with dl-alpha-tocopherol on cardiovascular structure in experimental renal failure. Kidney Int. 2002;62:877–884. doi: 10.1046/j.1523-1755.2002.00518.x. [DOI] [PubMed] [Google Scholar]
- 42.Noon JP, Walker BR, Webb DJ, Shore AC, Holton DW, Edwards HV, Watt GC. Impaired microvascular dilatation and capillary rarefaction in young adults with a predisposition to high blood pressure. J Clin Invest. 1997;99:1873–1879. doi: 10.1172/JCI119354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pipinos II, Nypaver TJ, Moshin SK, Careterro OA, Beierwaltes WH. Response to angiotensin inhibition in rats with sustained renovascular hypertension correlates with response to removing renal artery stenosis. J Vasc Surg. 1998;28:167–177. doi: 10.1016/s0741-5214(98)70212-5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure 1S. Phospho-myosin phosphatase targeting subunit (pMYPT1, A) and ZO-1 (B and C) expression detected in the myocardium by fluorescent immunostaining (Texas-Red), colorimetric immunostaining and Western blot, respectively, and quantified (D, E and F) at a magnification of ×40. The endothelial and perivascular expression of pMYPT1 was elevated in the HTN and HTN+bindarit. ZO-1 endothelial expression was upregulated in HTN and HTN+bindarit compared to normal. * p<0.05 vs. normal and † p<0.05 vs. HTN.
Figure 2S. Myocardial expression of eNOS and NAD(P)H oxidase (p47 and p67) detected with Western blotting (A) and superoxide production detected by dihydroethidium (DHE) staining (B) in normal, hypertension (HTN), and HTN+bindarit pigs. Densitometry was normalized by GAPDH and DHE staining by nuclei (C). The expression of eNOS was similarly and significantly decreased in both HTN and HTN+bindarit. NAD(P)H oxidase (p67 subunit) was increased, while p47 did not differ among the groups. Superoxide production was elevated in HTN and unaltered by bindarit. *p<0.05 vs. normal and † p<0.05 vs. HTN.
Figure 3S. The effects of MCP-1 on human cardiac fibroblasts (HCFs). A. Western blots, showing the expression of collagen I, collagen III, and TIMP-1 in HCFs treated with different concentration of MCP-1. B. Densitometry normalized by GAPDH. MCP-1 dose-dependently and significantly increased the production of both collagen I and collagen III, as well as the expression of TIMP-1. * p<0.05 vs. control (MCP-1 concentration is 0 ng/ml).