

Abstract
Human and animal studies demonstrate an untoward effect of excess dietary NaCl (salt) intake on cardiovascular function and life span. The endothelium in particular augments the production of transforming growth factor (TGF)-β, a fibrogenic growth factor, in response to excess dietary salt intake. This study explored the initiating mechanism that regulates salt-induced endothelial cell production of TGF-β. Male Sprague-Dawley rats were given diets containing different amounts of NaCl and potassium for 4 days. A bioassay for TGF-β demonstrated increased (35.2%) amounts of active TGF-β in the medium of aortic ring segments from rats on the high-salt diet compared with rats maintained on a 0.3% NaCl diet. Inhibition of the large-conductance, calcium-activated potassium channel inhibited dietary salt-induced vascular production of TGF-β but did not affect production of TGF-β by ring segments from rats on the low-salt diet. Immunohistochemical and Western analyses demonstrated the α subunit of the calcium-activated potassium channel in endothelial cells. Increasing medium [K+] inhibited production of dietary salt-induced vascular production levels of total and active TGF-β but did not alter TGF-β production by aortic rings from rats on the 0.3% NaCl diet. Increasing dietary potassium content decreased urinary active TGF-β in animals receiving the high-salt diet but did not change urinary active TGF-β in animals receiving the low-salt diet. The findings demonstrated an interesting interaction between the dietary intake of potassium and excess NaCl and further showed the fundamental role of the endothelial calcium-activated potassium channel in the vascular response to excess salt intake.
Keywords: dietary sodium chloride, potassium channel, endothelium, aorta, iberiotoxin, physiology
There is growing appreciation of the cardiovascular effects of excess NaCl (termed “salt” in this article) intake. It has been convenient to divide patients into those who are salt resistant or salt sensitive depending on their blood pressure responses to changes in dietary salt intake, because this distinction remains an interesting and valid assessment of cardiovascular risk. Patients found to have salt-sensitive hypertension subsequently had a greater incidence of left ventricular hypertrophy and rate of nonfatal and fatal cardiovascular events compared with hypertensive patients who were not salt sensitive.1 Weinberger et al2 expanded these observations by demonstrating that salt-sensitive patients who were initially normotensive at the time of study had a significant increase in mortality rate on follow-up evaluation compared with normotensive salt-resistant patients. More important, however, was the recent study by Cook et al,3 in which they performed a large prospective clinical trial involving adults aged 30 to 54 years with prehypertension and showed that, despite a small average reduction in blood pressure, chronic salt restriction to ≈2 to 2.6 g/d reduced the cardiovascular event rate by ≈25% in the intervention group. The combined data suggest that, whereas salt sensitivity is a cardiovascular risk factor, excess salt intake, per se, promotes vascular disease in humans.
Dietary salt intake directly impacts cardiovascular and renal function in animal models. Increasing dietary salt intake promoted a dose-dependent reduction in the life span of rodents.4 An 8.0% NaCl diet increased collagen deposition in the arteries, arterioles, glomeruli, and interstitium of the hearts and kidneys of both normotensive (Wistar Kyoto) and hypertensive (spontaneously hypertensive) rats; upregulation of TGF-β1 mRNA was observed in the kidney and heart during the administration of the high-salt diet in both strains.5 An 8.0% NaCl diet also increased albuminuria and accelerated progression of renal failure in a rodent model of chronic allograft nephropathy.6 In contrast, salt restriction reduced proteinuria and glomerulosclerosis in other rodent models of chronic kidney injury.7 Similarly, salt restriction resulted in reduced albuminuria among patients with hypertension.8
Excess salt intake has a direct effect on endothelial cell function. Sprague-Dawley rats, which did not demonstrate changes in blood pressure on an 8.0% NaCl diet during a 15-day period of observation, did increase expression of TGF-β in the renal cortex compared with tissues from rats maintained on a 0.3% NaCl diet.9 Increased expression was observed 1 day after institution of the diet and persisted over the course of the experiment. Urinary excretion of TGF-β1 increased on the 8.0% NaCl diet, indicating an intrarenal generation of TGF-β1 in response to salt. Increased salt intake promoted the production of total and active TGF-β1 throughout the arterial tree, including the aorta,10 intrarenal arteries and arterioles (unpublished observations), and glomeruli.9 Mechanical removal of the aortic endothelium confirmed that these cells were the source of excess TGF-β1 production during increased salt intake.
The opening of a tetraethylammonium-sensitive potassium channel was the initial event that stimulated TGF-β1 production by aortic ring and isolated glomerular preparations from Sprague-Dawley rats fed a high-salt diet.9,10 Activating these cell-surface molecules promoted the signal transduction mechanism that resulted in production of TGF-β.11-14 The present study was designed to address in greater detail the initiating mechanism by which endothelial cells are activated by excess salt intake and the role of extracellular potassium concentration in the regulation of salt-induced vascular production of TGF-β.
Methods
Animal and Tissue Preparation
The institutional animal care and use committee at the University of Alabama at Birmingham approved the project. Studies were conducted using male Sprague-Dawley rats (Harlan Sprague Dawley, Indianapolis, IN) that were 28 days of age at the start of study. The protocol that was followed has been standardized in our laboratory.9,10,15 The rats were housed under standard conditions and given 1 of 6 formulated diets (AIN-76A; Dyets, Inc) that contained different amounts of NaCl and K+. Three of the diets contained 0.3% NaCl (wt/wt) and 0%, 0.95%, or 1.99% K+ (wt/wt), and the other 3 diets contained 8.0% NaCl (wt/wt) and 0%, 0.95%, or 1.99% K+ (wt/wt). The 0.95% K+ (wt/wt) represented the standard potassium content of a rat diet. The diets were prepared specifically to be identical in protein composition and differed only in NaCl, K+, and sucrose content. On the fourth day of the diets, the rats were anesthetized by IP injection of pentobarbital sodium injection (Lundbeck Inc), 50 mg/kg of body weight, and the aorta was removed under sterile conditions for incubation studies, immunoblot analyses, or immunohistochemistry, as performed previously.9-13,15 In addition, blood was also collected for the determination of serum concentrations of potassium, urea, and creatinine.
In Vitro Incubation Studies
Initial studies used animals on a 0.3% NaCl (wt/wt), 0.95% K+ (wt/wt) diet and a 8.0% NaCl (wt/wt), 0.95% K+ (wt/wt) diet. After removal of adherent fat and connective tissue, the aortic tissue was rinsed in ice-cold PBS, cut into 3-mm ring segments, and placed in 48-well plates. Aortic ring segments were resuspended in serum-free medium (DMEM; Invitrogen Corporation) that contained vehicle alone, 3 mmol/L of tetraethylammonium chloride (TEA; Sigma Chemical), or 100 nM iberiotoxin (IB; Sigma). After a 30-minute incubation period, the medium was replaced and incubation continued at 37°C for 24 hours. In other experiments, aortic rings were incubated in medium with potassium concentration adjusted to 0, 10, 15, and 20 mEq/L; choline chloride was added to maintain a constant osmolality among the groups. After overnight incubation, the conditioned medium was harvested, centrifuged at 300g for 10 minutes at 4°C to remove cell debris, and then stored at -80°C until assayed for total and active TGF-β. TGF-β was determined using a bioassay, as published previously.14,16 Assays were performed in triplicate and averaged, and the results were factored by wet weight of aortic tissue in the sample.
Western Blotting
To obtain isolated endothelial cell lysates, the aorta was removed and opened with a longitudinal incision. The intimal surface was very gently scraped with a curved scalpel blade, and the endothelial cells that built up on the edge of the blade were rinsed into a centrifuge tube that contained serum-free DMEM. Each area was only scraped once, avoiding scraping close to any cut edge. The cell suspension was centrifuged at 500g for 5 minutes and supernatant discarded. The cell pellet was resuspended in 300 μL of modified radioimmunoprecipitation assay buffer that contained the following: 10 mmol/L of Tris-HCl (pH 7.4); 100 mm of NaCl; 1 mmol/L of EDTA and 1 mmol/L of EGTA; 0.5% sodium deoxycholate; 1% Triton-X100, 10% glycerol, and 0.1% SDS; 20 mmol/L of sodium pyrophosphate; and 2 mmol/L of Na3VO4, 1 mM NaF, 1 mmol/L of PMSF, and protease inhibitor mixture. Total protein concentration of the lysates was determined using a kit (BCA Protein Assay Reagent kit; Pierce Biotechnology), and the samples were processed for Western blotting, as we have performed previously.11,12 For Western blotting, samples containing 25 μg of total protein were used. The primary antibody, diluted 1:1000, recognized specifically the pore-forming α subunit of the Big K+ or Maxi K+ (designated “BKCa”) channel (Alomone Laboratories Ltd). Aortic tissue was also harvested in standard fashion for immunohistochemistry,11 performed using this same antibody.
In Vivo Studies
Rats on the 6 diets were anesthetized, and urine was collected from the bladder to determine TGF-β and creatinine, which was assayed using an autoanalyzer (Creatinine Analyzer 2; Beckman Coulter, Inc). Blood was also collected for the determination of serum potassium, serum urea nitrogen, and serum creatinine concentrations. To determine the active TGF-β activity, urine samples were diluted 1:5 in a total volume of 0.5 mL of DMEM containing 0.1% BSA and directly added to each well. Values were normalized using the creatinine concentration obtained in each sample.
Statistical Analysis
Data were expressed as the mean±SE. Significant differences among data sets were determined by unpaired t test or by ANOVA with posthoc testing (Fisher protected least-significant difference; Statview version 5.0; SAS Institute). Data that were not distributed normally were log-normal transformed. Significant differences among some data sets were determined using post hoc analysis by 2-way design with effect testing (Proc Mixed, SAS version 9.2; SAS Institute, Inc). Least-square means testing using the Tukey multiple comparisons test was used to investigate group effects in these analyses. A 2-tailed P<0.05 assigned statistical significance.
Results
TEA and IB Inhibited Dietary Salt-Induced Vascular Production of TGF-β
Aortic ring segments, which were obtained from rats maintained for 4 days on 0.3% and 8.0% NaCl containing the standard amount (0.95% wt/wt) of K+, were incubated in medium overnight. A bioassay for total and active TGF-β demonstrated increased (P<0.05) amounts of TGF-β in the medium of ring segments from rats on the high-salt diet compared with rats maintained on a 0.3% NaCl diet containing 0.95% K+ (Figure 1). Coincubation of the aortic rings from rats on the high-salt diet in medium containing TEA and IB, a specific inhibitor of the BKCa channel,17 inhibited (P<0.05) production of total and active TGF-β compared with vehicle-treated samples from the high-salt-treated rats. Neither TEA nor IB affected production of TGF-β by aortic ring segments from rats on the low-salt diet.
Figure 1.
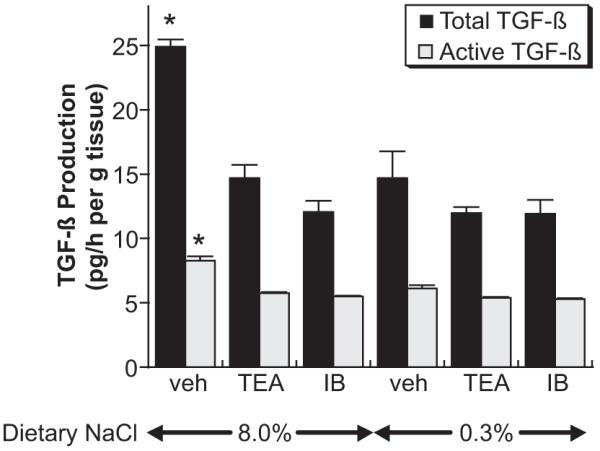
Effects of TEA (3 mmol/L) and IB (100 nM) on dietary salt-induced production of total and active TGF-β by aortic ring preparations from rats on 8.0% and 0.3% NaCl diets (n=4 rats in each group). Aortic rings from rats on the 8.0% NaCl diet produced more total and active TGF-β than ring preparations from animals on the low-salt diet. Both TEA and IB inhibited these increases but did not change production from rings of the low-salt animals. *P<0.05 vs the other 5 groups.
Endothelial Cells Express the BKCa Channel
Immunohistochemistry and Western blotting were performed using an antibody that recognized the α subunit of the BKCa channel. Immunohistochemistry identified endothelial cell expression of the BKCa channel in animals on both 0.3% and 8.0% NaCl diets (Figure 2). Expression of the channel was also observed in the adjacent vascular smooth muscle layers. Western blotting using lysates of endothelial cells demonstrated the presence of the α subunit at 125 kDa in every sample (Figure 3). The mean ratio of the density of the 125-kDa band to GAPDH did not differ between those animals on 0.3% NaCl versus the 8.0% NaCl diet (0.51±0.06 versus 0.62±0.14; P>0.05). Thus, dietary salt intake did not alter the protein expression of the subunit.
Figure 2.

Immunohistochemical stain of aortic tissue from rats on the 2 diets, showing expression of the pore-forming α subunit of the BKCa channel by endothelial cells (arrowheads) and adjacent vascular smooth muscle cells. A negative control, which did not include processing with the primary antibody, was provided for comparison. L indicates lumen. ■, 20 μm.
Figure 3.

Western analysis of endothelial cell lysates (n=4 rats in each group), showing the presence of the α subunit of the BKCa channel (top). Densitometric analysis showed that the mean ratio of the 125-kDa band to the GAPDH band did not differ between those animals on 0.3% NaCl vs 8.0% NaCl diets (0.51±0.06 vs 0.62±0.14; P>0.05).
Medium K+ Concentration Determined Dietary Salt-Induced TGF-β Production
The standard concentration of K+ in DMEM is 5.4 mmol/L. In these studies, the medium concentration of K+ was adjusted between 0 and 20 mmol/L. Choline chloride was added to maintain a constant osmolality and chloride concentration. An increase in the medium K+ concentration inhibited production of dietary salt-induced vascular production levels of total and active TGF-β but did not alter TGF-β production by aortic rings from rats on the 0.3% NaCl diet (Figure 4). Two-way ANOVA with effect analysis confirmed a positive effect of dietary salt intake on the production of both total and active TGF-β. In addition, a negative effect of medium potassium concentration on total and active TGF-β was also shown. Finally, a significant interaction between medium potassium concentration and salt intake was identified, but only in those experiments that used aortic rings from rats on the high-salt diet. The analysis suggested a trend in which those aortic rings derived from animals on high-salt diets produced less total and active TGF-β as the concentration of potassium increased in the medium.
Figure 4.
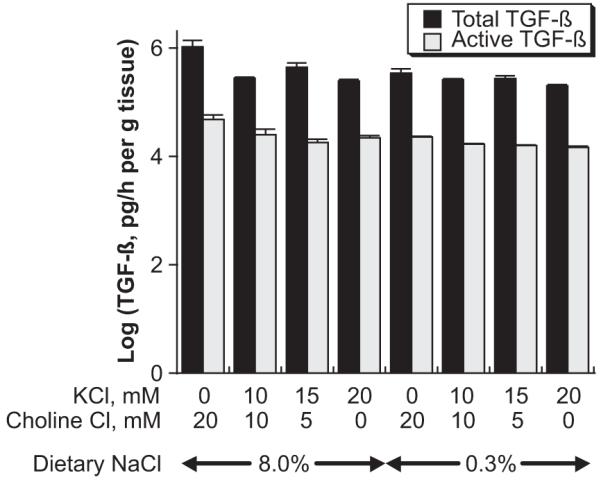
Effect of altering bath potassium concentration on production of total and active TGF-β by aortic ring preparations from rats on 8.0% and 0.3% NaCl diets (n=4 rats in each group). Increasing medium potassium concentration reduced total and active TGF-β production to levels observed using aortic rings from the low-salt animals. In these experiments, choline chloride was added to control for chloride concentration and osmolality. Two-way ANOVA with effect analysis demonstrated that both dietary salt intake and medium potassium concentration modulated the production of total and active TGF-β.
Dietary K+ Altered Salt-Induced Production of TGF-β
Groups of rats (n=6 to 12 in each group) received 1 of 6 different formulated diets that contained different amounts of NaCl and K+. Three of the diets contained 0.3% NaCl (wt/wt) and 0%, 0.95%, or 1.99% K+ (wt/wt), and the other 3 diets contained 8.0% NaCl (wt/wt) and 0%, 0.95%, or 1.99% K+ (wt/wt). After 4 days on the diet, urine was obtained to determine TGF-β. At this time, concentrations of serum urea nitrogen and creatinine did not differ among the 6 groups; serum K+ concentrations were decreased (P<0.05) in the 2 groups that received 0% K+ compared with the other groups (Table). In these studies, both NaCl and K intakes altered urinary active TGF-β excretion rates. A significant interaction between these parameters was observed only in the high-salt condition, suggesting that dietary K+ intake did not alter urinary active TGF-β in animals receiving the low-salt diet, and an increase in dietary K decreased urinary active TGF-β in animals receiving the high-salt diet (Figure 5).
Table. Whole Animal Data From the 6 Groups of Rats After 4 Days on the Study Diets.
| Group | n | Serum Potassium, mEq/L | Serum Urea Nitrogen, mg/dL | Serum Creatinine, mg/dL |
|---|---|---|---|---|
| 0.3% NaCl/0% K+ | 8 | 3.6±0.4* | 9.2±0.4 | 0.5±0.0 |
| 0.3% NaCl/0.95% K+ | 7 | 5.9±0.4 | 7.6±0.8 | 0.4±0.0 |
| 0.3% NaCl/1.9% K+ | 12 | 5.3±0.3 | 10.2±1.8 | 0.5±0.0 |
| 8.0% NaCl/0% K+ | 6 | 3.1±0.4* | 6.3±1.1 | 0.4±0.0 |
| 8.0% NaCl/0.95% K+ | 7 | 5.2±0.3 | 9.6±3.0 | 0.5±0.2 |
| 8.0% NaCl/1.9% K+ | 7 | 5.1±0.5 | 7.4±0.9 | 0.4±0.1 |
P<0.05 vs the other 2 groups on the same NaCl diet.
Figure 5.
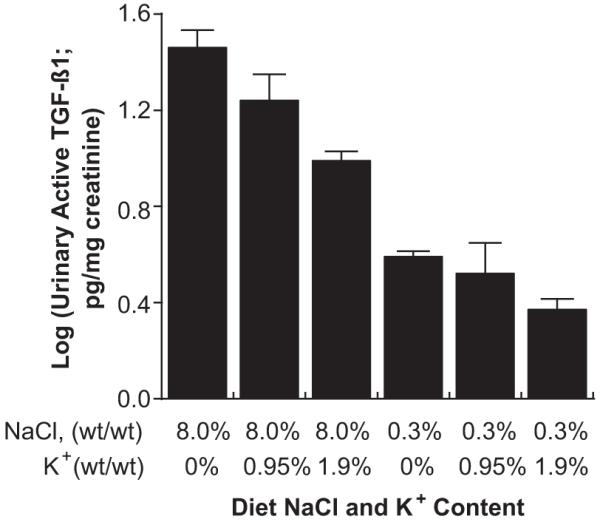
Urinary excretion rate of active TGF-β in rats on the 8.0% NaCl diet was modulated by dietary potassium intake. Changing potassium intake did not alter urinary TGF-β in rats on the 0.3% NaCl diet. In these in vivo experiments, both dietary salt and potassium intake modulated the urinary excretion rate of active TGF-β.
Discussion
The present study elucidated a role for potassium in dietary salt-mediated vascular production of TGF-β. Previous studies demonstrated that TEA, a nonselective potassium channel inhibitor, but not glibenclamide, a selective KATP channel inhibitor, inhibited salt-induced TGF-β production by aortic ring preparations and isolated glomeruli.9 The involvement of a TEA-sensitive potassium channel was again supported by the present observations (Figure 1). Also consistent with published studies,18 BKCa channels were identified on endothelial cells. Novel findings of this study include the pharmacological identification of this channel in endothelial cell TGF-β production induced by excess salt intake. Extracellular potassium concentration also modulated TGF-β production by aortic rings from high-salt-treated rats but not by aortic rings from the low-salt-treated rats. Finally, dietary potassium intake regulated urinary excretion of active TGF-β during ingestion of a high-salt diet. The combined findings demonstrated an interesting interaction between the dietary intake of potassium and excess sodium chloride and further showed the fundamental participation of the endothelial BKCa channel in the vascular response to excess salt intake.
Large-conductance BKCa channels are expressed in many tissues and cells and play an integral role in a variety of physiological events. Regulation of BKCa channels is complex and varies widely among cell types.18,19 Channel activity is generally involved in the regulation of membrane voltage and intracellular calcium concentrations.18 BKCa channels can respond to a variety of intracellular signaling events, as well as NO, CO, and phosphatidylinositol 4,5-biphosphate.20,21 Hypoxia, flow, and dietary potassium intake can regulate this channel.21-23 In the kidney, epithelial BKCa channels open in response to increases in tubular fluid flow, resulting in flow-mediated potassium secretion.23 The present studies demonstrated that excess salt intake is another stimulus that did not increase endothelial cell expression of the poreforming α subunit of the BKCa channel, as shown by Western analysis and immunohistochemistry, but did activate this channel, as revealed by IB, a highly selective pharmacological inhibitor.17 Activation of the channel, in turn, resulted in production of TGF-β. Although the present studies did not demonstrate the mechanism of activation of endothelial BKCa channels, the findings were compatible with flow-mediated generation of shear forces, which have been shown to activate potassium channels and stimulate production of TGF-β by cultured endothelial cells in vitro.24
Perspectives
The adverse effect of excess salt intake on the life span of rodents is impressive. These animals appear to succumb to cardiovascular disease induced by the high-salt diet.4,25 The same investigators experimented with changing the potassium content in the diet and found that supplementing dietary potassium favorably shifted the survival curves of rats on high-salt diets to the right. For example, the median duration of life of rats eating an 8.4% NaCl diet that contained a ratio of K+:Na+ of 0.8 increased to 24 months compared with a median life span of 16 months for rats consuming a diet containing 8.4% NaCl with a K+:Na+ ratio of 0.11.26 More recently, Cook et al27 showed that a higher urinary sodium:potassium excretion ratio was associated with an increased risk of cardiovascular disease. TGF-β has been linked to changes in compliance of conduit arteries through the promotion of vascular smooth muscle hypertrophy28 and by increasing the production of extracellular matrix proteins29 and inhibiting activity of those metalloproteinases involved in collagen degradation and remodeling.30 Altered vascular production of TGF-β has also been associated with hypertension from peripheral vasoconstriction.31 Although TGF-β alters the mechanics of conduit and resistance arteries, the interactions of the present findings with hypertension and salt sensitivity, per se, deserve consideration. Although the Sprague-Dawley rodent strain used in these experiments is relatively salt resistant, and blood pressure did not increase during the time frame of the present study,10 hypertension increases TGF-β production,13,32 and further dietary potassium intake attenuates the blood pressure increase that occurs with salt loading in susceptible humans and rats.33 Although there are several possible explanations for the protection from salt-induced cardiovascular disease, the findings of the present study suggest that 1 beneficial effect of potassium supplementation might relate to decreased salt-induced TGF-β production.
Acknowledgments
Sources of Funding
National Institutes of Health grants R01 DK046199 and P30 DK079337 (George M. O’Brien Kidney and Urologic Research Centers Program) and the Office of Research and Development, Medical Research Service, Department of Veterans’ Affairs, supported this research.
Footnotes
Disclosures
None.
References
- 1.Morimoto A, Uzu T, Fujii T, Nishimura M, Kuroda S, Nakamura S, Inenaga T, Kimura G. Sodium sensitivity and cardiovascular events in patients with essential hypertension. Lancet. 1997;350:1734–1737. doi: 10.1016/S0140-6736(97)05189-1. [DOI] [PubMed] [Google Scholar]
- 2.Weinberger MH, Fineberg NS, Fineberg SE, Weinberger M. Salt sensitivity, pulse pressure, and death in normal and hypertensive humans. Hypertension. 2001;37:429–432. doi: 10.1161/01.hyp.37.2.429. [DOI] [PubMed] [Google Scholar]
- 3.Cook NR, Cutler JA, Obarzanek E, Buring JE, Rexrode KM, Kumanyika SK, Appel LJ, Whelton PK. Long term effects of dietary sodium reduction on cardiovascular disease outcomes: observational follow-up of the trials of hypertension prevention (TOHP) BMJ. 2007;334:885. doi: 10.1136/bmj.39147.604896.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Meneely GR, Ball CO. Experimental epidemiology of chronic sodium chloride toxicity and the protective effect of potassium chloride. Am J Med. 1958;25:713–725. doi: 10.1016/0002-9343(58)90009-3. [DOI] [PubMed] [Google Scholar]
- 5.Yu HCM, Burrell LM, Black MJ, Wu LL, Dilley RJ, Cooper ME, Johnston CI. Salt induces myocardial and renal fibrosis in normotensive and hypertensive rats. Circulation. 1998;98:2621–2628. doi: 10.1161/01.cir.98.23.2621. [DOI] [PubMed] [Google Scholar]
- 6.Sanders PW, Gibbs CL, Akhi KM, MacMillan-Crow LA, Zinn KR, Chen Y-F, Young CJ, Thompson JA. Increased dietary salt accelerates chronic allograft nephropathy in rats. Kidney Int. 2001;59:1149–1157. doi: 10.1046/j.1523-1755.2001.0590031149.x. [DOI] [PubMed] [Google Scholar]
- 7.Benstein JA, Feiner HD, Parker M, Dworkin LD. Superiority of salt restriction over diuretics in reducing renal hypertrophy and injury in uninephrectomized SHR. Am J Physiol. 1990;258(6 pt 2):F1675–F1681. doi: 10.1152/ajprenal.1990.258.6.F1675. [DOI] [PubMed] [Google Scholar]
- 8.Swift PA, Markandu ND, Sagnella GA, He FJ, MacGregor GA. Modest salt reduction reduces blood pressure and urine protein excretion in black hypertensives: a randomized control trial. Hypertension. 2005;46:308–312. doi: 10.1161/01.HYP.0000172662.12480.7f. [DOI] [PubMed] [Google Scholar]
- 9.Ying W-Z, Sanders PW. Dietary salt modulates renal production of transforming growth factor-β in rats. Am J Physiol. 1998;274(4 pt 2):F635–F641. doi: 10.1152/ajprenal.1998.274.4.F635. [DOI] [PubMed] [Google Scholar]
- 10.Ying W-Z, Sanders PW. Dietary salt increases endothelial nitric oxide synthase and TGF-β1 in rat aortic endothelium. Am J Physiol. 1999;277(4 pt 2):H1293–H1298. doi: 10.1152/ajpheart.1999.277.4.H1293. [DOI] [PubMed] [Google Scholar]
- 11.Ying W-Z, Sanders PW. Increased dietary salt activates rat aortic endothelium. Hypertension. 2002;39:239–244. doi: 10.1161/hy0202.104142. [DOI] [PubMed] [Google Scholar]
- 12.Ying W-Z, Sanders PW. Dietary salt intake activates MAP kinases in the rat kidney. FASEB J. 2002;16:1683–1684. doi: 10.1096/fj.02-0982fje. [DOI] [PubMed] [Google Scholar]
- 13.Ying W-Z, Sanders PW. The interrelationship between TGF-β1 and nitric oxide is altered in salt-sensitive hypertension. Am J Physiol Renal Physiol. 2003;285:F902–F908. doi: 10.1152/ajprenal.00177.2003. [DOI] [PubMed] [Google Scholar]
- 14.Ying WZ, Aaron K, Sanders PW. Mechanism of dietary salt-mediated increase in intravascular production of TGF-β1. Am J Physiol Renal Physiol. 2008;295:F406–F414. doi: 10.1152/ajprenal.90294.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ying W-Z, Sanders PW. Dietary salt enhances glomerular endothelial nitric oxide synthase through TGF-β1. Am J Physiol. 1998;275(1 pt 2):F18–F24. doi: 10.1152/ajprenal.1998.275.1.F18. [DOI] [PubMed] [Google Scholar]
- 16.Abe M, Harpel JG, Metz CN, Nunes I, Loskutoff DJ, Rifkin DB. An assay for transforming growth factor-beta using cells transfected with a plasminogen activator inhibitor-1 promoter-luciferase construct. Anal Biochem. 1994;216:276–284. doi: 10.1006/abio.1994.1042. [DOI] [PubMed] [Google Scholar]
- 17.Galvez A, Gimenez-Gallego G, Reuben JP, Roy-Contancin L, Feigenbaum P, Kaczorowski GJ, Garcia ML. Purification and characterization of a unique, potent, peptidyl probe for the high conductance calcium-activated potassium channel from venom of the scorpion Buthus tamulus. J Biol Chem. 1990;265:11083–11090. [PubMed] [Google Scholar]
- 18.Nilius B, Droogmans G. Ion channels and their functional role in vascular endothelium. Physiol Rev. 2001;81:1415–1459. doi: 10.1152/physrev.2001.81.4.1415. [DOI] [PubMed] [Google Scholar]
- 19.Knaus HG, Eberhart A, Koch RO, Munujos P, Schmalhofer WA, Warmke JW, Kaczorowski GJ, Garcia ML. Characterization of tissue-expressed alpha subunits of the high conductance Ca(2+)-activated K+ channel. J Biol Chem. 1995;270:22434–22439. doi: 10.1074/jbc.270.38.22434. [DOI] [PubMed] [Google Scholar]
- 20.Lu R, Alioua A, Kumar Y, Eghbali M, Stefani E, Toro L. MaxiK channel partners: physiological impact. J Physiol. 2006;570:65–72. doi: 10.1113/jphysiol.2005.098913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cui J, Yang H, Lee US. Molecular mechanisms of BK channel activation. Cell Mol Life Sci. 2009;66:852–875. doi: 10.1007/s00018-008-8609-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Navarro-Antolin J, Levitsky KL, Calderon E, Ordonez A, Lopez-Barneo J. Decreased expression of maxi-K+ channel beta1-subunit and altered vasoregulation in hypoxia. Circulation. 2005;112:1309–1315. doi: 10.1161/CIRCULATIONAHA.104.529404. [DOI] [PubMed] [Google Scholar]
- 23.Woda CB, Bragin A, Kleyman TR, Satlin LM. Flow-dependent K+ secretion in the cortical collecting duct is mediated by a maxi-K channel. Am J Physiol Renal Physiol. 2001;280:F786–F793. doi: 10.1152/ajprenal.2001.280.5.F786. [DOI] [PubMed] [Google Scholar]
- 24.Ohno M, Cooke JP, Dzau VJ, Gibbons GH. Fluid shear stress induces endothelial transforming growth factor beta-1 transcription and production: modulation by potassium channel blockade. J Clin Invest. 1995;95:1363–1369. doi: 10.1172/JCI117787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Meneely GR, Tucker RG, Darby WJ, Auerbach SH. Chronic sodium chloride toxicity: hypertension, renal and vascular lesions. Ann Intern Med. 1953;39:991–998. doi: 10.7326/0003-4819-39-5-991. [DOI] [PubMed] [Google Scholar]
- 26.Meneely GR, Ball CO, Youmans JB. Chronic sodium chloride toxicity: the protective effect of added potassium chloride. Ann Intern Med. 1957;47:263–273. doi: 10.7326/0003-4819-47-2-263. [DOI] [PubMed] [Google Scholar]
- 27.Cook NR, Obarzanek E, Cutler JA, Buring JE, Rexrode KM, Kumanyika SK, Appel LJ, Whelton PK. Joint effects of sodium and potassium intake on subsequent cardiovascular disease: the Trials of Hypertension Prevention Follow-Up Study. Arch Intern Med. 2009;169:32–40. doi: 10.1001/archinternmed.2008.523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Owens GK, Geisterfer AA, Yang YW, Komoriya A. Transforming growth factor-beta-induced growth inhibition and cellular hypertrophy in cultured vascular smooth muscle cells. J Cell Biol. 1988;107:771–780. doi: 10.1083/jcb.107.2.771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ignotz RA, Endo T, Massague J. Regulation of fibronectin and type I collagen mRNA levels by transforming growth factor-beta. J Biol Chem. 1987;262:6443–6446. [PubMed] [Google Scholar]
- 30.Mozes MM, Bottinger EP, Jacot TA, Kopp JB. Renal expression of fibrotic matrix proteins and of transforming growth factor-beta (TGF-beta) isoforms in TGF-beta transgenic mice. J Am Soc Nephrol. 1999;10:271–280. doi: 10.1681/ASN.V102271. [DOI] [PubMed] [Google Scholar]
- 31.Zacchigna L, Vecchione C, Notte A, Cordenonsi M, Dupont S, Maretto S, Cifelli G, Ferrari A, Maffei A, Fabbro C, Braghetta P, Marino G, Selvetella G, Aretini A, Colonnese C, Bettarini U, Russo G, Soligo S, Adorno M, Bonaldo P, Volpin D, Piccolo S, Lembo G, Bressan GM. Emilin1 links TGF-beta maturation to blood pressure homeostasis. Cell. 2006;124:929–942. doi: 10.1016/j.cell.2005.12.035. [DOI] [PubMed] [Google Scholar]
- 32.Tamaki K, Okuda S, Nakayama M, Yanagida T, Fujishima M. Transforming growth factor-beta 1 in hypertensive renal injury in Dahl salt-sensitive rats. J Am Soc Nephrol. 1996;7:2578–2589. doi: 10.1681/ASN.V7122578. [DOI] [PubMed] [Google Scholar]
- 33.Tobian L. Dietary sodium chloride and potassium have effects on the pathophysiology of hypertension in humans and animals. Am J Clin Nutr. 1997;65:606S–611S. doi: 10.1093/ajcn/65.2.606S. [DOI] [PubMed] [Google Scholar]