

Abstract
There is a plethora of attractive drug targets in cancer, but their therapeutic exploitation proved more difficult than expected, and only rarely truly successful. One possibility not often considered in drug discovery is that cancer signaling pathways are not randomly arranged in cells, but orchestrated in specialized subcellular compartments. The identification of Heat Shock Protein-90 (Hsp90) chaperones in mitochondria of tumors, but not most normal tissues, provides an example of a compartmentalized network of cell survival, opening fresh prospects for novel, subcellularly-targeted cancer drug discovery.
Keywords: Hsp90, mitochondrial permeability transition, CypD, protein folding, cancer therapy
Cancer drug discovery in the post-Imatinib era
The improved knowledge of cancer genes (Vogelstein and Kinzler, 2004), coupled with the feasibility of disabling specific molecular lesion(s) in tumors (O'Dwyer and Druker, 2000), ushered the era of “targeted” drugs (Sawyers, 2004): molecular “magic bullets” that would change forever the landscape of cancer care. Unfortunately, this expectation met a much harsher reality. The extraordinary complexity of most malignancies with hundreds of mutated or deregulated gene pathways (Wood et al., 2007), the astronomical costs and low success rate (typically 0.0001%) of “target-centric” drug discovery (van der Greef and McBurney, 2005), and the herculean task of fulfilling hundreds of regulatory steps and decision points to bring new agents to the clinic (Steensma, 2009), have greatly hampered the advent of molecular cancer therapeutics. As a result, oncology trial design all too often pursues small, and at times clinically insignificant gains, over 50% of the largest (and costliest) oncology phase III trials fails to produce benefit, and new cancer drug registration has actually fallen by 40% over the past decade (Schein and Scheffler, 2006). These disappointments have not gone unnoticed, and patient advocacy groups and media organizations (Kolata, 2009; Leaf, 2004) are urging new venues to improve cancer drug discovery, and produce, if not the “cure”, at least meaningful advances in patient survival.
Pathway-oriented drug discovery and the Hsp90 chaperone network
A fresh approach to these challenges is to take advantage of systems biology tools, and model cancer pathways in their globality (Rajasethupathy et al., 2005). Connectivity maps (Lamb et al., 2006) linking multiple signaling pathways may more accurately recapitulate tumor heterogeneity, uncover redundancy and buffering, and ultimately identify nodal proteins, hub molecules integrating multiple downstream mechanisms of cell proliferation, survival, and adaptation (Butcher, 2005; Rajasethupathy et al., 2005). Nodal proteins, for instance the EGF receptor (Citri and Yarden, 2006), offer prime opportunities for cancer drug discovery, as their antagonists may provide genuine pathway inhibitors, disabling entire signaling network(s), instead of a single target, and thus suited to overcome tumor heterogeneity (Wood et al., 2007).
The molecular chaperone Hsp90 is another example of a nodal protein (McClellan et al., 2007), intersecting multiple signaling networks (Zhao and Houry, 2007). Hsp90 and Hsp90-like molecules (see below) oversee protein folding quality control (Young et al., 2001), trafficking of signaling molecules at the plasma membrane (Garcia-Cardena et al., 1998), delivery of preproteins to mitochondria (Young et al., 2003), assembly and disassembly of cytoskeletal proteins in the cytosol (Barral et al., 2002), and disassembly of transcriptional complexes in the nucleus (Freeman and Yamamoto, 2002). These functions require sequential cycles of ATPase activity (Prodromou et al., 1997), and recruitment of co-chaperones, including Hsp70, p50cdc37, Aha1, p60hop, and p23 (Pearl and Prodromou, 2000). Differently from Hsp70, which controls folding of all newly synthesized proteins (Schmid et al., 1994), Hsp90 oversees the maturation and/or stability of client proteins implicated in hormone and growth factor receptor signaling, cell cycle progression, and cell survival (Pearl and Prodromou, 2000; Young et al., 2003). This pathway is exploited in cancer, where Hsp90 chaperoning (Whitesell and Lindquist, 2005), contributes to drug resistance (Cowen and Lindquist, 2005), metastasis (Eustace et al., 2004), tumor cell survival (Rodina et al., 2007), and transcriptional mechanisms of transformation (Dai et al., 2007).
Targeting Hsp90 for novel cancer therapeutics: clinical experience and unanswered questions
In addition to these nodal properties, other features make Hsp90 an almost ideal cancer drug target (Isaacs et al., 2003). Its ATPase pocket can accommodate small molecule inhibitors (Isaacs et al., 2003), and Hsp90 protein levels (Neckers, 2002), as well as ATPase activity (Kamal et al., 2003) are increased in tumors, compared to normal tissues, suggesting that Hsp90 therapeutics may have desirable selectivity (Isaacs et al., 2003). Accordingly, multiple small molecule Hsp90 antagonists have been developed from 17-allylaminogeldanamycin (17-AAG), a less toxic derivative of the benzoquinone ansamycin antibiotic, Geldanamycin (GA) (Isaacs et al., 2003), or, more recently, from purine or resorcinol structures (Solit and Chiosis, 2008). These agents competitively inhibit the Hsp90 ATPase activity, shut off the chaperone function, and induce degradation of multiple client proteins (Isaacs et al., 2003). In turn, this exerts anticancer activity, predominantly through cell cycle arrest, followed by variable degrees of apoptosis, especially in “sensitive” cell types (Isaacs et al., 2003; Solit and Chiosis, 2008). Despite this promising background, the response to Hsp90 antagonists in the clinic has been in general disappointing (Table 1), certainly inferior to what was expected from potential “pathway inhibitors” (Drysdale et al., 2006). These less-than-impressive clinical data (Table 1) raised questions about drug efficacy, whether Hsp90 is as nodal a target as it was hoped, or, rather, whether other aspects of Hsp90 biology needed to be uncovered in order to unlock the full therapeutic potential of these agents.
Table 1.
Clinical experience with current small molecule Hsp90 antagonists.
| Drug | Trial | Patients | Regimen | Outcome | Citation |
|---|---|---|---|---|---|
| 17-AAG | Phase I | 21 | Single agent | NR | J Clin Oncol 23:1078 |
| 17-AAG | Phase I | 19 | Single agent | 3 SD | J Clin Oncol 23:1885 |
| 17-AAG | Phase I | 30 | Single agent | 2 SD | J Clin Oncol 23:4152 |
| 17-AAG | Phase I | 45 | Single agent | NR | Clin Cancer Res 11:3385 |
| 17-AAG | Phase I | 13 | Single agent | NR | Clin Cancer Res 12:6087 |
| 17-AAG | Phase II (RCC) |
20 | Single agent | NR | Invest. New Drugs 24:543 |
| 17-AAG | Phase I | 54 | Single agent | NR | Clin Cancer Res 13:1775 |
| 17-AAG | Phase I | 44 | Single agent | NR | Clin Cancer Res 13:1769 |
| 17-AAG | Phase I | 15 (Ped) | Single agent | NR | Clin Cancer Res 13:1783 |
| 17-AAG | Phase I | 17 (Ped) | Single agent | NR, 5 SD | Clin Cancer Res 13:1789 |
| 17-AAG | Phase I | 25 | plus Trastuzumab |
1 PR, 4 MR 4 SD |
J Clin Oncol 25:5410 |
| 17-AAG | Phase I | 25 | plus Paclitaxel | NR, 6 SD | Clin Cancer Res 14:3456 |
| 17-AAG | Phase I | 27 | plus Irinotecan | NR, 11 SD | Clin Cancer Res 14:6704 |
| 17-AAG | Phase II (HRPC) |
15 | Single agent | Trial discontinued |
Clin Cancer Res 14:7940 |
| 17-AAG | Phase II (MM) |
15 | Single agent | NR | Clin Cancer Res 14:8302 |
| IPI-504 | Phase III (GIST) |
Single agent | Trial discontinued |
http://investor.ipi.com/releasedetail.cfm?ReleaseID=377328 | |
| IPI-504 | Phase II (HRPC) |
Single agent | Trial discontinued |
http://biz.yahoo.com/pz/080714/146331.html |
Ped, pediatric patients; RCC, renal cell carcinoma; HRPC, hormone-refractory prostate cancer; MM, malignant melanoma; GIST, gastrointestinal stromal tumor; NR, no objective response; PR, partial response; MR, minor response; SD, stable disease.
Compartmentalization of Hsp90 chaperones in tumor mitochondria
At least one of the missing aspects of Hsp90 biology that may influence the response to therapy is its novel subcellular localization to mitochondria. Originally noted in a global survey of the mitochondrial proteome (Mootha et al., 2003), Hsp90 is abundantly present in mitochondria, and sorted to both the organelle intermembrane space and the matrix (Kang et al., 2007). An Hsp90-like chaperone, Tumor Necrosis Factor Receptor-Associated Protein-1, or TRAP-1 is also a mitochondrial protein (Cechetto and Gupta, 2000; Felts et al., 2000), localized to the organelle matrix (Kang et al., 2007), as well as the intermembrane space (Pridgeon et al., 2007), in vivo. This subcellular localization is selective, in that both Hsp90 chaperones are differentially expressed in mitochondria of tumor cells, while undetectable or expressed at very low levels in the organelles of most normal tissues (see below), in vivo (Kang et al., 2007). The basis for this tumor preference has not been determined, but it may have to do with changes in mitochondrial trafficking in transformed cells. Accordingly, deregulation of cancer signaling pathways, for instance Akt, has been associated with a more avid mitochondrial preprotein import machinery (Wright et al., 2008), and Ras transformation of model fibroblasts, but not glucose deprivation, was sufficient to upregulate Hsp90 levels in mitochondria (Kang et al., 2007).
Once in mitochondria, Hsp90 and TRAP-1 bind the matrix immunophilin, cyclophilin D (CypD) (Kang et al., 2007). These interactions are direct, recapitulated in cell-free systems, and non-overlapping, as TRAP-1-CypD complexes do not contain Hsp90, and vice versa (Kang et al., 2007). CypD is a peptidyl-prolyl cis, trans isomerase long recognized as a physical component of a mitochondrial permeability transition pore (Woodfield et al., 1998). Opening of the pore in response to cell death or stress stimuli is thought to mediate an acute increase in organelle conductance to solutes, a process called permeability transition (Green and Kroemer, 2004; Tsujimoto and Shimizu, 2007). In turn, this triggers a cascade of events, including swelling of the matrix, loss of inner membrane potential, remodeling of the cristae, and ultimately rupture of the outer membrane with release of apoptogenic proteins in the cytosol (Green and Kroemer, 2004; Tsujimoto and Shimizu, 2007). There is little doubt that mitochondrial permeability transition initiates cell death. What is less clear is the timing of these steps, how they are interconnected, or even the composition of a permeability transition pore (Halestrap, 2009). For instance, long-held constituents of the pore, such as the voltage-dependent anion channel (VDAC) (Baines et al., 2007), and the adenine nucleotide translocator (ANT) (Kokoszka et al., 2004) turned out to be dispensable for apoptosis, in vivo, and only CypD was required for some, but not all, forms of cell death, in particular those triggered by oxidative stress and Ca2+ overload (Baines et al., 2005; Nakagawa et al., 2005).
Hsp90 chaperones as novel regulators of mitochondrial permeability transition
To reconcile these seemingly contradictory views, an alternative model for a dynamic permeability transition pore was proposed, in which clusters of misfolded proteins generated in response to oxidative stress acquire the ability to function as a CypD-regulated pore (He and Lemasters, 2002). A prediction of this model was that yet-to-be-discovered molecular chaperones opposed the pore-forming functions of CypD via protein (re)folding mechanisms, and thus preserve mitochondrial integrity (He and Lemasters, 2002). The fact that Hsp90 and TRAP-1 are novel CypD-associated chaperones (Kang et al., 2007) fits well with this scenario. Functional data are also consistent with the model, as acute knockdown of TRAP-1 triggered CypD-dependent apoptosis (Kang et al., 2007; Masuda et al., 2004), whereas overexpression of TRAP-1 protected against oxidative cell death (Hua et al., 2007; Kang et al., 2007; Montesano Gesualdi et al., 2007). As far as pathophysiological relevance, this cytoprotective pathway emerged as an important regulator of neuronal homeostasis. Accordingly, TRAP-1 functions as a survival factor for neurons (Xu et al., 2008), and astrocytes (Voloboueva et al., 2007), via an activating phosphorylation by the PTEN-induced kinase, PINK1 (Pridgeon et al., 2007). In line with this model, PINK1 is also anti-apoptotic in neurons (MacKeigan et al., 2005), countering oxidative damage (Petit et al., 2005), and loss-of-function mutations in this kinase are associated with certain types of familial Parkinson’s disease characterized by neuronal wasting (Henchcliffe and Beal, 2008). Whether CypD is the downstream target of TRAP-1 neuroprotection (Pridgeon et al., 2007) is currently not known. What is known, however, is that CypD mediates neuronal cell death after focal cerebral ischemia (Schinzel et al., 2005), and neurodegenerative conditions (Forte et al., 2007), including Alzheimer’s disease (Du et al., 2008). As one of only two exceptions in normal tissues, mitochondrial Hsp90 is present in the brain (Kang et al., 2007), but whether it has protective functions for neurons has not yet been tested. Although a PINK1-TRAP-1-CypD axis may regulate the balance of neuronal apoptosis, the converse approach of selectively disabling this pathway in mitochondria may have intriguing implications for cancer therapeutics. Therefore, an obvious question is whether the Hsp90 antagonists currently in the clinic (Table 1) accumulate in mitochondria, and inhibit this compartmentalized pool of the chaperones. Surprisingly, neither GA- (Kang et al., 2007), nor non-GA (Kang et al., 2009)-based antagonists accumulated in tumor mitochondria, suggesting that these subcellular pools of Hsp90 and TRAP-1 escaped inhibition by current Hsp90 therapeutics.
Targeting mitochondrial Hsp90 chaperones for compartmentalized, pathway-oriented cancer drug discovery
The concept of “mitochondrial medicine”, aimed at manipulating cell death in human diseases has long been pursued for experimental therapeutics (Armstrong, 2007). Much of the challenge resides in how best to deliver cargos to mitochondria, either to dampen oxidative stress for improved cell survival (Szeto, 2008), or, conversely, trigger permeability transition and cell death (Armstrong, 2007). The latter strategy has appeal for cancer therapeutics because it may directly lower a general survival threshold in tumors (Pilkington et al., 2008), regardless of their heterogeneity or genetic makeup (Fig. 1). This approach is a conceptual advance over conventional anticancer regimens, which also activate mitochondrial cell death (Johnstone et al., 2002), but do so indirectly as a result of checkpoint activation, DNA damage, etc., and are thus susceptible to compensatory and redundant survival signals (Igney and Krammer, 2002) (Fig. 1). The concept of truly “mitochondriotoxic” agents, directly inducing organelle collapse (Fantin and Leder, 2006) (Fig. 1) materialized with the development of small molecule mimetics of pro-apoptotic Bcl-2 proteins (Zeitlin et al., 2008), designed to directly permeabilize the mitochondrial outer membrane, and trigger permeability transition (Green and Kroemer, 2004; Tsujimoto and Shimizu, 2007). These compounds are showing encouraging responses in certain hematologic malignancies (Zeitlin et al., 2008), but the sheer redundancy of Bcl-2 proteins requires simultaneous blockade of multiple members for optimal efficacy (Vogler et al., 2008), and examples of compensatory mechanisms executed by other pro-survival Bcl-2 proteins have been reported (Deng et al., 2007).
Fig. 1. Mitochondriotoxic agents for cancer therapy.
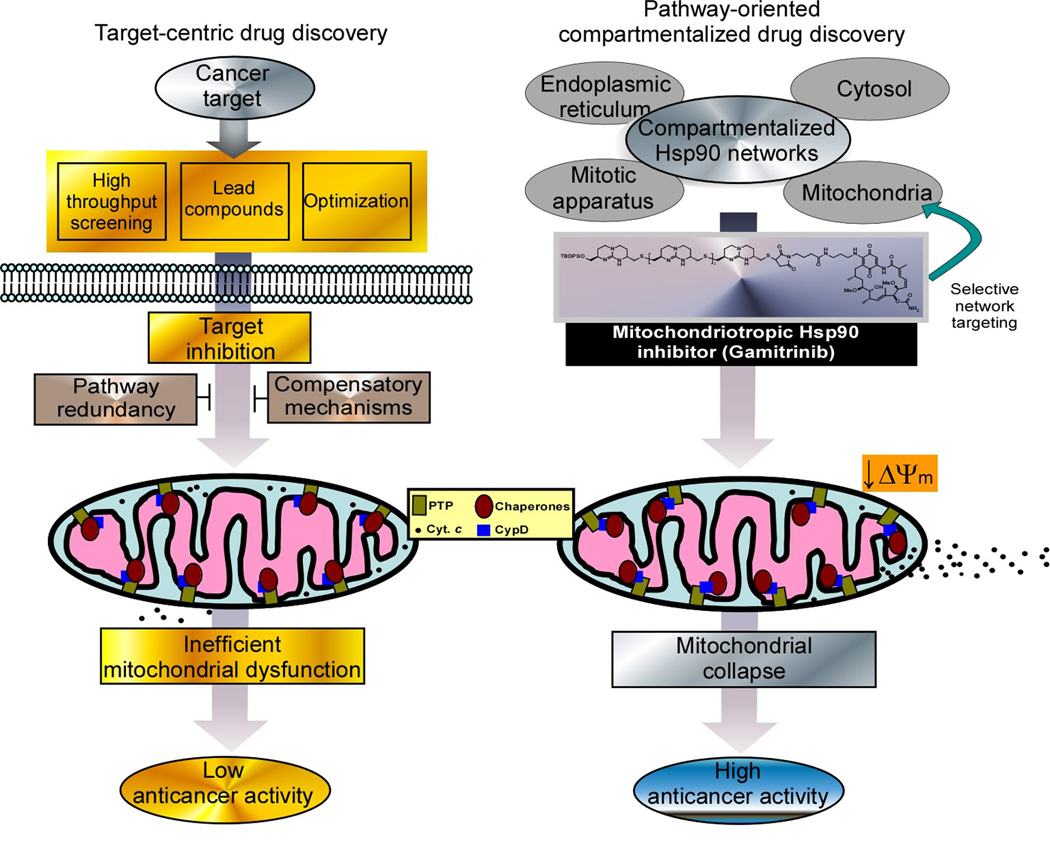
‘Target-centric’ cancer drug discovery (left panel) involves the selection of a target gene, and the generation of inhibitors by high throughput screening, and lead optimization. These agents may inhibit the target, and cause indirect activation of mitochondrial cell death, in vivo, but anticancer activity is often reduced by pathway redundancy and compensatory prosurvival signals. Conversely, combinatorial engineering of pathway inhibitors, for instance 17-AAG, to target a subcellular cancer network, i.e. mitochondria (right panel), generates antagonists that directly induce organelle collapse, and enhanced anticancer activity, bypassing compensatory survival mechanisms. ΔΨm, mitochondrial membrane potential, PTP, permeability transition pore; cyt c, cytochrome c, CypD, cyclophilin D.
To develop a direct mitochondriotoxic strategy targeting Hsp90 chaperones (Fig. 1), proof-of-concept experiments were carried out with Shepherdin, a peptidomimetic inhibitor of the interaction between Hsp90 and one of its client proteins, survivin (Plescia et al., 2005). Shepherdin crosses the plasma membrane via a positively charged Antennapedia helix III sequence (Torchilin, 2006) fused at the amino terminus of the peptidomimetic (Plescia et al., 2005). The ability of this moiety to deliver cargos to mitochondria has been debated (Li et al., 2007; Ross et al., 2004), and in any case likely reflects a general property of cross-membrane transfer, rather than a specific mitochondriotropic signal. Through its Antennapedia sequence, Shepherdin readily accumulated in all mitochondrial compartments (Kang et al., 2007), and within minutes triggered all the pathophysiological hallmarks of permeability transition, including membrane blebbing, loss of membrane potential, discharge of cytochrome c, and massive cell death (Gyurkocza et al., 2006; Kang et al., 2007). This pathway was mediated by CypD, independently of p53 status or expression of pro-survival Bcl-2 (Kang et al., 2007; Plescia et al., 2005), and correlated with physical binding of Shepherdin to Hsp90 and TRAP-1 inside mitochondria (Kang et al., 2007). In preclinical studies, systemic administration of Shepherdin was feasible, safe (see below), and resulted in inhibition of tumor growth in multiple xenograft models, in vivo (Gyurkocza et al., 2006; Plescia et al., 2005).
Prompted by these results, an approach to deliver a non-peptidyl, small molecule Hsp90 inhibitor selectively to mitochondria was undertaken. This involved combinatorial chemistry, linking the Hsp90 inhibitor, 17-AAG to lipophilic cations acting as mitochondrial targeting moieties. These were provided either by 1 to 4 tandem repeats of cyclic guanidinium (Fernandez-Carneado et al., 2005), or, alternatively, triphenylphosphonium (TPP) (Armstrong, 2007). The derived combinatorial compounds, designated Gamitrinibs (GA mitochondrial matrix inhibitors), accumulated in mitochondria, abolished Hsp90 ATPase activity (Kang et al., 2009), triggered permeability transition, and caused complete tumor cell killing, in vitro (Kang et al., 2009) (Fig. 1). For concentrations of unconjugated 17-AAG that had no effect in vivo, Gamitrinibs suppressed tumor growth in various xenograft models in mice, with evidence of mitochondrial dysfunction and apoptosis, in situ (Kang et al., 2009). Although Gamitrinibs likely do not discriminate between the two Hsp90 isoforms (α and β), these compounds differ sharply from all other non-subcellularly targeted Hsp90 antagonists for mechanism of action, that is cytotoxic instead of cytostatic (i), direct mitochondriotoxic activity (ii), and lack of effect on Hsp90 outside mitochondria (iii) (Kang et al., 2009). The latter property may reflect a rapid cytosolic transfer of these agents mediated by their mitochondrial-targeting moiety. For instance, suboptimal concentrations of Shepherdin induce a dual phenotype of mitochondrial dysfunction (Kang et al., 2007), as well as inhibition of cytosolic Hsp90 (Plescia et al., 2005), whereas Gamitrinibs are solely mitochondriotoxic, without affecting Hsp90 client protein stability in the cytosol. Because of this subcellular selectivity, Gamitrinibs do not cause compensatory elevation of anti-apoptotic Hsp70 levels (Beere et al., 2000), and are not expected to induce Src phosphorylation (Koga et al., 2006), which may enhance tumor cell invasion and metastasis (Price et al., 2005), two unwanted effects of non-subcellularly targeted Hsp90 inhibitors, i.e. 17-AAG.
Qualitatively different mechanisms of mitochondrial homeostasis in tumor versus normal tissues?
A distinctive feature of mitochondria-directed Hsp90 antagonists, Shepherdin (Gyurkocza et al., 2006; Plescia et al., 2005), and Gamitrinibs (Kang et al., 2009), is their safety for normal tissues. Although these agents do accumulate in normal mitochondria (Kang et al., 2009), they have no effect on permeability transition or cell viability, in vitro, and cause no signs of systemic toxicity, alterations in tissue histology, blood chemistry parameters, or bone marrow function, in vivo (Gyurkocza et al., 2006; Kang et al., 2007; Kang et al., 2009; Plescia et al., 2005). Several possibilities may explain this high degree of selectivity, which bodes well for a desirable therapeutic window of these agents. First, mitochondria of most normal tissues do not contain Hsp90 or TRAP-1 (Kang et al., 2007), and this may sharply reduce their sensitivity to Gamitrinib or Shepherdin (Fig. 2). Similar to their cytosolic counterparts (Kamal et al., 2003), it is also possible that tumor-associated mitochondrial Hsp90s exhibit a 100-fold increase in ATPase activity compared to normal tissues, further broadening the selectivity of these antagonists. Different kinetics of mitochondrial accumulation may also play a role, and the higher mitochondrial membrane potential of tumor cells compared to normal tissues (Chen, 1988), may facilitate a 10-fold preferential accumulation of Gamitrinibs, especially Gamitrinib-TPP, in mitochondria of tumor versus normal cells (Murphy, 2008). And, finally, there is the possibility that additional regulatory molecules, perhaps co-chaperones, be similarly recruited to tumor mitochondria and cooperate with Hsp90 protein (re)folding to oppose CypD (Fig. 2). The identity of these putative accessory molecules is unknown, but this model may explain why Gamitrinibs are apparently safe even for normal tissues that do contain mitochondrial Hsp90, for instance the brain (Kang et al., 2007). In this context, it would be the lack of other tumor-associated regulators of the mitochondrial Hsp90 network that reduce the sensitivity of these tissue(s) to the compounds.
Fig. 2. Qualitative differences in mitochondrial homeostasis in normal and tumor cells.
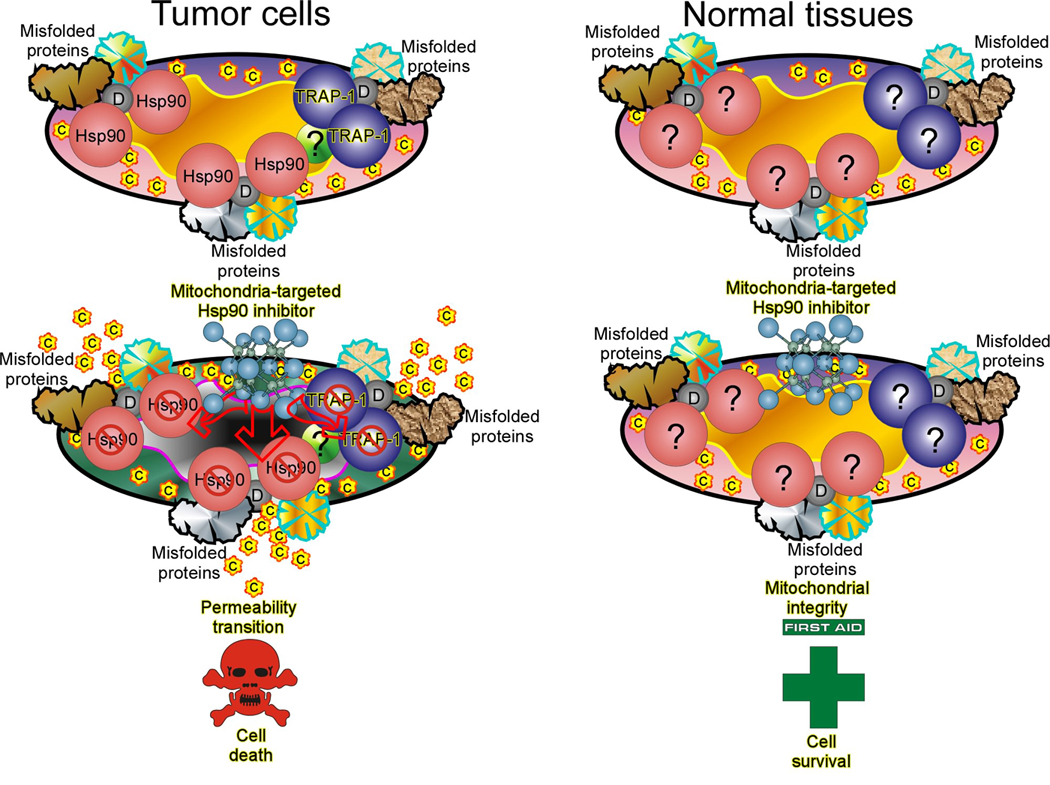
Mitochondria of tumor cells (left panel) exploit an Hsp90 chaperone protein refolding network to suppress Cyclophilin D (CypD)-mediated permeability transition. This may involve CypD participation in an organized permeability pore, or as a regulator of clustered, misfolded proteins formed in response to oxidative damage that acquire pore conductance (shown in the figure). Inhibition of organelle Hsp90 ATPase activity by mitochondria-directed Hsp90 antagonists (Shepherdin, Gamitrinibs) results in permeability pore opening, and CypD-dependent cell death. Conversely, mitochondria of most normal cells (right panel) are devoid of Hsp90 chaperones, and potentially also of regulators/co-chaperones in this pathway, suggesting that alternative mechanisms control CypD pore functions. This makes normal tissues insensitive to the action of mitochondria-targeted Hsp90 antagonists. C, cytochrome c; D, CypD.
Regardless of the mechanism, or mechanisms underlying tissue selectivity, one general hypothesis emerges from these findings, that the machinery controlling CypD pore-forming properties may be qualitatively different in tumor versus normal mitochondria (Fig. 2). In this context, it is possible that tumor cells commandeer a protective network of mitochondrial Hsp90 chaperones operative in selected normal tissues, where CypD function must be tightly controlled, for instance the brain (Forte et al., 2007; Schinzel et al., 2005). For tumor cells, the survival advantage conferred by this pathway would be almost ideal, specifically preventing CypD-mediated apoptosis triggered by oxidative stress, a condition invariably associated with tumor growth, in vivo (Fig. 2). This model may also explain a second key feature of mitochondria-directed Hsp90 inhibitors, namely their broad efficacy in so many unrelated tumor cell types. This suggests that transformed cells may become dependent, or “addicted” (Weinstein and Joe, 2006) to a steady-state, anti-oxidant survival threshold maintained by mitochondrial Hsp90 chaperones. Following this logic, and as observed experimentally, acute loss of this protective mechanism by mitochondria-targeted Hsp90 inhibitors cannot be compensated for, resulting in sudden organelle collapse and cell death, regardless of the genetic makeup of the tumor (Fig. 2).
Concluding remarks -mitochondrial medicine revisited
Taken together, these recent observations open intriguing opportunities. From a mechanistic perspective, there has been a tremendous effort to map the regulators of mitochondrial permeability transition (Green and Kroemer, 2004; Tsujimoto and Shimizu, 2007), and the role of some of the key players, for instance pro-apoptotic Bcl-2 family proteins, is now firmly established. Are now chaperones, co-chaperones and overall protein (re)folding mechanisms new chapters in the evolving saga of mitochondrial homeostasis (He and Lemasters, 2002), especially when it comes to oxidative stress? And if this is, in fact, a cancer signaling network, rather than the activity of individual molecules, which other players or co-chaperones cooperate with Hsp90s in taming CypD-mediated pore formation in tumor mitochondria? From a disease-relevant perspective, it is encouraging to see that a long pursuit of “mitochondrial medicine” (Armstrong, 2007), is finally paying off with attractive drugs in the clinic (Zeitlin et al., 2008), and other new agents, i.e. Shepherdin and Gamitrinibs (Gyurkocza et al., 2006; Kang et al., 2009; Plescia et al., 2005) on the horizon. Because of the flexible combinatorial platform of Gamitrinibs, a similar approach could be envisioned to direct smaller, purine- or resorcinol-based Hsp90 antagonists (Solit and Chiosis, 2008) to mitochondria, thus potentially further improving on their anticancer activity. In addition, the concept of directing therapeutic agents to specialized subcellular compartments need not be exclusively limited to mitochondria. As shown by the proof-of-concept data with Gamitrinibs (Kang et al., 2009), compartmentalized inhibition of signaling pathways could dramatically expand the repertoire of cancer drug discovery as a whole. This may mean fewer costly and low-yield screening efforts, generation of agents with new specificity and mechanism of action, and concrete therapeutic prospects for targeting cancer signaling pathways that are currently considered not “drugable”.
ACKNOWLEDGMENTS
We apologize to all the colleagues, whose work could not be cited due to space limitations. This work was supported by National Institutes of Health grants CA78810, CA90917 and CA118005.
Footnotes
CONFLICT OF INTEREST
The authors declare that no conflict of interest exists.
REFERENCES
- Armstrong JS. Mitochondrial medicine: pharmacological targeting of mitochondria in disease. Br J Pharmacol. 2007;151:1154–1165. doi: 10.1038/sj.bjp.0707288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baines CP, Kaiser RA, Purcell NH, Blair NS, Osinska H, Hambleton MA, et al. Loss of cyclophilin D reveals a critical role for mitochondrial permeability transition in cell death. Nature. 2005;434:658–662. doi: 10.1038/nature03434. [DOI] [PubMed] [Google Scholar]
- Baines CP, Kaiser RA, Sheiko T, Craigen WJ, Molkentin JD. Voltage-dependent anion channels are dispensable for mitochondrial-dependent cell death. Nat Cell Biol. 2007;9:550–555. doi: 10.1038/ncb1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barral JM, Hutagalung AH, Brinker A, Hartl FU, Epstein HF. Role of the myosin assembly protein UNC-45 as a molecular chaperone for myosin. Science. 2002;295:669–671. doi: 10.1126/science.1066648. [DOI] [PubMed] [Google Scholar]
- Beere HM, Wolf BB, Cain K, Mosser DD, Mahboubi A, Kuwana T, et al. Heat-shock protein 70 inhibits apoptosis by preventing recruitment of procaspase-9 to the Apaf-1 apoptosome. Nat Cell Biol. 2000;2:469–475. doi: 10.1038/35019501. [DOI] [PubMed] [Google Scholar]
- Butcher EC. Can cell systems biology rescue drug discovery? Nat Rev Drug Discov. 2005;4:461–467. doi: 10.1038/nrd1754. [DOI] [PubMed] [Google Scholar]
- Cechetto JD, Gupta RS. Immunoelectron microscopy provides evidence that tumor necrosis factor receptor-associated protein 1 (TRAP-1) is a mitochondrial protein which also localizes at specific extramitochondrial sites. Exp Cell Res. 2000;260:30–39. doi: 10.1006/excr.2000.4983. [DOI] [PubMed] [Google Scholar]
- Chen LB. Mitochondrial membrane potential in living cells. Annu Rev Cell Biol. 1988;4:155–181. doi: 10.1146/annurev.cb.04.110188.001103. [DOI] [PubMed] [Google Scholar]
- Citri A, Yarden Y. EGF-ERBB signalling: towards the systems level. Nat Rev Mol Cell Biol. 2006;7:505–516. doi: 10.1038/nrm1962. [DOI] [PubMed] [Google Scholar]
- Cowen LE, Lindquist S. Hsp90 potentiates the rapid evolution of new traits: drug resistance in diverse fungi. Science. 2005;309:2185–2189. doi: 10.1126/science.1118370. [DOI] [PubMed] [Google Scholar]
- Dai C, Whitesell L, Rogers AB, Lindquist S. Heat shock factor 1 is a powerful multifaceted modifier of carcinogenesis. Cell. 2007;130:1005–1018. doi: 10.1016/j.cell.2007.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng J, Carlson N, Takeyama K, Dal Cin P, Shipp M, Letai A. BH3 profiling identifies three distinct classes of apoptotic blocks to predict response to ABT-737 and conventional chemotherapeutic agents. Cancer Cell. 2007;12:171–185. doi: 10.1016/j.ccr.2007.07.001. [DOI] [PubMed] [Google Scholar]
- Drysdale MJ, Brough PA, Massey A, Jensen MR, Schoepfer J. Targeting Hsp90 for the treatment of cancer. Curr Opin Drug Discov Devel. 2006;9:483–495. [PubMed] [Google Scholar]
- Du H, Guo L, Fang F, Chen D, Sosunov AA, McKhann GM, et al. Cyclophilin D deficiency attenuates mitochondrial and neuronal perturbation and ameliorates learning and memory in Alzheimer's disease. Nat Med. 2008;14:1097–1105. doi: 10.1038/nm.1868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eustace BK, Sakurai T, Stewart JK, Yimlamai D, Unger C, Zehetmeier C, et al. Functional proteomic screens reveal an essential extracellular role for hsp90 alpha in cancer cell invasiveness. Nat Cell Biol. 2004;6:507–514. doi: 10.1038/ncb1131. [DOI] [PubMed] [Google Scholar]
- Fantin VR, Leder P. Mitochondriotoxic compounds for cancer therapy. Oncogene. 2006;25:4787–4797. doi: 10.1038/sj.onc.1209599. [DOI] [PubMed] [Google Scholar]
- Felts SJ, Owen BA, Nguyen P, Trepel J, Donner DB, Toft DO. The hsp90-related protein TRAP1 is a mitochondrial protein with distinct functional properties. J Biol Chem. 2000;275:3305–3312. doi: 10.1074/jbc.275.5.3305. [DOI] [PubMed] [Google Scholar]
- Fernandez-Carneado J, Van Gool M, Martos V, Castel S, Prados P, de Mendoza J, et al. Highly efficient, nonpeptidic oligoguanidinium vectors that selectively internalize into mitochondria. J Am Chem Soc. 2005;127:869–874. doi: 10.1021/ja044006q. [DOI] [PubMed] [Google Scholar]
- Forte M, Gold BG, Marracci G, Chaudhary P, Basso E, Johnsen D, et al. Cyclophilin D inactivation protects axons in experimental autoimmune encephalomyelitis, an animal model of multiple sclerosis. Proc Natl Acad Sci. 2007;104:7558–7563. doi: 10.1073/pnas.0702228104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freeman BC, Yamamoto KR. Disassembly of transcriptional regulatory complexes by molecular chaperones. Science. 2002;296:2232–2235. doi: 10.1126/science.1073051. [DOI] [PubMed] [Google Scholar]
- Garcia-Cardena G, Fan R, Shah V, Sorrentino R, Cirino G, Papapetropoulos A, et al. Dynamic activation of endothelial nitric oxide synthase by Hsp90. Nature. 1998;392:821–824. doi: 10.1038/33934. [DOI] [PubMed] [Google Scholar]
- Green DR, Kroemer G. The pathophysiology of mitochondrial cell death. Science. 2004;305:626–629. doi: 10.1126/science.1099320. [DOI] [PubMed] [Google Scholar]
- Gyurkocza B, Plescia J, Raskett CM, Garlick DS, Lowry PA, Carter BZ, et al. Antileukemic activity of shepherdin and molecular diversity of hsp90 inhibitors. J Natl Cancer Inst. 2006;98:1068–1077. doi: 10.1093/jnci/djj300. [DOI] [PubMed] [Google Scholar]
- Halestrap AP. What is the mitochondrial permeability transition pore? J Mol Cell Cardiol. 2009;46:821–831. doi: 10.1016/j.yjmcc.2009.02.021. [DOI] [PubMed] [Google Scholar]
- He L, Lemasters JJ. Regulated and unregulated mitochondrial permeability transition pores: a new paradigm of pore structure and function? FEBS Lett. 2002;512:1–7. doi: 10.1016/s0014-5793(01)03314-2. [DOI] [PubMed] [Google Scholar]
- Henchcliffe C, Beal MF. Mitochondrial biology and oxidative stress in Parkinson disease pathogenesis. Nat Clin Pract Neuro. 2008;4:600–609. doi: 10.1038/ncpneuro0924. [DOI] [PubMed] [Google Scholar]
- Hua G, Zhang Q, Fan Z. Heat shock protein 75 (TRAP1) antagonizes reactive oxygen species generation and protects cells from granzyme M-mediated apoptosis. J Biol Chem. 2007;282:20553–20560. doi: 10.1074/jbc.M703196200. [DOI] [PubMed] [Google Scholar]
- Igney FH, Krammer PH. Death and anti-death: tumour resistance to apoptosis. Nat Rev Cancer. 2002;2:277–288. doi: 10.1038/nrc776. [DOI] [PubMed] [Google Scholar]
- Isaacs JS, Xu W, Neckers L. Heat shock protein 90 as a molecular target for cancer therapeutics. Cancer Cell. 2003;3:213–217. doi: 10.1016/s1535-6108(03)00029-1. [DOI] [PubMed] [Google Scholar]
- Johnstone RW, Ruefli AA, Lowe SW. Apoptosis: a link between cancer genetics and chemotherapy. Cell. 2002;108:153–164. doi: 10.1016/s0092-8674(02)00625-6. [DOI] [PubMed] [Google Scholar]
- Kamal A, Thao L, Sensintaffar J, Zhang L, Boehm MF, Fritz LC, et al. A high-affinity conformation of Hsp90 confers tumour selectivity on Hsp90 inhibitors. Nature. 2003;425:407–410. doi: 10.1038/nature01913. [DOI] [PubMed] [Google Scholar]
- Kang BH, Plescia J, Dohi T, Rosa J, Doxsey SJ, Altieri DC. Regulation of tumor cell mitochondrial homeostasis by an organelle-specific Hsp90 chaperone network. Cell. 2007;131:257–270. doi: 10.1016/j.cell.2007.08.028. [DOI] [PubMed] [Google Scholar]
- Kang BH, Plescia J, Song HY, Meli M, Colombo G, Beebe K, et al. Combinatorial drug design targeting multiple cancer signaling networks controlled by mitochondrial Hsp90. J Clin Invest. 2009;119:454–464. doi: 10.1172/JCI37613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koga F, Xu W, Karpova TS, McNally JG, Baron R, Neckers L. Hsp90 inhibition transiently activates Src kinase and promotes Src-dependent Akt and Erk activation. Proc Natl Acad Sci U S A. 2006;103:11318–11322. doi: 10.1073/pnas.0604705103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kokoszka JE, Waymire KG, Levy SE, Sligh JE, Cai J, Jones DP, et al. The ADP/ATP translocator is not essential for the mitochondrial permeability transition pore. Nature. 2004;427:461–465. doi: 10.1038/nature02229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolata G. In long drive to cure cancer, advances have been elusive. The New York Times. 2009 Apr 24; Health Section. [Google Scholar]
- Lamb J, Crawford ED, Peck D, Modell JW, Blat IC, Wrobel MJ, et al. The Connectivity Map: using gene-expression signatures to connect small molecules, genes, and disease. Science. 2006;313:1929–1935. doi: 10.1126/science.1132939. [DOI] [PubMed] [Google Scholar]
- Leaf C. Why we're losing the war on cancer (and how to win it) Fortune. 2004;149:76–97. [PubMed] [Google Scholar]
- Li R, Boehm AL, Miranda MB, Shangary S, Grandis JR, Johnson DE. Targeting antiapoptotic Bcl-2 family members with cell-permeable BH3 peptides induces apoptosis signaling and death in head and neck squamous cell carcinoma cells. Neoplasia. 2007;9:801–811. doi: 10.1593/neo.07394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacKeigan JP, Murphy LO, Blenis J. Sensitized RNAi screen of human kinases and phosphatases identifies new regulators of apoptosis and chemoresistance. Nat Cell Biol. 2005;7:591–600. doi: 10.1038/ncb1258. [DOI] [PubMed] [Google Scholar]
- Masuda Y, Shima G, Aiuchi T, Horie M, Hori K, Nakajo S, et al. Involvement of tumor necrosis factor receptor-associated protein 1 (TRAP1) in apoptosis induced by beta-hydroxyisovalerylshikonin. J Biol Chem. 2004;279:42503–42515. doi: 10.1074/jbc.M404256200. [DOI] [PubMed] [Google Scholar]
- McClellan AJ, Xia Y, Deutschbauer AM, Davis RW, Gerstein M, Frydman J. Diverse cellular functions of the Hsp90 molecular chaperone uncovered using systems approaches. Cell. 2007;131:121–135. doi: 10.1016/j.cell.2007.07.036. [DOI] [PubMed] [Google Scholar]
- Montesano Gesualdi N, Chirico G, Pirozzi G, Costantino E, Landriscina M, Esposito F. Tumor necrosis factor-associated protein 1 (TRAP-1) protects cells from oxidative stress and apoptosis. Stress. 2007;10:342–350. doi: 10.1080/10253890701314863. [DOI] [PubMed] [Google Scholar]
- Mootha VK, Bunkenborg J, Olsen JV, Hjerrild M, Wisniewski JR, Stahl E, et al. Integrated Analysis of Protein Composition, Tissue Diversity, and Gene Regulation in Mouse Mitochondria. Cell. 2003;115:629–640. doi: 10.1016/s0092-8674(03)00926-7. [DOI] [PubMed] [Google Scholar]
- Murphy MP. Targeting lipophilic cations to mitochondria. Biochim Biophys Acta. 2008;1777:1028–1031. doi: 10.1016/j.bbabio.2008.03.029. [DOI] [PubMed] [Google Scholar]
- Nakagawa T, Shimizu S, Watanabe T, Yamaguchi O, Otsu K, Yamagata H, et al. Cyclophilin D-dependent mitochondrial permeability transition regulates some necrotic but not apoptotic cell death. Nature. 2005;434:652–658. doi: 10.1038/nature03317. [DOI] [PubMed] [Google Scholar]
- Neckers L. Hsp90 inhibitors as novel cancer chemotherapeutic agents. Trends Mol Med. 2002;8:S55–S61. doi: 10.1016/s1471-4914(02)02316-x. [DOI] [PubMed] [Google Scholar]
- O'Dwyer ME, Druker BJ. STI571: an inhibitor of the BCR-ABL tyrosine kinase for the treatment of chronic myelogenous leukaemia. Lancet Oncol. 2000;1:207–211. doi: 10.1016/s1470-2045(00)00149-2. [DOI] [PubMed] [Google Scholar]
- Pearl LH, Prodromou C. Structure and in vivo function of Hsp90. Curr Opin Struct Biol. 2000;10:46–51. doi: 10.1016/s0959-440x(99)00047-0. [DOI] [PubMed] [Google Scholar]
- Petit A, Kawarai T, Paitel E, Sanjo N, Maj M, Scheid M, et al. Wild-type PINK1 Prevents Basal and Induced Neuronal Apoptosis, a Protective Effect Abrogated by Parkinson Disease-related Mutations. J Biol Chem. 2005;280:34025–34032. doi: 10.1074/jbc.M505143200. [DOI] [PubMed] [Google Scholar]
- Pilkington GJ, Parker K, Murray SA. Approaches to mitochondrially mediated cancer therapy. Semin Cancer Biol. 2008;18:226–235. doi: 10.1016/j.semcancer.2007.12.006. [DOI] [PubMed] [Google Scholar]
- Plescia J, Salz W, Xia F, Pennati M, Zaffaroni N, Daidone MG, et al. Rational design of shepherdin, a novel anticancer agent. Cancer Cell. 2005;7:457–468. doi: 10.1016/j.ccr.2005.03.035. [DOI] [PubMed] [Google Scholar]
- Price JT, Quinn JM, Sims NA, Vieusseux J, Waldeck K, Docherty SE, et al. The heat shock protein 90 inhibitor, 17-allylamino-17-demethoxygeldanamycin, enhances osteoclast formation and potentiates bone metastasis of a human breast cancer cell line. Cancer Res. 2005;65:4929–4938. doi: 10.1158/0008-5472.CAN-04-4458. [DOI] [PubMed] [Google Scholar]
- Pridgeon JW, Olzmann JA, Chin LS, Li L. PINK1 Protects against Oxidative Stress by Phosphorylating Mitochondrial Chaperone TRAP1. PLoS Biol. 2007;5:e172. doi: 10.1371/journal.pbio.0050172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prodromou C, Roe SM, O'Brien R, Ladbury JE, Piper PW, Pearl LH. Identification and structural characterization of the ATP/ADP-binding site in the Hsp90 molecular chaperone. Cell. 1997;90:65–75. doi: 10.1016/s0092-8674(00)80314-1. [DOI] [PubMed] [Google Scholar]
- Rajasethupathy P, Vayttaden SJ, Bhalla US. Systems modeling: a pathway to drug discovery. Curr Opin Chem Biol. 2005;9:400–406. doi: 10.1016/j.cbpa.2005.06.008. [DOI] [PubMed] [Google Scholar]
- Rodina A, Vilenchik M, Moulick K, Aguirre J, Kim J, Chiang A, et al. Selective compounds define Hsp90 as a major inhibitor of apoptosis in small-cell lung cancer. Nat Chem Biol. 2007;3:498–507. doi: 10.1038/nchembio.2007.10. [DOI] [PubMed] [Google Scholar]
- Ross MF, Filipovska A, Smith RA, Gait MJ, Murphy MP. Cell-penetrating peptides do not cross mitochondrial membranes even when conjugated to a lipophilic cation: evidence against direct passage through phospholipid bilayers. Biochem J. 2004;383:457–468. doi: 10.1042/BJ20041095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawyers C. Targeted cancer therapy. Nature. 2004;432:294–297. doi: 10.1038/nature03095. [DOI] [PubMed] [Google Scholar]
- Schein PS, Scheffler B. Barriers to efficient development of cancer therapeutics. Clin Cancer Res. 2006;12:3243–3248. doi: 10.1158/1078-0432.CCR-06-0329. [DOI] [PubMed] [Google Scholar]
- Schinzel AC, Takeuchi O, Huang Z, Fisher JK, Zhou Z, Rubens J, et al. Cyclophilin D is a component of mitochondrial permeability transition and mediates neuronal cell death after focal cerebral ischemia. Proc Natl Acad Sci U S A. 2005;102:12005–12010. doi: 10.1073/pnas.0505294102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmid D, Baici H, Gehring H, Christen P. Kinetics of molecular chaperon action. Science. 1994;263:971–973. doi: 10.1126/science.8310296. [DOI] [PubMed] [Google Scholar]
- Solit DB, Chiosis G. Development and application of Hsp90 inhibitors. Drug Discov Today. 2008;13:38–43. doi: 10.1016/j.drudis.2007.10.007. [DOI] [PubMed] [Google Scholar]
- Steensma DP. The Ordinary Miracle of Cancer Clinical Trials. J Clin Oncol. 2009;27:1737–1739. doi: 10.1200/JCO.2008.20.6292. [DOI] [PubMed] [Google Scholar]
- Szeto HH. Development of mitochondria-targeted aromatic-cationic peptides for neurodegenerative diseases. Ann N Y Acad Sci. 2008;1147:112–121. doi: 10.1196/annals.1427.013. [DOI] [PubMed] [Google Scholar]
- Torchilin VP. Recent approaches to intracellular delivery of drugs and DNA and organelle targeting. Annu Rev Biomed Eng. 2006;8:343–375. doi: 10.1146/annurev.bioeng.8.061505.095735. [DOI] [PubMed] [Google Scholar]
- Tsujimoto Y, Shimizu S. Role of the mitochondrial membrane permeability transition in cell death. Apoptosis. 2007;12:835–840. doi: 10.1007/s10495-006-0525-7. [DOI] [PubMed] [Google Scholar]
- van der Greef J, McBurney RN. Innovation: Rescuing drug discovery: in vivo systems pathology and systems pharmacology. Nat Rev Drug Discov. 2005;4:961–967. doi: 10.1038/nrd1904. [DOI] [PubMed] [Google Scholar]
- Vogelstein B, Kinzler KW. Cancer genes and the pathways they control. Nat Med. 2004;10:789–799. doi: 10.1038/nm1087. [DOI] [PubMed] [Google Scholar]
- Vogler M, Dinsdale D, Dyer MJS, Cohen GM. Bcl-2 inhibitors: small molecules with a big impact on cancer therapy. Cell Death Differ. 2008;16:360–367. doi: 10.1038/cdd.2008.137. [DOI] [PubMed] [Google Scholar]
- Voloboueva LA, Duan M, Ouyang Y, Emery JF, Stoy C, Giffard RG. Overexpression of mitochondrial Hsp70//Hsp75 protects astrocytes against ischemic injury in vitro. J Cereb Blood Flow Metab. 2007;28:1009–1016. doi: 10.1038/sj.jcbfm.9600600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinstein IB, Joe AK. Mechanisms of disease: Oncogene addiction--a rationale for molecular targeting in cancer therapy. Nat Clin Pract Oncol. 2006;3:448–457. doi: 10.1038/ncponc0558. [DOI] [PubMed] [Google Scholar]
- Whitesell L, Lindquist SL. HSP90 and the chaperoning of cancer. Nat Rev Cancer. 2005;5:761–772. doi: 10.1038/nrc1716. [DOI] [PubMed] [Google Scholar]
- Wood LD, Parsons DW, Jones S, Lin J, Sjoblom T, Leary RJ, et al. The genomic landscapes of human breast and colorectal cancers. Science. 2007;318:1108–1113. doi: 10.1126/science.1145720. [DOI] [PubMed] [Google Scholar]
- Woodfield K, Ruck A, Brdiczka D, Halestrap AP. Direct demonstration of a specific interaction between cyclophilin-D and the adenine nucleotide translocase confirms their role in the mitochondrial permeability transition. Biochem J. 1998;336(Pt 2):287–290. doi: 10.1042/bj3360287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright GL, Maroulakou IG, Eldridge J, Liby TL, Sridharan V, Tsichlis PN, et al. VEGF stimulation of mitochondrial biogenesis: requirement of AKT3 kinase. FASEB J. 2008;22:3264–3275. doi: 10.1096/fj.08-106468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu L, Voloboueva LA, Ouyang Y, Emery JF, Giffard RG. Overexpression of mitochondrial Hsp70//Hsp75 in rat brain protects mitochondria, reduces oxidative stress, and protects from focal ischemia. J Cereb Blood Flow Metab. 2008;29:365–374. doi: 10.1038/jcbfm.2008.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young JC, Hoogenraad NJ, Hartl FU. Molecular chaperones Hsp90 and Hsp70 deliver preproteins to the mitochondrial import receptor Tom70. Cell. 2003;112:41–50. doi: 10.1016/s0092-8674(02)01250-3. [DOI] [PubMed] [Google Scholar]
- Young JC, Moarefi I, Hartl FU. Hsp90: a specialized but essential protein-folding tool. J Cell Biol. 2001;154:267–273. doi: 10.1083/jcb.200104079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeitlin BD, Zeitlin IJ, Nor JE. Expanding circle of inhibition: small-molecule inhibitors of Bcl-2 as anticancer cell and antiangiogenic agents. J Clin Oncol. 2008;26:4180–4188. doi: 10.1200/JCO.2007.15.7693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao R, Houry WA. Molecular interaction network of the Hsp90 chaperone system. Adv Exp Med Biol. 2007;594:27–36. doi: 10.1007/978-0-387-39975-1_3. [DOI] [PubMed] [Google Scholar]