

Abstract
AIM: To investigate the effect of stable c-Fos overexpression on immortalized human hepatocyte (IHH) proliferation.
METHODS: IHHs stably transfected with c-Fos (IHH-Fos) or an empty vector (IHH-C) were grown in medium supplemented with 1% serum or stimulated with 10% serum. Cell proliferation was assessed by cell counts, 3H-thymidine uptake and flow cytometry analyses. The levels of cell cycle regulatory proteins (Cyclin D1, E, A) cyclin dependent kinases (cdk) cdk2, cdk4, cdk6, and their inhibitors p15, p16, p21, p27, total and phosphorylated GSK-3β and epidermal growth factor receptor (EGF-R) were assayed by Western blotting. Analysis of Cyclin D1 mRNA levels was performed by reverse transcription-polymerase chain reaction and real-time polymerase chain reaction (PCR) analysis. Stability of Cyclin D1 was studied by cycloheximide blockade experiments.
RESULTS: Stable c-Fos overexpression increased cell proliferation under low serum conditions and resulted in a two-fold increase in [3H]-thymidine incorporation following serum addition. Cell cycle analysis by flow cytometry showed that c-Fos accelerated the cell cycle kinetics. Following serum stimulation, Cyclin D1 was more abundantly expressed in c-Fos overexpressing cells. Cyclin D1 accumulation did not result from increased transcriptional activation, but from nuclear stabilization. Overexpression of c-Fos correlated with higher nuclear levels of inactive phosphorylated GSK-3β, a kinase involved in Cyclin D1 degradation and higher levels of EGF-R mRNA, and EGF-R protein compared to IHH-C both in serum starved, and in serum stimulated cells. Abrogation of EGF-R signalling in IHH-Fos by treatment with AG1478, a specific EGF-R tyrosine kinase inhibitor, prevented the phosphorylation of GSK-3β induced by serum stimulation and decreased Cyclin D1 stability in the nucleus.
CONCLUSION: Our results clearly indicate a positive role for c-Fos in cell cycle regulation in hepatocytes. Importantly, we delineate a new mechanism by which c-Fos could contribute to hepatocarcinogenesis through stabilization of Cyclin D1 within the nucleus, evoking a new feature to c-Fos implication in hepatocellular carcinoma.
Keywords: c-Fos, Cyclin D1, GSK-3, Cell growth, Cell cycle, Hepatoma, Epidermal growth factor
INTRODUCTION
Hepatocellular carcinoma (HCC) is the fifth most common cancer in the world, with an increasing number of new cases emerging each year. Etiologically it is linked to chronic viral infections (hepatitis B and C viruses), alcohol-related cirrhosis or aflatoxin B1 exposure, which all cause disruptions in signal transduction cascades leading to abnormalities in gene expression.
The proto-oncogene c-fos is an important member of the activating protein 1 (AP-1) transcription factor involved in major cellular functions such as transformation, proliferation, differentiation and apoptosis[1,2]. Such a large variety of functions is achieved by the combination of different Jun (c-Jun, JunB or JunD) and Fos (c-Fos, FosB, ∆fosB, Fra-1, Fra-2) family members forming various AP-1 homo and heterodimers. c-fos is an immediate early gene whose expression is rapidly and transiently induced after mitogenic stimulation[3]. The role of c-Fos in cell proliferation and transformation remains controversial. c-Fos is required during all phases of the cell cycle in exponentially growing cells and is a potent inducer of cell proliferation[4]. However, some studies have suggested that c-Fos poorly contributes to proliferation[5], was totally dispensable for[6], or even down-regulated, cell growth[7,8]. Overexpression of c-Fos leads to morphological transformation of fibroblasts[9,10], and to osteosarcoma formation in transgenic mice[11,12]. Apart from one study describing a negative role for c-Fos in hepatocellular tumorigenesis[8], several reports rather support a potential positive role for c-Fos in this process. High expression levels of c-Fos were determined in tumour tissue compared to the adjacent non-tumour liver in human HCC[13-15], as well as in several models of HCC in rodents[16-18]. A recent study in humans identified a subtype of HCC sharing gene expression patterns with foetal hepatoblasts which can be distinguished from another HCC subtype closer to adult hepatocytes[19]. Interestingly, c-Fos, but not c-Jun expression was higher in the foetal subtype which displayed a poorer prognosis and a greater tendency to invasion than the adult subtype. In addition, the expression of DNA 5-methylcytosine transferase, a c-Fos target gene involved in DNA methylation[20] is increased in human tumour cells and in HCCs[21]. Despite these studies showing that c-Fos overexpression might be an important step towards the development of liver cancer, its precise role in hepatocarcinogenesis remains ill-defined.
In order to clarify c-Fos implication in hepatocarcinogenesis, we examined the effect on proliferation of stable c-Fos overexpression in immortalized human hepatocytes (IHH). We show, for the first time, that a positive role for c-Fos on hepatocyte proliferation can be attained by stabilization of Cyclin D1 in the nuclear compartment, a mechanism which has not been described as a c-Fos related process in any cell type to date.
MATERIALS AND METHODS
Cell culture and reagents
IHH were cultured in Williams’ medium E (Invitrogen, Cergy Pontoise, France) supplemented with 100 mL/L fetal calf serum (FCS) (Biochrom AG, Cambridge, UK), 1% penicillin-streptomycin, 1% Glutamax and 1% DMEM sodium pyruvate (Invitrogen). Specific reagents were AG1478 (Calbiochem, San Diego, CA) and cycloheximide (CHX) (Euromedex, Souffelweyersheim, France).
Generation of stably transfected cells
The human c-fos cDNA was inserted into the cytomegalovirus driven pCIneo expression vector (Promega, Charbonnières, France) containing a neomycin resistance gene to obtain the pCIneo-cfos vector. Cells were stably transfected by electroporation (230V, 960 μF) in PBS-Hepes Buffer 10 mmol/L, pH 7.4 with the empty vector (pCIneo) or with pCIneo-cfos. Two days post-transfection, stable clones were selected in media containing 500 μg/mL of G418 (Invitrogen). The resistant clones were pooled after 3 wk of selection, and maintained with G418. c-Fos overexpression was verified by Western blot analysis as shown in Figure 1A.
Figure 1.

Overexpression of c-Fos accelerates the cell cycle. A: IHH-C and IHH-Fos were grown in 1% FCS, cultured for 5 d and counted daily. Cell growth was determined by counting the number of attached cells every day. Results are the mean ± SE of three independent experiments; B: [3H] thymidine incorporation into DNA. Non-synchronized IHH-C or IHH-Fos serum starved for 24 h then serum stimulated for 4 h were incubated with [3H] thymidine for 4 h. DNA was extracted as described in materials and methods, and [3H] thymidine incorporation into DNA was assessed by scintillation counting. Results are expressed as percentage of increase of [3H] thymidine incorporation in serum-stimulated cells over that of quiescent cells for each cell population. Results are the mean ± SE of six independent experiments; C: Flow cytometry analysis for quantification of cell cycle phase distribution and progression through cell cycle. IHH-C or IHH-Fos serum starved for 24 h were incubated with BrdU for 1 h and stained with propidium iodide 0, 4, 8 and 24 h after serum stimulation. The percentage of cells in each phase is plotted against time. Results of a representative experiment are shown (out of 3); D: IHH-C or IHH-Fos serum starved for 24 h were serum stimulated for 12 h, BrdU pulsed for 1 h, chased with fresh medium for 0, 3, 6, 9, 12 h, and then stained with propidium iodide. The percentage of BrdU-negative cells in the G1 phase (G1 exit) (left panel) and of the BrdU-positive cells in the G1 phase (G1 entry) (right panel) of the cell cycle is plotted against time. Results are representative of four independent experiments.
Growth curve analysis
Cells were plated in triplicate at a density of 1.0 × 105 per well in six-well plates, and cultured in low serum (1% FCS) conditions for 5 d. Triplicate cultures were trypsinized and diluted in an equal volume of trypan blue solution (Invitrogen). Viable cells were counted daily in a haemocytometer counting chamber.
[3H]-thymidine incorporation
DNA synthesis was determined by measuring [3H]-thymidine incorporation. Cells were plated onto 24-well plates at a density of 1.0 × 105 cells/well in quadruplets. Cells were serum deprived for 24 h, and serum stimulated in culture media containing 1.5 μCi/mL tritiated thymidine ([3H]dT) (specific activity of 740 GBq/mmol) (Perkin-Elmer, Waltham, Ma) for 4 h. Cells were fixed and washed in ice-cold 10% trichloroacetic acid. DNA was solubilized in 0.1 mol/L NaOH for 1 h at 37°C. [3H]dT incorporated into the DNA was measured using liquid scintillation counting.
Flow cytometry
DNA cell cycle analysis was measured by 5-bromodeoxyuridine (BrdU) incorporation and propidium iodide staining of the nuclei by flow cytometry (FACScalibur, BD Biosciences, Mansfield, MA) and analyzed with the ProCellQuest software provided by the manufacturer. Cell cycle progression was measured by pulse/chase experiments. Cells plated at a density of 5 × 105 per 6-cm dish were serum starved for 24 h, serum stimulated for 12 h and stained with BrdU (30 μg/mL) for 1 h. Cells were then chased with BrdU free medium for 0, 3, 6, 9, 12 h, stained with propidium iodide and harvested in 70% ethanol. Cells were then treated with 2 N HCl and pepsin (0.2 mg/mL) for 30 min. BrdU content was analyzed using a FITC-labeled monoclonal antibody to BrdU (BD Pharmingen, Le-Pont-de-Claix, France). Labeled cells were washed and resuspended in PBS containing propidium iodide (10 μg/mL) for 30 min prior to flow cytometric analysis.
Western blot analysis
Nuclear proteins were extracted as described[22]. Total proteins were extracted with lysis buffer [1% (v/v) SDS, 1 mmol/L Na3VO4, 10 mmol/L Tris pH 7.4, 1% benzonase) for 10 min at room temperature and heated for 5 min at 100°C. Equal quantities of nuclear proteins were fractionated on a SDS-polyacrylamide gel and transferred onto nitrocellulose membranes by electroblotting. The antibodies used in this study were as follows: c-Fos, Cyclin D1, Cyclin E, Cyclin A, cdk2, cdk4, cdk6, p15, p16, p21, p27 and EGF-R (Santa Cruz Biotechnology, Santa Cruz, CA), GSK3β (Affinity BioReagents, Golden, CO), Phospho-GSK3β (Serine9) (Abcam, Paris, France). Immunoreactive bands were visualized using the ECL kit (Amersham BioSciences, Saclay, France) according to the manufacturer's instructions.
Reverse transcription-polymerase chain reaction (RT-PCR) and real-time quantitative PCR analysis
mRNA was isolated from cells by Nucleospin RNA II kit (Macherey-Nagel, Hoerdt, France) following the manufacturer’s instructions. RNA (1 μg) was reverse transcribed using ThermoScriptTM RT-PCR System (Invitrogen, Cergy-Pontoise, France). Real-time quantitative PCR was performed using the following primers: Cyclin D1: forward 5'-GCATGTTCGTGGCCTCTAAGA-3'; reverse 5'-CGGTGTAGATGCACAGCTTCTC-3', EGF-R: forward 5'-GCGTCTCTTGCCGGAATGT-3' and reverse 5'-GGCTCACCCTCCAGAAGGTT-3'. Real-time quantitative PCR was performed with an ABI PRISM 7700 instrument (Applied Biosystems, Foster City, CA)) using SYBRGreen PCR core reagents (Applied Biosystems). Fold changes in mRNA were calculated by the ∆∆Ct method using cyclophilin A (forward: 5'-CAAATGCTGGACCCAACACA-3'; reverse: 5'-TGCCATCCAACCACTCAGTCT-3') as a standard. All PCR reactions were done in triplicate.
Statistical analysis
Data were expressed as means ± SE. Student’s t-test was performed and statistical significance was considered as P < 0.05.
RESULTS
Growth rate and cell cycle regulation by c-Fos overexpression
To determine whether c-Fos could modulate hepatocyte growth, we carried out growth curve assays. While cell growth was similar in the presence of 10% FCS in IHH-C and IHH-Fos (data not shown), we observed that the growth pattern of the two cell lines differed in low serum conditions (1% FCS). While the number of IHH-Fos increased exponentially over 5 d in culture, IHH-C number increased slowly during the first 3 d of culture, and then reached a plateau, due to the induction of cell death by serum deprivation (Figure 1A and data not shown). Thus, c-Fos overexpression correlated with a more rapid growth in low serum conditions. The effect of c-Fos overexpression on cell proliferation was further established by measuring [3H]dT incorporation following 4 h serum stimulation of cells deprived of serum for 24 h. The increase of [3H]dT incorporation induced by serum was 2.2 times higher in IHH-Fos than in IHH-C (231% and 107%, respectively), and the difference was statistically significant (P < 0.001) (Figure 1B). To further analyze the role of c-Fos on the cell cycle, cell cycle phase distribution and cell cycle kinetics were analyzed by flow cytometry. Following serum stimulation, the percentage of cells in the G1 phase decreased, while the percentage of cells in the S-phase increased 24 h after serum stimulation. However, the percentage of cells in the different stages of the cell cycle was comparable in IHH-Fos and IHH-C (Figure 1C). Cell cycle progression was measured by BrdU pulse/chase experiments. The rate at which BrdU positive cells progress into G1 indicates the rate of transit through S, G2 and M phases. Similarly, the rate at which BrdU negative cells become depleted from the G1 pool indicates the transit rate through G1. We show that IHH-Fos quit (Figure 1D, left panel) and enter (Figure 1D, right panel) G1 faster than IHH-C, which reflects a global increase in cell cycle kinetics. The fact that the cell cycle profile was not altered by c-Fos indicates that the acceleration is proportional in all phases of the cycle. These data taken together indicate that c-Fos overexpression increases the growth of exponentially growing cells cultured in low serum medium as well as the proliferation response induced by serum refeeding.
Induction of cell cycle regulatory proteins by c-Fos
The levels of various cell cycle regulatory proteins before and after serum stimulation of IHH-C and IHH-Fos were analyzed by Western blotting experiments. In both cell lines, serum addition induced an increase in the nuclear levels of Cyclin A, cdk2 and cdk4, but no change in Cyclin E. Of interest, the nuclear levels of Cyclin D1 were increased after 8 h of stimulation in IHH-Fos, but not in IHH-C (Figure 2A). In addition, the levels of p27 were higher in the absence of serum stimulation or following serum stimulation in IHH-Fos than in IHH-C (Figure 2B).
Figure 2.
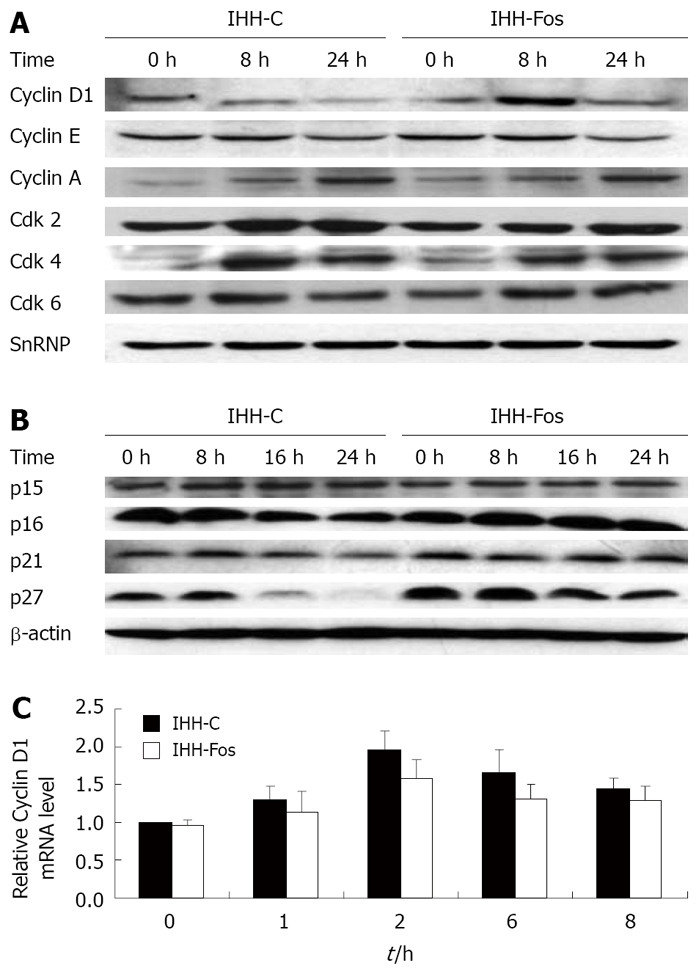
Induction of cell cycle regulatory proteins after serum refeeding. IHH-C or IHH-Fos were serum starved for 24 h. Nuclear (A) or total (B) extracts prepared before or after serum stimulation for 8 h or 24 h were immunoblotted with antibodies, as indicated. Loading of nuclear or total extracts was normalized using a SnRNP or a β-actin antibody, respectively. Results of a representative experiment are shown (out of 3); C: Quantitative real time PCR of Cyclin D1 mRNA levels in quiescent IHH-C or IHH-Fos serum stimulated for the indicated times. Bars indicate mean ± SE of three independent experiments each performed in triplicate.
Quantitative RT-PCR analysis was performed to determine whether the increase of Cyclin D1 at 8 h of serum stimulation in IHH-Fos was controlled transcriptionally. Interestingly, a similar 2-fold increase in Cyclin D1 mRNA 2 h following serum stimulation was observed in IHH-Fos and IHH-C, without any significant differences at any of the time points (Figure 2C), indicating that the higher levels of Cyclin D1 in the nucleus in IHH-Fos are not due to transcriptional mechanisms.
Cyclin D1 stabilization in the nucleus
We next determined whether post-translational regulations could explain the increase of nuclear Cyclin D1 in serum-stimulated IHH-Fos. CHX, a translational inhibitor, was used to block protein synthesis. While in IHH-C, nuclear Cyclin D1 protein levels started to decline as from 1 h, and decreased by 85% after 2 h of CHX treatment, Cyclin D1 nuclear levels were decreased by only 20% upon 2 h of CHX treatment in IHH-Fos (Figure 3), indicating that c-Fos overexpression correlates with increased stability of nuclear Cyclin D1.
Figure 3.
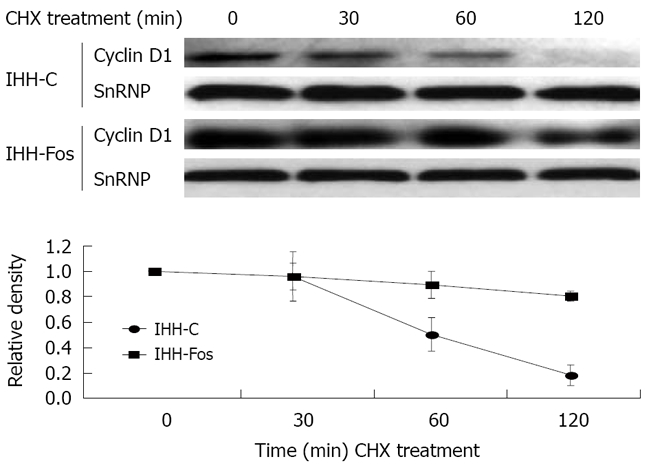
Nuclear Cyclin D1 stability is increased by c-Fos overexpression. IHH-C or IHH-Fos serum starved for 24 h were serum stimulated for 6 h and then treated with CHX (30 μg/mL) for 30 min, 1 h or 2 h. Nuclear extracts were immunoblotted with an antibody against Cyclin D1, and normalized with a SnRNP antibody, as indicated (Upper panels). The Cyclin D1 over SnRNP ratios were quantified by densitometric analysis of the immunoreactive bands (Lower panel). The results are the mean ± SE of three independent experiments.
Inactivation of GSK-3β in IHH-Fos contributes to Cyclin D1 stabilization
Previous studies have indicated that Cyclin D1 degra-dation is triggered by GSK-3β-induced phosphorylation on a single threonine residue (Thr-286)[23]. Of note, phosphorylated GSK-3β is the inactive form of the protein[24]. We, therefore, compared the level of phosphorylated GSK-3β between the two cell lines by Western blot analysis. As shown in Figure 4A, the levels of GSK3β phosphorylation were much higher in the nucleus of IHH-Fos than in IHH-C, both in unstimulated and in serum stimulated conditions, indicating higher basal, and induced levels of inactive GSK-3β in IHH-Fos (Figure 4A). Therefore, a decrease in active GSK-3β in IHH-Fos could be responsible for the increased stability of nuclear Cyclin D1 after serum stimulation.
Figure 4.
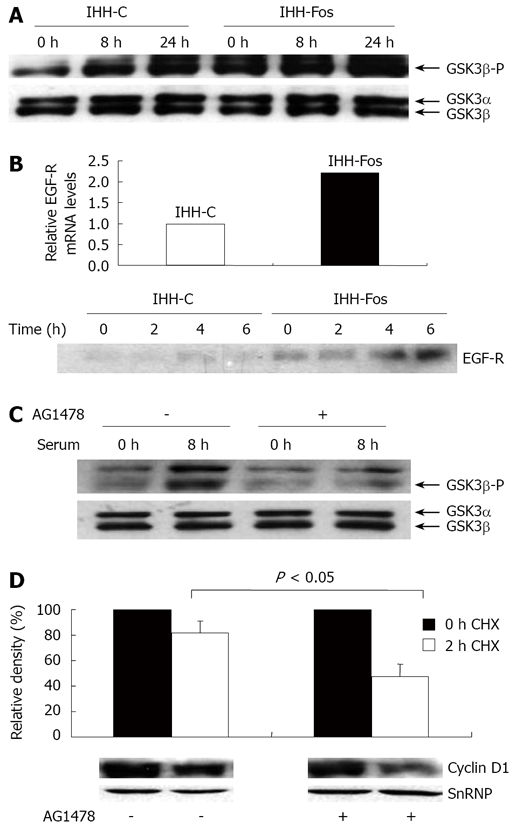
Stimulation of EGF-R signaling by c-Fos overexpression. A: Total and phosphorylated levels of nuclear GSK-β. IHH-C or IHH-Fos were serum starved for 24 h. Nuclear extracts prepared from unstimulated (0 h), 8 h or 24 h serum stimulated IHH-C or IHH-Fos, were immunoblotted with an antibody against phosphorylated or total GSK3β. B: Upper panel, detection of EGF-R mRNA in IHH-C and IHH-Fos by quantitative real time PCR analysis of mRNA isolated from cells grown in the presence of serum. Lower panel, Western blot analysis of EGF-R in total cell extracts from IHH-C and IHH-Fos cells serum starved for 24 h (0) or stimulated with serum for the indicated times; C: Serum deprived IHH-Fos were pre-treated (+) or not (-) with AG1478 (10 μmol/L) for 1 h. Nuclear proteins were prepared from non stimulated and 8 h serum-stimulated cells. Phosphorylated and total GSK3β levels were detected by Western blot; D: Serum-deprived IHH-Fos were pretreated or not with AG1478 (10 μmol/L) for 1 h, then serum-stimulated for 6 h. Nuclear proteins were extracted before (0 h, filled columns) or after 2 h (empty columns) of CHX treatment (30 µg/mL). Cyclin D1 levels were quantified by Western blotting. The immunoreactive bands were quantified by densitometric analysis after loading normalization of the blot using a SnRNP antibody. The results are expressed as the % of Cyclin D1/SnRNP expression and are the mean ± SE of 3 independent experiments. The lower panel illustrates one representative experiment.
Several signaling pathways are able to induce GSK-3β phosphorylation, including the two main cascades targeted by tyrosine kinase receptors: the phosphatidyl inositol 3-kinase (PI3K) and the Ras/mitogen activated protein kinase pathways[24]. Since the epidermal growth factor receptor (EGF-R) is a known transcriptional target of AP-1[25-27], we tested the hypothesis that overexpression of EGF-R might contribute to high levels of GSK-3β phosphorylation in IHH-Fos. Quantitative RT-PCR analysis revealed a 2.2-fold increase in the basal level of EGF-R mRNA in IHH-Fos compared to IHH-C (Figure 4B). Higher levels of EGF-R protein were also observed in serum starved and serum-stimulated IHH-Fos compared to IHH-C (Figure 4B), strongly indicative of increased EGF-R signaling in c-Fos-overexpressing cells. Altogether, these data suggest that increased EGF-R signaling might contribute, at least partly, to increased levels of GSK-3β phosphorylation in IHH-Fos cells.
To demonstrate the implication of EGF-R in GSK-3β-mediated Cyclin D1 stabilization, IHH-Fos cells were treated with AG1478, a specific inhibitor of the EGF-R tyrosine kinase before serum stimulation. Western blot analysis indicated that AG1478 treatment did block the phosphorylation of GSK-3β induced by serum (Figure 4C). Interestingly, the decrease in the nuclear level of Cyclin D1 protein observed after a 2 h-CHX treatment of IHH-Fos cells stimulated by serum was more important in cells treated with AG1478 (50% decrease) than in untreated cells (10% decrease), and the difference was statistically significant (P < 0.05, Figure 4D), confirming that blockade of EGF-R induced signaling in IHH-Fos leads to a more rapid nuclear Cyclin D1 degradation.
DISCUSSION
Our results indicate that c-Fos overexpression accelerates cell growth under reduced serum concentration suggesting that hepatocytes overexpressing c-Fos become relatively independent of the presence of growth factors. We show that c-Fos enhances DNA synthesis after serum stimulation, and accelerates hepatocyte cell cycle progression without altering the overall distribution of cells in each phase due to a proportional acceleration of cell cycle kinetics in all phases.
Our results are at variance with those obtained in immortalized murine hepatocytes[8]. In this model, c-Fos conditional expression for 48 h was shown to decrease cell growth and [3H]dT incorporation of cells grown in serum-supplemented medium. Besides species differences (murine vs human cells), the discrepancy in results can be explained by the use of very different cellular models which cannot be compared. The human hepatocytes used in our study were immortalized by SV40 T antigen while murine hepatocytes were immortalized using truncated c-Met[28]. Overexpression of c-Fos in our model was permanently established as the result of stable transfection, while in Mikula’s study a c-Fos-estrogen receptor fusion protein was expressed for a limited period (1-3 d) following estradiol treatment of the cultures[8]. Furthermore, the function of the conditionally expressed c-Fos protein may have been modified, since gene fusion has been shown to alter the function of Fos family proteins[29].
We aimed to determine whether the positive role of c-Fos on hepatocyte proliferation depicted in our study was mediated through changes in cell cycle regulation. Different studies have reported an effect of c-fos gene deletion or c-Fos protein overexpression on Cyclin D1[9,30,31], Cyclin E[31] or Cyclin A[32] expression, depending on the cell type studied. In our study, while the levels of Cyclin E and A and their associated kinases varied with a similar pattern in both cell types following serum stimulation, nuclear Cyclin D1 levels were higher in IHH-Fos compared to IHH-C 8 h after serum re-feeding. Contrary to previous reports describing c-Fos as a transcriptional activator of Cyclin D1[30], the higher levels of nuclear Cyclin D1 in IHH-Fos than in IHH-C were not due to differences in transcriptional regulation, but to increased protein stability in the nucleus. A similar lack of correlation between Cyclin D1 mRNA and protein expression has been previously described in an in vivo experimental model of HCC[33]. Our results strongly suggest a mechanism whereby c-Fos induces nuclear accumulation of Cyclin D1 without affecting the total cellular amount of the protein.
The Cyclin D1 protein is quite unstable, with a half-life of less than 30 min[34]. It accumulates in the nucleus during the G1 phase and exits into the cytoplasm during the S phase. Nuclear export of Cyclin D1, and its subsequent ubiquitination and proteolysis, are dependent on phosphorylation on a single threonine residue (Thr-286) performed mainly by GSK-3β[23], a protein kinase active only when dephosphorylated. In contrast to Cyclin D1, GSK-3β is predominantly cytoplasmic during G1 phase, but a considerable amount becomes nuclear during S phase[23]. We show herein that phosphorylated levels of nuclear GSK-3β are higher in IHH-Fos than in IHH-C. Lower levels of active GSK-3β would consequently lead to a decrease in Cyclin D1 phosphorylation, resulting in its nuclear accumulation in IHH-Fos. Since EGF-R is a known transcriptional target of AP-1[25-27], we tested the possibility that c-Fos overexpression increases the activation of the pathways downstream to EGF signaling. EGF-R activates both the PI3K and the mitogen-activated protein kinase cascades[35], two upstream activators of GSK-3β phosphorylation[24,36]. In support of an involvement of EGF-R signaling in GSK-3β inactivation and nuclear cylin D1 stabilization, we show that IHH-Fos display increased levels of expression of EGF-R mRNA and protein than IHH-C. Furthermore, blocking the activation of the EGF-R tyrosine kinase significantly accelerates the rate of Cyclin D1 degradation assessed in CHX experiments. Upregulated expression of EGF-R is a frequent finding in HCC[37-39], and increased EGF-R signaling has been associated with a poorer prognosis[40]. c-Fos is also frequently overexpressed in HCC tumoral tissues[13-15,41]. Our data, therefore, suggest that a causal relationship could exist between c-Fos and EGF-R overexpression in HCC.
Our finding of high levels of nuclear Cyclin D1 associated with c-Fos overexpression adds further support for a contributing effect of c-Fos on HCC development. Indeed, Cyclin D1 exit from the nucleus during S phase is essential for regulated cell division, and its retention in the nucleus is a cancer promoting or predisposing event[42]. Thus, expression of a Cyclin D1 mutant that cannot be phosphorylated by GSK-3β, and remains nuclear throughout the cell cycle is highly transforming and induces tumour growth in nude mice[43].
In accordance with previous reports[32,44], we also found that p27 protein levels were higher in c-Fos overexpressing cells. It is now well recognized that the family of p21/p27 proteins plays a dual role in cell cycle regulation. On one hand, they bind to cdk2 complexes and inhibit their kinase activities. On the other hand, they are able to promote the activation of Cyclin D1/cdk4-6 by complex stabilization, and by facilitating the nuclear import of these complexes, without inhibiting Cyclin D-associated kinase activity[45-48]. In our study, higher levels of p27 in IHH-Fos could, therefore, represent another mechanism contributing to the increase in nuclear levels of Cyclin D1, although the precise mechanisms linking c-Fos and p27 overexpression are currently unknown. Nevertheless, the mechanism is not at the level of transcription, as indicated by our quantitative PCR analysis (data not shown).
To conclude, our results clearly indicate a positive role for c-Fos in cell cycle regulation in hepatocytes. Importantly, we delineate a new mechanism by which c-Fos could contribute to hepatocarcinogenesis through stabilization of Cyclin D1 within the nucleus, evoking a new feature to c-Fos implication in HCC.
COMMENTS
Background
Human hepatocellular carcinoma (HCC) is the fifth most common cancer in the world. Among the numerous genes potentially implicated in hepatocarcinogenesis, the proto-oncogene c-Fos, a member of activating protein 1 (AP-1) transcription factor is a good candidate. Apart from one study reporting a negative role for c-Fos in hepatocellular tumorigenesis, several papers rather support a positive role in this process. High expression levels of c-Fos were determined in tumor tissue compared to the adjacent non-tumor liver in human HCC. However, in different cell types or tissues, the role of c-Fos in cell proliferation and/or transformation remains controversial. This study was designed to determine whether c-Fos could contribute to hepatocarcinogenesis by increasing cell proliferation.
Research frontiers
The role of c-Fos on hepatocyte proliferation has never been studied in human cells, but only in murine hepatocytes. These cells had been immortalized and stably transfected by c-Fos using different techniques than those reported in the present study. The authors showed that c-Fos overexpression led to decreased hepatocyte proliferation. However, these results did not appear consistent with most studies suggesting a positive role for c-Fos in hepatocarcinogenesis.
Innovations and breakthroughs
This study shows for the first time that c-Fos deregulates hepatocyte proliferation by stabilizing Cyclin D1 in the nucleus which is a cancer promoting or predisposing event.
Applications
Strategies designed to suppress c-Fos expression in HCC could contribute reducing hepatocyte proliferation and thereby cancer development.
Terminology
Human immortalized hepatocytes are hepatocytes which have been transfected by SV40 T antigen, allowing them to proliferate when cultured contrary to normal hepatocytes. However these immortalized cells are not tumorigenic in vitro and in vivo.
Peer review
This is an interesting study. Authors investigated the effect of stable c-Fos overexpression on IHH proliferation.
Acknowledgments
The IHH cell line was kindly provided by Dr. H Moshage (Groningen, The Netherlands). We gratefully acknowledge the technical assistance of J André and C Tacheau. This project was supported by INSERM, and Meryem Güller by a doctoral fellowship from the Ministry of Research and Technologies (MRT) and a grant from the Association pour la Recherche contre le Cancer (ARC).
Footnotes
Peer reviewer: Adrian G Cummins, PhD, Department of Gastroenterology and Hepatology, (DX 465384), 28 Woodville Road, Woodville South, South Australia 5011, Australia
S- Editor Li DL E- Editor Ma WH
References
- 1.Shaulian E, Karin M. AP-1 as a regulator of cell life and death. Nat Cell Biol. 2002;4:E131–E136. doi: 10.1038/ncb0502-e131. [DOI] [PubMed] [Google Scholar]
- 2.Eferl R, Wagner EF. AP-1: a double-edged sword in tumorigenesis. Nat Rev Cancer. 2003;3:859–868. doi: 10.1038/nrc1209. [DOI] [PubMed] [Google Scholar]
- 3.Angel P, Karin M. The role of Jun, Fos and the AP-1 complex in cell-proliferation and transformation. Biochim Biophys Acta. 1991;1072:129–157. doi: 10.1016/0304-419x(91)90011-9. [DOI] [PubMed] [Google Scholar]
- 4.Pai SR, Bird RC. c-fos expression is required during all phases of the cell cycle during exponential cell proliferation. Anticancer Res. 1994;14:985–994. [PubMed] [Google Scholar]
- 5.Kovary K, Bravo R. The jun and fos protein families are both required for cell cycle progression in fibroblasts. Mol Cell Biol. 1991;11:4466–4472. doi: 10.1128/mcb.11.9.4466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brusselbach S, Mohle-Steinlein U, Wang ZQ, Schreiber M, Lucibello FC, Muller R, Wagner EF. Cell proliferation and cell cycle progression are not impaired in fibroblasts and ES cells lacking c-Fos. Oncogene. 1995;10:79–86. [PubMed] [Google Scholar]
- 7.Balsalobre A, Jolicoeur P. Fos proteins can act as negative regulators of cell growth independently of the fos transforming pathway. Oncogene. 1995;11:455–465. [PubMed] [Google Scholar]
- 8.Mikula M, Gotzmann J, Fischer AN, Wolschek MF, Thallinger C, Schulte-Hermann R, Beug H, Mikulits W. The proto-oncoprotein c-Fos negatively regulates hepatocellular tumorigenesis. Oncogene. 2003;22:6725–6738. doi: 10.1038/sj.onc.1206781. [DOI] [PubMed] [Google Scholar]
- 9.Miao GG, Curran T. Cell transformation by c-fos requires an extended period of expression and is independent of the cell cycle. Mol Cell Biol. 1994;14:4295–4310. doi: 10.1128/mcb.14.6.4295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hennigan RF, Hawker KL, Ozanne BW. Fos-transformation activates genes associated with invasion. Oncogene. 1994;9:3591–3600. [PubMed] [Google Scholar]
- 11.Grigoriadis AE, Schellander K, Wang ZQ, Wagner EF. Osteoblasts are target cells for transformation in c-fos transgenic mice. J Cell Biol. 1993;122:685–701. doi: 10.1083/jcb.122.3.685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang ZQ, Grigoriadis AE, Mohle-Steinlein U, Wagner EF. A novel target cell for c-fos-induced oncogenesis: development of chondrogenic tumours in embryonic stem cell chimeras. EMBO J. 1991;10:2437–2450. doi: 10.1002/j.1460-2075.1991.tb07783.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Feng DY, Zheng H, Tan Y, Cheng RX. Effect of phosphorylation of MAPK and Stat3 and expression of c-fos and c-jun proteins on hepatocarcinogenesis and their clinical significance. World J Gastroenterol. 2001;7:33–36. doi: 10.3748/wjg.v7.i1.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yuen MF, Wu PC, Lai VC, Lau JY, Lai CL. Expression of c-Myc, c-Fos, and c-jun in hepatocellular carcinoma. Cancer. 2001;91:106–112. doi: 10.1002/1097-0142(20010101)91:1<106::aid-cncr14>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- 15.Tabor E. Tumor suppressor genes, growth factor genes, and oncogenes in hepatitis B virus-associated hepatocellular carcinoma. J Med Virol. 1994;42:357–365. doi: 10.1002/jmv.1890420406. [DOI] [PubMed] [Google Scholar]
- 16.Masui T, Nakanishi H, Inada K, Imai T, Mizoguchi Y, Yada H, Futakuchi M, Shirai T, Tatematsu M. Highly metastatic hepatocellular carcinomas induced in male F344 rats treated with N-nitrosomorpholine in combination with other hepatocarcinogens show a high incidence of p53 gene mutations along with altered mRNA expression of tumor-related genes. Cancer Lett. 1997;112:33–45. doi: 10.1016/s0304-3835(96)04543-0. [DOI] [PubMed] [Google Scholar]
- 17.Yao X, Hu JF, Daniels M, Yien H, Lu H, Sharan H, Zhou X, Zeng Z, Li T, Yang Y, et al. A novel orthotopic tumor model to study growth factors and oncogenes in hepatocarcinogenesis. Clin Cancer Res. 2003;9:2719–2726. [PubMed] [Google Scholar]
- 18.Borlak J, Meier T, Halter R, Spanel R, Spanel-Borowski K. Epidermal growth factor-induced hepatocellular carcinoma: gene expression profiles in precursor lesions, early stage and solitary tumours. Oncogene. 2005;24:1809–1819. doi: 10.1038/sj.onc.1208196. [DOI] [PubMed] [Google Scholar]
- 19.Lee JS, Heo J, Libbrecht L, Chu IS, Kaposi-Novak P, Calvisi DF, Mikaelyan A, Roberts LR, Demetris AJ, Sun Z, et al. A novel prognostic subtype of human hepatocellular carcinoma derived from hepatic progenitor cells. Nat Med. 2006;12:410–416. doi: 10.1038/nm1377. [DOI] [PubMed] [Google Scholar]
- 20.Bakin AV, Curran T. Role of DNA 5-methylcytosine transferase in cell transformation by fos. Science. 1999;283:387–390. doi: 10.1126/science.283.5400.387. [DOI] [PubMed] [Google Scholar]
- 21.Saito Y, Kanai Y, Nakagawa T, Sakamoto M, Saito H, Ishii H, Hirohashi S. Increased protein expression of DNA methyltransferase (DNMT) 1 is significantly correlated with the malignant potential and poor prognosis of human hepatocellular carcinomas. Int J Cancer. 2003;105:527–532. doi: 10.1002/ijc.11127. [DOI] [PubMed] [Google Scholar]
- 22.Sadowski HB, Gilman MZ. Cell-free activation of a DNA-binding protein by epidermal growth factor. Nature. 1993;362:79–83. doi: 10.1038/362079a0. [DOI] [PubMed] [Google Scholar]
- 23.Diehl JA, Cheng M, Roussel MF, Sherr CJ. Glycogen synthase kinase-3beta regulates cyclin D1 proteolysis and subcellular localization. Genes Dev. 1998;12:3499–3511. doi: 10.1101/gad.12.22.3499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cohen P, Frame S. The renaissance of GSK3. Nat Rev Mol Cell Biol. 2001;2:769–776. doi: 10.1038/35096075. [DOI] [PubMed] [Google Scholar]
- 25.Johnson AC, Murphy BA, Matelis CM, Rubinstein Y, Piebenga EC, Akers LM, Neta G, Vinson C, Birrer M. Activator protein-1 mediates induced but not basal epidermal growth factor receptor gene expression. Mol Med. 2000;6:17–27. [PMC free article] [PubMed] [Google Scholar]
- 26.Zenz R, Scheuch H, Martin P, Frank C, Eferl R, Kenner L, Sibilia M, Wagner EF. c-Jun regulates eyelid closure and skin tumor development through EGFR signaling. Dev Cell. 2003;4:879–889. doi: 10.1016/s1534-5807(03)00161-8. [DOI] [PubMed] [Google Scholar]
- 27.Mialon A, Sankinen M, Soderstrom H, Junttila TT, Holmstrom T, Koivusalo R, Papageorgiou AC, Johnson RS, Hietanen S, Elenius K, et al. DNA topoisomerase I is a cofactor for c-Jun in the regulation of epidermal growth factor receptor expression and cancer cell proliferation. Mol Cell Biol. 2005;25:5040–5051. doi: 10.1128/MCB.25.12.5040-5051.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Amicone L, Spagnoli FM, Spath G, Giordano S, Tommasini C, Bernardini S, De Luca V, Della Rocca C, Weiss MC, Comoglio PM, et al. Transgenic expression in the liver of truncated Met blocks apoptosis and permits immortalization of hepatocytes. EMBO J. 1997;16:495–503. doi: 10.1093/emboj/16.3.495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schuermann M, Hennig G, Muller R. Transcriptional activation and transformation by chimaeric Fos-estrogen receptor proteins: altered properties as a consequence of gene fusion. Oncogene. 1993;8:2781–2790. [PubMed] [Google Scholar]
- 30.Brown JR, Nigh E, Lee RJ, Ye H, Thompson MA, Saudou F, Pestell RG, Greenberg ME. Fos family members induce cell cycle entry by activating cyclin D1. Mol Cell Biol. 1998;18:5609–5619. doi: 10.1128/mcb.18.9.5609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sunters A, McCluskey J, Grigoriadis AE. Control of cell cycle gene expression in bone development and during c-Fos-induced osteosarcoma formation. Dev Genet. 1998;22:386–397. doi: 10.1002/(SICI)1520-6408(1998)22:4<386::AID-DVG8>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 32.Sunters A, Thomas DP, Yeudall WA, Grigoriadis AE. Accelerated cell cycle progression in osteoblasts overexpressing the c-fos proto-oncogene: induction of cyclin A and enhanced CDK2 activity. J Biol Chem. 2004;279:9882–9891. doi: 10.1074/jbc.M310184200. [DOI] [PubMed] [Google Scholar]
- 33.Ramljak D, Calvert RJ, Wiesenfeld PW, Diwan BA, Catipovic B, Marasas WF, Victor TC, Anderson LM, Gelderblom WC. A potential mechanism for fumonisin B(1)-mediated hepatocarcinogenesis: cyclin D1 stabilization associated with activation of Akt and inhibition of GSK-3beta activity. Carcinogenesis. 2000;21:1537–1546. [PubMed] [Google Scholar]
- 34.Diehl JA, Zindy F, Sherr CJ. Inhibition of cyclin D1 phosphorylation on threonine-286 prevents its rapid degradation via the ubiquitin-proteasome pathway. Genes Dev. 1997;11:957–972. doi: 10.1101/gad.11.8.957. [DOI] [PubMed] [Google Scholar]
- 35.Normanno N, De Luca A, Bianco C, Strizzi L, Mancino M, Maiello MR, Carotenuto A, De Feo G, Caponigro F, Salomon DS. Epidermal growth factor receptor (EGFR) signaling in cancer. Gene. 2006;366:2–16. doi: 10.1016/j.gene.2005.10.018. [DOI] [PubMed] [Google Scholar]
- 36.Roux PP, Shahbazian D, Vu H, Holz MK, Cohen MS, Taunton J, Sonenberg N, Blenis J. RAS/ERK signaling promotes site-specific ribosomal protein S6 phosphorylation via RSK and stimulates cap-dependent translation. J Biol Chem. 2007;282:14056–14064. doi: 10.1074/jbc.M700906200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nicholson RI, Gee JM, Harper ME. EGFR and cancer prognosis. Eur J Cancer. 2001;37 Suppl 4:S9–S15. doi: 10.1016/s0959-8049(01)00231-3. [DOI] [PubMed] [Google Scholar]
- 38.Ito Y, Takeda T, Sakon M, Tsujimoto M, Higashiyama S, Noda K, Miyoshi E, Monden M, Matsuura N. Expression and clinical significance of erb-B receptor family in hepatocellular carcinoma. Br J Cancer. 2001;84:1377–1383. doi: 10.1054/bjoc.2000.1580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Breuhahn K, Longerich T, Schirmacher P. Dysregulation of growth factor signaling in human hepatocellular carcinoma. Oncogene. 2006;25:3787–3800. doi: 10.1038/sj.onc.1209556. [DOI] [PubMed] [Google Scholar]
- 40.Daveau M, Scotte M, Francois A, Coulouarn C, Ros G, Tallet Y, Hiron M, Hellot MF, Salier JP. Hepatocyte growth factor, transforming growth factor alpha, and their receptors as combined markers of prognosis in hepatocellular carcinoma. Mol Carcinog. 2003;36:130–141. doi: 10.1002/mc.10103. [DOI] [PubMed] [Google Scholar]
- 41.Moghaddam SJ, Haghighi EN, Samiee S, Shahid N, Keramati AR, Dadgar S, Zali MR. Immunohistochemical analysis of p53, cyclinD1, RB1, c-fos and N-ras gene expression in hepatocellular carcinoma in Iran. World J Gastroenterol. 2007;13:588–593. doi: 10.3748/wjg.v13.i4.588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gladden AB, Diehl JA. Location, location, location: the role of cyclin D1 nuclear localization in cancer. J Cell Biochem. 2005;96:906–913. doi: 10.1002/jcb.20613. [DOI] [PubMed] [Google Scholar]
- 43.Alt JR, Cleveland JL, Hannink M, Diehl JA. Phosphorylation-dependent regulation of cyclin D1 nuclear export and cyclin D1-dependent cellular transformation. Genes Dev. 2000;14:3102–3114. doi: 10.1101/gad.854900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kobayashi K, Phuchareon J, Inada K, Tomita Y, Koizumi T, Hatano M, Miyatake S, Tokuhisa T. Overexpression of c-fos inhibits down-regulation of a cyclin-dependent kinase-2 inhibitor p27Kip1 in splenic B cells activated by surface Ig cross-linking. J Immunol. 1997;158:2050–2056. [PubMed] [Google Scholar]
- 45.LaBaer J, Garrett MD, Stevenson LF, Slingerland JM, Sandhu C, Chou HS, Fattaey A, Harlow E. New functional activities for the p21 family of CDK inhibitors. Genes Dev. 1997;11:847–862. doi: 10.1101/gad.11.7.847. [DOI] [PubMed] [Google Scholar]
- 46.Soos TJ, Kiyokawa H, Yan JS, Rubin MS, Giordano A, DeBlasio A, Bottega S, Wong B, Mendelsohn J, Koff A. Formation of p27-CDK complexes during the human mitotic cell cycle. Cell Growth Differ. 1996;7:135–146. [PubMed] [Google Scholar]
- 47.Cheng M, Olivier P, Diehl JA, Fero M, Roussel MF, Roberts JM, Sherr CJ. The p21(Cip1) and p27(Kip1) CDK 'inhibitors' are essential activators of cyclin D-dependent kinases in murine fibroblasts. EMBO J. 1999;18:1571–1583. doi: 10.1093/emboj/18.6.1571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sherr CJ, Roberts JM. CDK inhibitors: positive and negative regulators of G1-phase progression. Genes Dev. 1999;13:1501–1512. doi: 10.1101/gad.13.12.1501. [DOI] [PubMed] [Google Scholar]