

Abstract
In pemphigus foliaceus (PF), autoantibodies against desmoglein 1 (Dsg1) cause blisters. Using antibody phage display we have cloned monoclonal antibodies (mAbs) from a PF patient. These mAbs, like those from a previous patient, were directed against mature (mat) Dsg1 on the cell surface of keratinocytes and precursor (preDsg1) in the cytoplasm. To determine if individuals without pemphigus have B cell tolerance to Dsg1 we cloned mAbs from two patients with thrombotic thrombocytopenic purpura and a healthy person. We found mAbs against pre but not mat Dsg1. All but one of the 23 anti-preDsg1 mAbs from PF patients and those without PF used the VH3-09 (or closely related VH3-20) heavy chain gene, whereas no PF anti-matDsg1 used these genes. VH cDNA encoding anti-preDsg1 had significantly fewer somatic mutations than did anti-matDsg1 cDNA, consistent with chronic antigen driven hypermutation of the latter compared to the former. These data indicate that individuals without PF do not have B cell tolerance to preDsg1 and that loss of tolerance to matDsg1 is not due to epitope shifting of anti-preDsg1 B cells (because of different VH gene usage). However, presentation of peptides from Dsg1 by preDsg1-specific B cells may be one step in developing autoimmunity in PF.
Keywords: Adhesion Molecules, Autoantibodies, Autoimmunity, B Cells, Skin
Introduction
Pemphigus foliaceus (PF) is a tissue-specific autoimmune disease in which antibodies against the desmosomal cadherin desmoglein 1 (Dsg1) cause loss of keratinocyte adhesion in the superficial epidermis, resulting in skin blisters (1-3). Desmogleins are thought to function as cell-cell adhesion molecules in the epidermis and maintain its integrity. Dsg1 is synthesized in the endoplasmic reticulum as an inactive precursor protein with an amino-terminal propeptide (preDsg1), which is thought to prevent intracellular aggregation with other newly synthesized cadherins within the secretory pathway. The propeptide is cleaved by a Golgi proprotein convertase such as furin to yield a biologically active mature cadherin (matDsg1) that is assembled into desmosomes on the cell surface (4,5).
Polyclonal anti-Dsg1 antibodies from PF patients have been shown to be pathogenic in organ culture of normal human skin and by passive transfer to neonatal mice. In both models these autoantibodies cause blisters from loss of cell-cell adhesion with the typical histology of PF (6-8).
Because previous studies of pathogenic PF autoantibodies were performed with polyclonal antibodies from sera, we recently isolated monoclonal antibodies (mAbs) as single-chain variable fragments (scFvs) from a PF patient using phage display in order to understand the pathogenicity of individual anti-Dsg1 mAbs (9). These scFvs included pathogenic anti-Dsg1 mAbs which bound the keratinocyte cell surface by indirect immunofluorescence (IIF) and induced blisters in the epidermis, as do the sera from PF patients. However most of the isolated anti-Dsg1 mAbs were non-pathogenic. These non-pathogenic mAbs could be divided into two groups by IIF and immunoprecipitation antigen mapping: one group showing typical keratinocyte cell surface staining and binding of matDsg1, and the other showing no, or very weak intracellular, staining, with binding of preDsg1 (9,10). The reason such anti-preDsg1 mAbs can be isolated by phage display is because the antibody phage libraries are panned on Dsg1-coated ELISA plates to isolate anti-Dsg1 antibodies. Such plates are made from recombinant Dsg1 produced by baculovirus in insect cells, and this recombinant protein contains both matDsg1 and preDsg1 (9-11).
The unexpected finding of antibodies specific for intracellular preDsg1 in PF patients led us to hypothesize that individuals without PF might also have B cells that express antibodies specific for preDsg1 because preDsg1, being intracellular and not normally exposed to the immune system, would not necessarily induce B cell tolerance (and, of course, such intracellular antigens can under certain circumstances, e.g. in lupus erythematosus, induce autoimmunity). On the other hand, we hypothesized that only patients with PF would have antibodies against extracellular matDsg1, which, being exposed to the immune system, would normally induce tolerance. Stated differently, the specific autoimmune defect in PF would be loss of tolerance only to the matDsg1. To test these hypotheses experimentally, we cloned anti-Dsg1 mAbs from another PF patient and from three controls: a healthy individual and two patients with another unrelated autoimmune disease, thrombotic thrombocytopenic purpura (TTP). We chose patients with TTP because, like PF, it is an autoimmune disease with a specific autoantigen target (ADAMTS13 metalloprotease). We found that mAbs reacting with preDsg1 were isolated not only from this second PF patient but also from all three controls, while mAbs against matDsg1 were isolated only from the PF patients. Our results suggest that lack of tolerance to matDsg1 is specific to PF, while autoreactivity to preDsg1 is not limited to PF patients. We discuss a potential role for autoreactivity to preDsg1 in the onset of autoimmunity in PF patients.
Materials and Methods
Construction of phage library
Using previously described methods(12), we constructed separate IgG-κ and IgG-λ phage libraries from 4 × 107 mononuclear cells isolated from 30 ml of peripheral blood collected from a PF patient with clinically active disease. Briefly, RT-PCR was used to amplify the immunoglobulin variable regions of the heavy (VH) and light chains (VL), and the gene fragments were then cloned into the phagemid vector pComb3X (Scripps Institute). The phagemid library was electroporated into XL-1 Blue suppressor strain of E. coli (Stratagene) with superinfection by VCSM13 helper phage (Stratagene). In this system, filamentous phage particles express scFv antibody fragments (with a carboxy-terminal 6× histidine tag and a hemagglutinin [HA] tag) fused to the pIII bacteriophage coat protein. Recombinant phage were purified from culture supernatants by polyethylene glycol precipitation and resuspended in PBS, pH 7.4 with 1% BSA containing 1 mM CaCl2. The library comprised more than 1 × 108 independent transformants as determined by titering on E. coli XL1-Blue. To validate library diversity prior to selection on Dsg1, we analyzed the sequences of 20 phage antibody clones from the unpanned library. We found no duplicate sequences and marked heterogeneity in VH and VL gene usage similar to that found in normal human peripheral blood lymphocytes (data not shown). We also selected anti-Dsg1 mAbs from previously constructed libraries derived from the peripheral blood lymphocytes of two patients with TTP and a healthy person donor.
These studies have been approved by the University of Pennsylvania Institutional Review Board for human research.
Panning of phage libraries
ELISA plates coated with recombinant Dsg1 (Medical and Biological Laboratories (MBL)) were used to isolate phage clones that express anti-Dsg1 scFv as previously described (9,13). Briefly, 4 microtiter plate wells were incubated with blocking buffer (PBS with 3% skim milk) at room temperature for 1 hour. The phage library was diluted into blocking buffer and was incubated with Dsg1 on the wells for 2 hours at room temperature. After 5 to 10 washes with PBS-Ca containing 0.1% Tween 20, adherent phage were eluted with 76 mM citric acid, pH 2.0, incubated for 10 minutes at room temperature, and then neutralized with 2M unbuffered Tris. The eluted phage were amplified in XL1-Blue E. coli and rescued by superinfection with VCSM13 helper phage. Phage were harvested from bacterial culture supernatant and then re-panned on Dsg1 ELISA plates for 3 additional rounds. Individual phage clones were isolated from each round of panning and analyzed for their binding to Dsg1 by ELISA using horseradish peroxidase (HRP)-conjugated anti-M13 antibody (GE Healthcare).
Sequence analysis of scFv antibodies
Recombinant phagemids were purified with a plasmid preparation system (Qiagen) and the VH and VL inserts were sequenced using pComb3X specific primers previously described (12). The nucleotide sequences were compared with the germline sequences in V Base sequence directory (http://vbase.mrc-cpe.cam.ac.uk/) to determine their germline gene origins and interrelatedness.
Production and purification of soluble scFvs
The Top10 F′ non-suppressor strain of E. coli (Invitrogen) was infected with monoclonal phage, and soluble scFvs were purified using Fastbreak lysis reagent (Promega) or osmotic lysis and Talon or nickel metal affinity resin (Clontech Laborarories) as previously described (9,13).
Dsg1 scFv ELISA
The reactivity of scFv against human Dsg1 was measured by Dsg1 ELISA (Medical and Biological Laboratories) using HRP-conjugated anti-HA monoclonal antibody (clone 3F10, 1:1000 dilution, Roche Diagnostics) as a secondary antibody as described (9,13). In some experiments, to increase the ratio of the mature form of Dsg1 on ELISA plates, we pretreated the plates with 10U/well of furin (New England Biolabs) in 20 mM Tris, 500 mM sodium chloride, pH7.5 [TBS], with 1mM CaCl2 at room temperature overnight.
Direct and indirect immunofluorescence
Binding of scFvs to human skin was visualized by immunofluroescence as previously described (9,13). Binding was detected with rat monoclonal anti-HA antibody (3F10, 1:100 dilution, Roche Diagnostics) followed by Alexa Fluor 568-conjugated anti-rat IgG (1:200 dilution, Invitrogen).
Immunoprecipitation-immunoblotting analysis
The ectodomain of human Dsg1 fused to E-tag and a 6×histidine tag (Dsg1-EHis) produced by a baculovirus expression system was used as substrate (14). One hundred μL of baculovirus-infected insect cell culture supernatants containing recombinant Dsg1 were incubated with scFvs for 30 minutes and then immunoprecipitated with 100 ng of anti-HA antibody (3F10, Roche Diagnostics) and Protein G Sepharose (GE Healthcare) at 4°C overnight with gentle rotation. After washing with TBS-Ca, the immunoprecipitates were resuspended in Laemmli sample buffer (Bio-Rad Labratories), separated by SDS-PAGE, and transferred to nitrocellulose membranes (Bio-Rad Labratories). Membranes were probed with monoclonal mouse anti- E-tag antibody (1:2000 dilution, GE Healthcare) followed by HRP-conjugated anti-mouse IgG (1:2000 dilution, Bio-Rad Labratories). To increase the ratio of matDsg1 to preDsg1 in some experiments, 100 μL of the culture supernatants containing recombinant molecules were pretreated with 10U of furin (New England Biolabs) at room temperature overnight.
Human skin organ culture injection
Specimens were obtained from residual healthy skin after excisional surgery. The specimens were defatted and cut into 5 mm sections. After intradermal injection of 50 μL of purified scFv (1000 ng/μL) using an insulin syringe, skin specimens were incubated on transwell inserts (Corning) with defined keratinocyte SFM (Invitrogen) containing 1.2 mM CaCl2 in the outer compartment. After 24 hours at 37°C, the skin was harvested for direct immunofluorescence and histology.
Statistical analysis
All parameters were compared by the Mann-Whitney U test as appropriate.
Results
Phage cloning of antibodies from a PF patient (PF2) identifies pathogenic and non-pathogenic anti-matDsg1 antibodies and non-pathogenic anti-preDsg1 antibodies
In a previous study we isolated mAbs by phage display cloning from a PF patient (PF1) (9). To determine the similarity of the antibody repertoire among PF patients, we cloned mAbs from a second PF patient (PF2) and obtained 6 unique mAbs from this patient (Table I). Each of these mAbs was encoded for by a different heavy-chain gene (VH1-08, VH3-09, VH3-30, VH3-53, VH3-66, and VH4-30). Even though all of these mAbs had a positive reaction on Dsg1 ELISA, we found that 4 of them (F24-2, F24-15, F23-5, and F24-9) bound the cell surface of keratinocytes as determined by IIF on normal human skin (Fig. 1A), while 2 mAbs (F24-1 and F23-6) did not show cell surface staining but showed no, or very weak cytoplasmic, staining (Fig. 1B).
Table I.
Monoclonal antibodies from PF1 and PF2 libraries
| Source | Clone name | VH gene | D gene | J gene | VL gene | Human skin IIF | Immunoprecipitation | Pathogenicity |
|---|---|---|---|---|---|---|---|---|
| PF2 | F24-1 | VH4-30 | D3-10/DXP′1 | JH3b | 3r | cytoplasm | preDsg1 | (-) |
| F23-6 | VH3-09 | D3-22/D21-9 | JH3b | L1 | cytoplasm | preDsg1 | (-) | |
| F24-2 | VH3-30 | D6-13/DN1 | JH4d | 3j | cell surface | pre & matDsg1 | (-) | |
| F24-15 | VH3-66 | D7-27/DHQ52 | JH4b | 1c | cell surface | matDsg1 | (-) | |
| F23-5 | VH1-08 | D3-10/DXP′1 | JH6b | 4b | cell surface | matDsg1 | (-) | |
| F24-9 | VH3-53 | D4 | JH4b | 1c | cell surface | matDsg1 | (+) | |
| PF1a | PF1-2-17 (3-093/O18) | VH3-09 | D2-2 | JH6b | O18/O8 | cytoplasm | preDsg1 | (-) |
| PF1-2-10 (3-094/O18) | VH3-09 | D3-3/DXP4 | JH3a | O18/O8 | cytoplasm | preDsg1 | (-) | |
| PF1-2-9 (3-095/O18) | VH3-09 | D3-3/DXP4 | JH3a | O18/O8 | cytoplasm | preDsg1 | (-) | |
| PF1-26 (3-096/L12) | VH3-09 | D3-3/DXP4 | JH3a | L12 | cytoplasm | preDsg1 | (-) | |
| PF1-2-3 (3-097/1c) | VH3-09 | D3-3/DXP4 | JH3b | 1c | cytoplasm | preDsg1 | (-) | |
| PF1-2-18 (3-098/L11) | VH3-09 | D3-22/D21-9 | JH4b | L11 | cytoplasm | preDsg1 | (-) | |
| PF1-1-7 (3-099/L8) | VH3-09 | D2-8/DLR1 | JH2 | L8 | cytoplasm | preDsg1 | (-) | |
| PF1-2-5 (1-18/L1) | VH1-18 | D3-10/DXP′1 | JH4b | L1 | cell surface | pre & matDsg1 | (-) | |
| PF1-2-22 (1-08/O12) | VH1-08 | D3-3/DXP4 | JH6b | O12/O2 | cell surface | matDsg1 | (-) | |
| PF1-8-2 (3-07/1e) | VH3-07 | D3-10/DXP′1 | JH4b | 1e | cell surface | matDsg1 | Weak (+) | |
| PF1-8-15 (3-30/3h) | VH3-30 | D5-24 | JH4b | 3h | cell surface | matDsg1 | (+) | |
a The parenthesis of each antibody from PF1 indicates the names used in the previous study (9).
Figure 1.
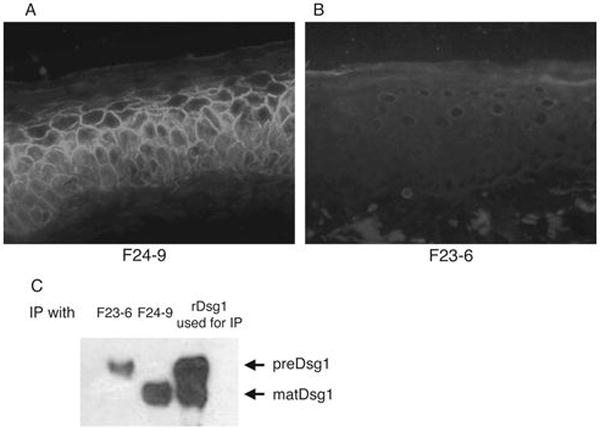
Immunohistological and immunochemical analysis of two major types of anti-Dsg1 mAbs isolated from PF patient 2 (PF2). A and B, IIF on normal human skin with anti-HA of F24-9 and F23-6 mAbs (each cloned with an HA tag). F24-9 stains the cell surface of keratinocytes whereas F23-6 does not stain the cell surface but, if it stains at all, weakly stains the cytoplasm, suggesting binding of mat and pre Dsg1, respectively. C, Immunoblotting with anti-E-tag. Left 2 lanes show immunoblot of an immunoprecipitation of recombinant human Dsg1Ehis. F23-6 precipitates a higher molecular weight band, preDsg1, while F24-9 binds to lower molecular weight matDsg1. Right lane shows immunoblot of recombinant Dsg1Ehis used for the immunoprecipitation and shows both the high and lower molecular weight species.
To further characterize these mAbs, we used immunoprecipitation experiments of recombinant human Dsg1 produced by a baculovirus expression system. This system produces both preDsg1 and matDsg1, which are seen as two bands on SDS-PAGE, the former with slightly slower migration (Fig. 1C, lane 3) (11). The two mAbs with no, or weak cytoplasmic, staining patterns on IIF (F24-1 and F23-6) immunoprecipitated only the higher molecular weight band (i.e. preDsg1) (Fig. 1C, lane 1) The 4 mAbs with cell-surface IIF staining patterns precipitated the lower molecular weight band (matDsg1). One of those mAbs (F24-2) that bound matDsg1 also precipitated preDsg1, presumably because its antigen on matDsg1 is also present on preDsg1, as previously shown with a mAb from PF1 (10). In summary, the cell surface mAbs bound matDsg1 and those that did not bind the cell surface bound only preDsg1.
Binding to preDsg1 is consistent with the very weak, or even undetectable, intracellular staining by IIF because preDsg1, normally inside the cell, is rapidly transported to the cell surface where it is processed to the mature form which is stabilized and accumulates in desmosomes (5). Finally, monoclonal antibodies cloned from patient PF1, with similar IIF and immunochemical characteristics have been shown to bind to preDsg1 (9,10).
To examine the pathogenicity of the cloned mAbs, we injected them into the dermis of normal human skin in organ culture. F24-9 was the only pathogenic mAb cloned from PF2. It showed a cell surface-staining pattern in direct immunofluorescence (DIF) of the injected skin and histology showed blister formation from acantholysis in the granular layer of the epidermis (Fig. 2A and 2D), typical of the histology of blisters in PF patients. When injected intradermally into normal human skin organ culture, the other three mAbs that bound matDsg1 (F24-2, F24-15, and F23-5) showed the cell surface-staining pattern by DIF that is typical of PF, but, in three separate experiments, these mAbs did not cause a blister in the injected skin tissue at the same concentrations as the pathogenic antibody (Fig. 2B and 2E). The mAbs F24-1 and F23-6, which showed very weak cytoplasmic staining by IIF, were negative by DIF when injected into human skin, presumably because they cannot access the intracellular antigen in living cells (Fig. 2C and 2F).
Figure 2.
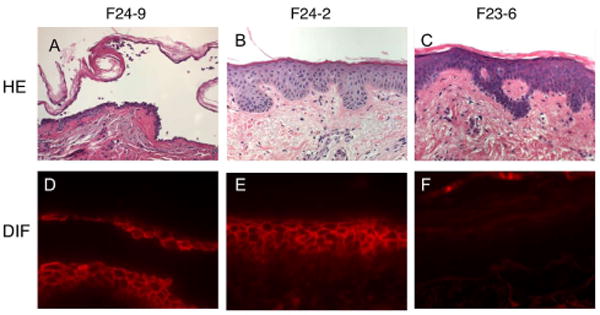
Pathogenicity and binding of mAbs from patient PF2 as determined by injection into normal human skin in organ culture. A and D, F24-9 causes a blister in the superficial epidermis typical of PF (as shown by histology with routine hematoxylin and eosin staining) and shows typical keratinocyte cell surface staining by direct immunofluorescence (DIF) of the injected skin. B and E, F24-2 does not cause an epidermal blister but binds the cell surface of keratinocytes. C and F, F23-6 is non-pathogenic and shows no binding by DIF. These results demonstrate the three types of antibodies cloned from patient PF2: pathogenic anti-matDsg1 (F24-9), non-pathogenic anti-matDsg1 (F24-2), and non-pathogenic anti-preDsg1 (F23-6).
Comparison of antibodies cloned from PF2 with previously cloned antibodies from patient PF1
Previous studies have indicated that the immunologic properties of mAbs cloned from pemphigus patients are mainly dependent on the heavy chain variable region with light chain usage being promiscuous (9,13,15). In other words the same heavy chain with different light chains had similar antigen binding characteristics and pathogenicity. Therefore, in comparing mAbs from the two PF patients, we sorted them by their variable heavy chain usage (Table I). We isolated a total of 17 clones with unique heavy chains from the two PF patients (Table I). These mAbs could be categorized into three major groups, 1) anti-matDsg1 mAbs that are pathogenic (Fig. 2A and 2D); 2) mAbs that are non-pathogenic but that bind matDsg1 (Fig. 2B and 2E), and 3) anti-preDsg1 mAbs that are all non-pathogenic (Fig. 2C and 2F). Both PF patients had mAbs in each of these three groups.
The properties of these mAbs indicate that pathogenic anti-Dsg1 antibodies do not necessarily use the same VH gene in different patients (compare PF1-8-15 and F24-9). Furthermore, pathogenic and non-pathogenic anti-Dsg1 mAbs can use the same VH gene (compare PF1-8-15 and F24-2). These data suggest that simply targeting antibodies by virtue of the specific VH gene used for their expression in various patients will not necessarily be a reasonable strategy to pursue for therapy of PF. Interestingly, ELISA studies showed that F24-9 and PF1-81-15 inhibited each others binding to Dsg1, whereas a non-pathogenic anti-Dsg1 mAb did not inhibit pathogenic antibody binding (unpublished). Thus, idiotypes and/or epitopes might be shared by pathogenic antibodies from different patients.
Remarkably, unlike anti-matDsg1 antibodies, almost all anti-preDsg1 antibodies use heavy chain gene VH3-09 (as do other cloned anti-preDsg1 antibodies—see below), indicating marked genetic restriction. Furthermore, anti-matDsg1 mAbs use different VH genes than do anti-preDsg1 antibodies, demonstrating that one set is not derived from the other through B cell maturation (also see Discussion).
Phage cloning of anti-Dsg1 antibodies from a healthy person and two patients with TTP identifies only non-pathogenic anti-preDsg1 mAbs
Our results revealed that PF patients had antibodies not only against matDsg1 on the cell surface but also against preDsg1. PreDsg1 is intracellular, and under non-traumatic conditions may be inaccessible to the immune system, allowing B cells to escape tolerance. If this is the case, then those without pemphigus should also have anti-preDsg1 antibodies. However, we did not expect individuals without pemphigus to have anti-matDsg1 because tolerance should normally be developed to a protein exposed to the immune system. We, therefore, used phage display to clone anti-Dsg1 antibodies from patients with TTP (control patients without pemphigus but with an autoantigen-specific autoantibody-mediated disease) and one healthy person. We isolated anti-Dsg1 mAbs from each of the non-PF libraries by panning on Dsg1-coated ELISA plates which contain both matDsg1 and preDsg1 (9,10). Ten unique anti-Dsg1 mAbs were isolated from the two TTP patient libraries (designated TTP2 and TTP3), and 4 anti-Dsg1 clones were isolated from the library constructed from a healthy person (designated K2) (Table II). Remarkably, all of these mAbs used variable heavy chain gene VH3-09 except one clone that used the VH3-20 gene, a human VH germline sequence very similar to that of the VH3-09 gene (91% nucleotide identity). Even though all of these mAbs bound to Dsg1 on ELISA, they showed no staining or a very weak cytoplasmic staining pattern by IIF of normal human skin. None showed cell surface staining (Fig. 3A). Furthermore, these mAbs immunoprecipitated a Dsg1 protein from baculovirus-produced recombinant Dsg1 with slightly higher molecular weight than that precipitated by an anti-matDsg1 mAb from patient PF2 (Fig. 3B). Therefore, these anti-Dsg1 mAbs isolated from non-pemphigus patients showed the same immunochemical characteristics as anti-preDsg1 mAbs from PF patients.
Table II.
Monoclonal antibodies from TTP and K2 libraries
| Source | Clone name | VH gene | D gene | J gene | VL gene |
|---|---|---|---|---|---|
| TTP2 | T2D13-5 | VH3-09 | D3-22/D21-9 | JH6b | L11 |
| T2D13-8 | VH3-09 | D3-10/DXP′1 | JH5b | 1c | |
| T2D14-2 | VH3-09 | D4-17 | JH4b | 1 | |
| T2D14-6 | VH3-20 | D3-10/DXP′1 | JH5b | O12/O2 | |
| T2D14-12 | VH3-09 | D2-15/D2 | JH6b | 3h | |
| T2D14-15 | VH3-09 | D4-11/DA1 | JH3b | 3h | |
| TTP3 | T3D14-1 | VH3-09 | D1-26 | JH4b | O18/O8 |
| T3D14-2 | VH3-09 | not found | JH4b | O18/O8 | |
| T3D14-3 | VH3-09 | D4-17 | JH3b | 1c | |
| T3D14-4 | VH3-09 | D2-8/DLR1 | JH4b | 1e | |
| K2 (normal) | K2D14-1 | VH3-09 | D2-15/D2 | JH4b | L12 |
| K2D14-3 | VH3-09 | D3-10/DXP′1 | JH4b | O18/O8 | |
| K2D14-4 | VH3-09 | D6-19 | JH4b | O12/O2 | |
| K2D14-7 | VH3-09 | not found | JH4b | L12 | |
Figure 3.
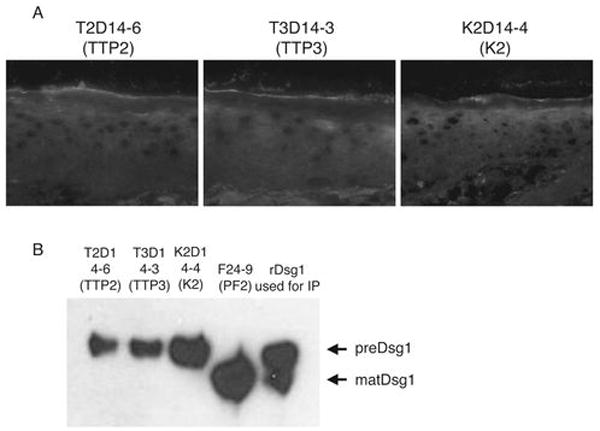
IIF and immunoprecipitation of mAbs cloned from TTP patients (TTP2, TTP3) and a normal individual (K2). A, All of the anti-Dsg1 mAbs isolated from non-pemphigus individuals show either no staining or very weak cytoplasmic staining in IIF of normal human skin. None show cell surface staining. B, These mAbs immunoprecipitate recombinant preDsg1, in contrast to F24-9, a mAb cloned from patient PF2, that binds the keratinocyte cell surface (Fig. 1A) and immunoprecipitates matDsg1.
To confirm that these mAbs bound preDsg1, we treated Dsg1 ELISA plates with furin proprotein convertase to convert adsorbed preDsg1 antigen to matDsg1. This approach would be expected to decrease, but not completely eliminate, binding of anti-preDsg1 antibodies, as the enzyme is probably not 100% efficient on plate-bound substrate and the cleaved propeptide may still remain adsorbed to plate wells. These ELISA experiments showed that the binding of an anti-Dsg1 mAb (K2D14-4) cloned from a healthy individual was, indeed, decreased on a furin-treated plate by 69% compared to a control buffer-treated plate, while an anti-matDsg1 mAb from a PF patient (F24-9) increased binding by 222% on furin-treated plates, compared to controls (Fig. 4A). In addition, mAbs cloned from PF and non-pemphigus patients were tested for their ability to immunoprecipitate furin-treated recombinant Dsg1. Immunoblotting of baculovirus-produced Dsg1 indicates a higher and a lower molecular weight band, preDsg1 and matDsg1, respectively (Fig 4B, lane 7). After furin treatment of this baculovirus-produced Dsg1 the preDsg1 band is decreased and the matDsg1 band is increased (Fig. 4B, Lane 6 and 7). T2D14-6, a mAb cloned from one of the TTP patients immunoprecipitates the higher molecular weight preDsg1 from the baculovirus-produced Dsg1, but no longer precipitates a band from the furin-treated Dsg1, since the preDsg1 is processed to the mature form (Fig. 4B, lanes 1, 2). Conversely, F24-9 (anti-matDsg1 mAb from patient PF2) immunoprecipitates the lower molecular weight matDsg1 before and after furin-treatment of recombinant Dsg1, although it precipitated more Dsg1 after furin treatment, presumably because more matDsg1 is present (Fig. 4B, lanes 3, 4).
Figure 4.
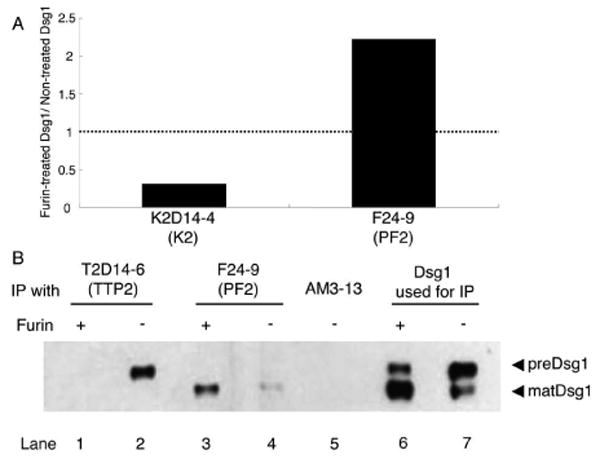
Furin, which processes preDsg1 to matDsg1, causes decreased binding of anti-Dsg1 antibodies cloned from individuals without pemphigus. A, Furin treatment of Dsg1 ELISA plates causes decreased binding of K2D14-4, a mAb from a normal individual, but increased binding of F24-9, a pathogenic cell surface mAb from patient PF2. B, Immunoblot with anti-E-tag. Dsg1Ehis recombinant protein without furin treatment shows a high and lower molecular weight band (lane 7). Treatment with furin shows decreased intensity of the higher molecular weight band from processing of preDsg1 to matDsg1 (lane 6). T2D14-6 immunoprecipitates preDsg1 (lane 2) that is not longer detectable after furin treatment (lane 1). However, F24-9 precipitates only matDsg1 with or without furin treatment (lanes 3,4).
Taken together, these results suggest that anti-Dsg1 antibodies derived from individuals without pemphigus identified only non-pathogenic anti-preDsg1 mAbs, and interestingly, these anti-preDsg1 mAbs are predominantly encoded by an identical human VH germline gene (VH3-09), as are most anti-preDsg1 mAbs cloned from PF patients.
Analysis of somatic hypermutation indicates greater affinity maturation for anti-matDsg1 than for anti-preDsg1 antibodies
When antigen-specific B cells are activated by antigen stimulation, DNA encoding their immunoglobulin receptors undergo somatic mutation (16). B cells whose receptors develop higher affinities for an antigen are preferentially selected through a process referred to as affinity maturation (17). In those B cells that undergo affinity maturation, replacement mutations (i.e. mutations that result in a new amino acid) within the variable complementarity determining regions (CDRs) are positively selected because these sequences make contact with antigen(18,19). Thus, B cells that are chronically stimulated by antigen are selected by affinity maturation and show increased replacement to silent mutations compared to their germline-encoded CDRs. Therefore, we predicted that anti-matDsg1 mAbs from PF patients undergo affinity maturation from chronic stimulation by the exposed antigen and thus should have more somatic mutations in their variable regions with increased replacement to silent mutation ratio in their CDRs compared to anti-preDsg1 mAbs which are derived from B cells not chronically stimulated by intracellular preDsg1.
To test this hypothesis, we analyzed the rates of somatic mutation of the VH regions of the anti-matDsg1 and anti-preDsg1 mAbs compared to their original VH germline sequences. Figure 5A shows the average mutation rates of anti-matDsg1 mAbs from PF patients (n=8) and anti-preDsg1 mAbs using VH3-09 and VH3-20 genes from PF patients and those without pemphigus (n=22). Rate of mutation in anti-preDsg1 mAbs was 1.8% in the VH framework regions (FRs) and 3.0% in CDRs. Conversely, mutation rates in anti-matDsg1 mAbs were 5.3% in FRs and 13.4% in CDRs (vs. CDRs of anti-preDsg1, p<0.0001), respectively. As expected, the somatic mutations in anti-matDsg1 antibodies occurred more often in the CDRs than in the FRs (CDR vs. FR of anti-matDsg1, P=0.0045) because the B cells encoding the former group can be positively selected by increasing antibody affinity whereas the latter group may be negatively selected by destabilizing the FR (18). Finally, consistent with affinity maturation of anti-matDsg1 antibodies, we found an increase in the ratio of mutations causing amino acid replacement to those that are silent in the CDR compared to that in the FR (Fig. 5B). The replacement to silent ratios of anti-matDsg1 mAbs were 1.79 in FRs and 3.23 in CDRs.
Figure 5.
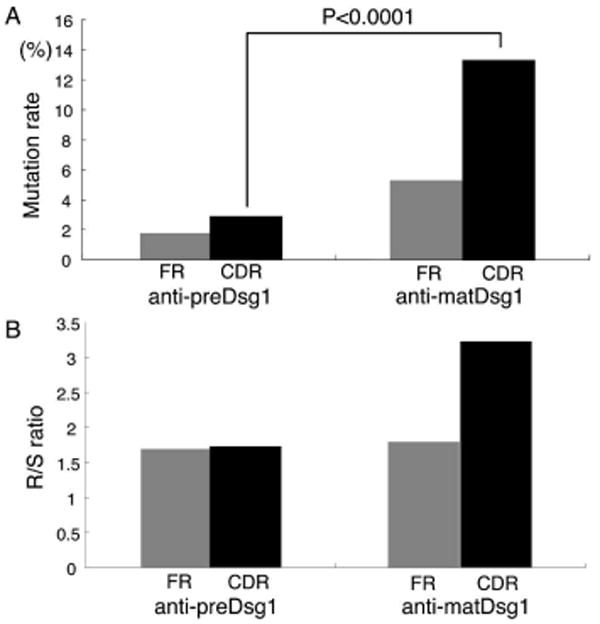
Variable heavy chains of anti-matDsg1 mAbs have higher mutation rates compared to the VH germline sequences and a higher replacement to silent (R/S) ratio of those mutations than anti-preDsg1 mAbs. A, Comparison of the average mutation rates shows that anti-matDsg1 antibodies have more mutations than anti-preDsg1 antibodies throughout the heavy chain variable regions and that these mutations occur more often in CDRs than in FR regions. B, The replacement to silent ratio of somatic mutations is elevated in the CDRs compared to FRs of anti-matDsg1 antibodies.
Finally, we also compared the rate of somatic mutations in the anti-preDsg1 mAbs from the PF patients and from the individuals without PF, because we thought that in PF there might be some exposure to preDsg1 released from damaged epidermis, therefore, there might be a somewhat higher mutation rate in PF patients due to some antigen stimulation. This was, in fact, the case. Anti-preDsg1 mAbs from PF patients showed higher mutation rates (n=8, 4.7%) than anti-preDsg1 mAbs from non-PF individuals (n=14, 1.9%) in the CDRs (P=0.0158), and the former group also showed higher replacement to silent ratios than the latter group.
Discussion
In this and a previous study (9), we cloned a cohort of monoclonal antibodies from two PF patients (PF1 and PF2) that could be categorized into three groups based on pathogenicity and autoantigen specificity: pathogenic anti-matDsg1 mAbs, non-pathogenic anti-matDsg1 mAbs, and non-pathogenic anti-preDsg1 mAbs.
To determine if individuals without pemphigus have B cell tolerance to Dsg1, we cloned anti-Dsg1 mAbs from two patients with an unrelated autoimmune disease and a healthy individual. In contrast to anti-Dsg1 mAbs isolated from PF patients, we only found mAbs against the precursor but not mature form of Dsg1. All but one of the 23 anti-preDsg1 mAbs from PF patients and those without PF used the VH3-09 (or closely related VH3-20) heavy chain gene, whereas no PF anti-matDsg1 used these genes. Although no antibody repertoire cloning method, including phage display, may necessary reflect the exact frequency of VH gene usage in a given individual, the finding of so many independent anti-preDsg1 clones with VH3-09 (or a closely related heavy chain) in two PF patients and 3 individuals without PF, and the absence of the use of VH3-09 genes in all anti-matDsg1 antibodies, implies the importance of VH3-09 in forming anti-preDsg1 antibodies. Furthermore, although methods used to construct phage display libraries may generate heavy and light chain pairings not present in vivo, previous studies employing phage display to study human immune repertoires (9,15,20,21), and this study, show that the immunological properties of many antigen-specific auto- and alloantibodies are mainly carried by their heavy chains. That is, the same immunological properties are found when the same heavy chain is paired with multiple different light chains.
Anti-preDsg1 antibodies cloned from pemphigus and non-pemphigus patients suggest lack of B cell tolerance to this intracellular antigen and have similarities to so-called “natural autoantibodies”. These natural autoantibodies (NAA) have been long recognized as self-reactive antibodies in healthy people (22,23). The specificity of NAA is mostly carried by the heavy chain, which has few somatic mutations, and the light chain contributes minimally to the specificity of these antibodies (22,24). NAA are often directed against nuclear or cytoplasmic antigens, and some of the B cells that express such antibodies are thought to survive because the antigens are mostly hidden (25-27). The mAbs against preDsg1 isolated in this study share many of these features. They are directed against a cytoplasmic antigen, they show restricted VH gene usage but variable VL gene usage, and their heavy chains have few somatic mutations. These anti-preDsg1 B cells would not usually be stimulated because preDsg1 is intracellular, and they, therefore, would be expected to be low producers of antibodies. In some PF patients, anti-preDsg1 is detected in serum (10) possibly because epidermal damage releases preDsg1.
One theory of autoimmunity is that B cells with NAA receptors become stimulated either through molecular mimicry or tissue injury with resultant somatic mutation causing pathologic autoantibody production (22,25,28). Such a switch to pathologic antibodies resulting from somatic mutation could be from a process called epitope shift or epitope migration in which somatic mutations cause an antibody to bind a different part of the original antigen (20,29,30). However, in the case of PF autoantibodies against Dsg1, this mechanism does not seem to be operative. The B cells in PF that express anti-matDsg1 antibodies are not derived through somatic mutation from the B cells expressing anti-preDsg1 antibodies, because our clonal analysis indicates that pathologic (and non-pathologic anti-matDsg1) PF autoantibodies are derived from different VH genes than are the anti-preDsg1 B cell receptors.
Although it is possible that the presence of anti-preDsg1 B cells is unrelated to, and independent of, the pathogenicity of PF, anti-preDsg1 B cells could still be involved in the initiation of autoimmunity. For instance, they could present peptides representing parts of the matDsg1, derived from the processing of preDsg1, to T cells that have lost tolerance to peptides from matDsg1, thereby stimulating those cells (Fig. 6). In the context of tissue destruction, these B cells could take up preDsg1 then present peptides that are found in matDsg1 in the context of the MHC molecule (31). In this model, B cells would also have to lose tolerance to matDsg1 as a last step, and they would be stimulated to produce anti-matDsg1 antibodies by these T cells. This multiple step model requires many so-called check point failures and thus would be consistent with the rarity of PF.
Figure 6.
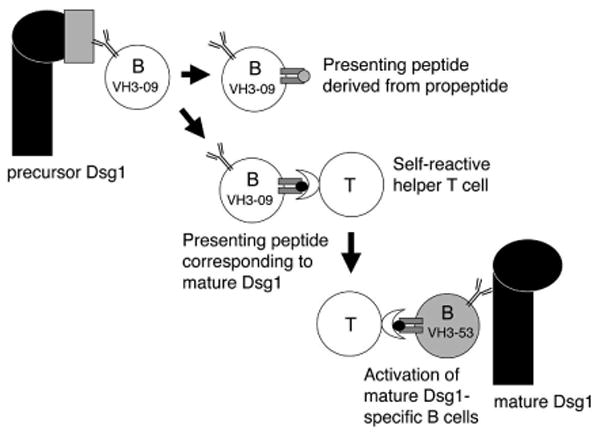
A model of PF development in which, as a first step, anti-preDsg1 B cells present peptides from matDsg1. These B cells could present antigen to T cells that recognize matDsg1 peptides. Once such T cells proliferate they can then provide help, not only to the original anti-preDsg1 B cells but also to any B cells that have escaped from tolerance to matDsg1. Such a model requires both T cells and B cells to escape tolerance to matDsg1, unlikely events consistent with the rarity of PF. Note that preDsg1 reacting B cells and matDsg1-specific B cells have different clonal origins, consistent with our data showing that anti-preDsg1 and anti-matDsg1 cloned mAbs derive from different B cell clones.
One method that has been postulated for induction of tolerance to self-antigens is light chain receptor editing, in which an autoreactive B cell during its development switches its light chain to become unreactive (32). Although this particular light chain from the autoreactive B cell could be genetically excised, phage display cloning allows artificial pairing in vitro of heavy chains with the entire light chain repertoire, which might in turn allow “re-expression” of these autoimmune receptors that had been deleted in vivo. Our results that we do not find anti-matDsg1 autoantibodies by phage display in individuals without pemphigus suggest that such a light chain receptor editing probably does not take place to induce tolerance to matDsg1. Alternatively, if tolerance is induced by light chain editing, the edited light chain could be essentially unique in the entire light chain repertoire, which seems unlikely, especially as the properties of anti-Dsg1 antibodies in PF is heavy chain dependent with light chains being more promiscuous (i.e. mAbs with different light chains paired with the same heavy chain have similar immunological properties) (9).
Though the potential physiologic role of anti-preDsg1-expressing B cells is not well understood, the existence of anti-preDsg1 in patient sera is of practical importance when interpreting certain aspects of pemphigus biology. For example, there are pemphigus patients with significant anti-preDsg1 antibodies in their sera that give high titer ODs by ELISA assays but minimal clinical disease (10). Furthermore the existence of such B cells is important when interpretating T cell stimulation studies in normals and pemphigus patients in which recombinant desmoglein, which comprises an admixture of precursor and mature forms, is used (33-35).
The findings of anti-preDsg1, but not anti-matDsg1, in individuals without pemphigus suggests a potential initiating event in the immunopathogenesis of PF and points out the necessity of studying loss of tolerance to matDsg1, and not preDsg1 in studies of the immunology of this disease.
Acknowledgments
We thank Chenyan Lin for technical expertise.
This work was supported by grants AR052672 (JRS) and K08 AR053505 (ASP) from the National Institute of Arthritis, Musculoskeletal and Skin Diseases and P50-HL81012 (DLS) from the National Heart, Lung and Blood Institute, respectively.
Abbreviations used in this paper
- CDR
complementarity determining region
- DIF
direct immunofluorescence
- Dsg
desmoglein
- FR
framework region
- IIF
indirect immunofluorescence
- matDsg1
mature Dsg
- PF
pemphigus foliaceus
- preDsg1
precursor Dsg1
- scFv
single-chain variable fragment
- TTP
thrombotic thrombocytopenic purpura
Footnotes
Disclosures: The authors declare no conflict of interest.
References
- 1.Stanley JR, Amagai M. Pemphigus, bullous impetigo, and staphylococcal scalded skin syndrome. N Engl J Med. 2006;355:1800–1810. doi: 10.1056/NEJMra061111. [DOI] [PubMed] [Google Scholar]
- 2.Eyre RW, Stanley JR. Human autoantibodies against a desmosomal protein complex with a calcium-sensitive epitope are characteristic of pemphigus foliaceus patients. J Exp Med. 1987;165:1719–1724. doi: 10.1084/jem.165.6.1719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Koulu L, Kusumi A, Steinberg MS, Klaus Kovtun V, Stanley JR. Human autoantibodies against a desmosomal core protein in pemphigus foliaceus. J Exp Med. 1984;160:1509–1518. doi: 10.1084/jem.160.5.1509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Posthaus H, Dubois CM, Muller E. Novel insights into cadherin processing by subtilisin-like convertases. FEBS Lett. 2003;536:203–208. doi: 10.1016/s0014-5793(02)03897-8. [DOI] [PubMed] [Google Scholar]
- 5.Wahl JK, III, Kim YJ, Cullen JM, Johnson KR, Wheelock MJ. N-cadherin-catenin complexes form prior to cleavage of the proregion and transport to the plasma membrane. J Biol Chem. 2003;278:17269–17276. doi: 10.1074/jbc.M211452200. [DOI] [PubMed] [Google Scholar]
- 6.Hashimoto K, Shafran KM, Webber PS, Lazarus GS, Singer KH. Anti-cell surface pemphigus autoantibody stimulates plasminogen activator activity of human epidermal cells. J Exp Med. 1983;157:259–272. doi: 10.1084/jem.157.1.259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Roscoe JT, Diaz L, Sampaio SA, Castro RM, Labib RS, Takahashi Y, Patel H, Anhalt GJ. Brazilian pemphigus foliaceus autoantibodies are pathogenic to BALB/c mice by passive transfer. J Invest Dermatol. 1985;85:538–541. doi: 10.1111/1523-1747.ep12277362. [DOI] [PubMed] [Google Scholar]
- 8.Rock B, Martins CR, Theofilopoulos AN, Balderas RS, Anhalt GJ, Labib RS, Futamura S, Rivitti EA, Diaz LA. The pathogenic effect of IgG4 autoantibodies in endemic pemphigus foliaceus (fogo selvagem) N Engl J Med. 1989;320:1463–1469. doi: 10.1056/NEJM198906013202206. [DOI] [PubMed] [Google Scholar]
- 9.Ishii K, Lin C, Siegel DL, Stanley JR. Isolation of pathogenic monoclonal anti-desmoglein 1 human antibodies by phage display of pemphigus foliaceus autoantibodies. J Invest Dermatol. 2008;128:939–948. doi: 10.1038/sj.jid.5701132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yokouchi M, Saleh MA, Kuroda K, Hachiya T, Stanley JR, Amagai M, Ishii K. Pathogenic Epitopes of Autoantibodies in Pemphigus Reside in the Amino-Terminal Adhesive Region of Desmogleins Which Are Unmasked by Proteolytic Processing of Prosequence. J Invest Dermatol. 2009 doi: 10.1038/jid.2009.61. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hanakawa Y, Schechter N, Lin C, Garza L, Li H, Yamaguchi T, Fudaba Y, Nishifuji K, Sugai M, Amagai M, Stanley JR. Molecular mechanisms of blister formation in bullous impetigo and staphylococcal scalded skin syndrome. J Clin Invest. 2002;110:53–60. doi: 10.1172/JCI15766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Barbas CFI, Burton DR, Scott JK, Silverman GJ. Phage Display: A Laboratory Manual. Cold Spring Harbor Laboratory Press; Cold Spring Harbor: 2001. [Google Scholar]
- 13.Payne AS, Ishii K, Kacir S, Lin C, Li H, Hanakawa Y, Tsunoda K, Amagai M, Stanley JR, Siegel DL. Genetic and functional characterization of human pemphigus vulgaris monoclonal autoantibodies isolated by phage display. J Clin Invest. 2005;115:888–899. doi: 10.1172/JCI24185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ishii K, Amagai M, Hall RP, Hashimoto T, Takayanagi A, Gamou S, Shimizu N, Nishikawa T. Characterization of autoantibodies in pemphigus using antigen-specific enzyme-linked immunosorbent assays with baculovirus-expressed recombinant desmogleins. J Immunol. 1997;159:2010–2017. [PubMed] [Google Scholar]
- 15.Payne AS, Siegel DL, Stanley JR. Targeting pemphigus autoantibodies through their heavy-chain variable region genes. J Invest Dermatol. 2007;127:1681–1691. doi: 10.1038/sj.jid.5700790. [DOI] [PubMed] [Google Scholar]
- 16.Basu U, Chaudhuri J, Alpert C, Dutt S, Ranganath S, Li G, Schrum JP, Manis JP, Alt FW. The AID antibody diversification enzyme is regulated by protein kinase A phosphorylation. Nature. 2005;438:508–511. doi: 10.1038/nature04255. [DOI] [PubMed] [Google Scholar]
- 17.Baumgarth N, Herman OC, Jager GC, Brown L, Herzenberg LA, Herzenberg LA. Innate and acquired humoral immunities to influenza virus are mediated by distinct arms of the immune system. Proc Natl Acad Sci U S A. 1999;96:2250–2255. doi: 10.1073/pnas.96.5.2250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.McKean D, Huppi K, Bell M, Staudt L, Gerhard W, Weigert M. Generation of antibody diversity in the immune response of BALB/c mice to influenza virus hemagglutinin. Proc Natl Acad Sci U S A. 1984;81:3180–3184. doi: 10.1073/pnas.81.10.3180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yau KY, Dubuc G, Li S, Hirama T, Mackenzie CR, Jermutus L, Hall JC, Tanha J. Affinity maturation of a V(H)H by mutational hotspot randomization. J Immunol Methods. 2005;297:213–224. doi: 10.1016/j.jim.2004.12.005. [DOI] [PubMed] [Google Scholar]
- 20.Chang TY, Siegel DL. Genetic and immunological properties of phage-displayed human anti-Rh(D) antibodies: implications for Rh(D) epitope topology. Blood. 1998;91:3066–3078. [PubMed] [Google Scholar]
- 21.Roark JH, Bussel JB, Cines DB, Siegel DL. Genetic analysis of autoantibodies in idiopathic thrombocytopenic purpura reveals evidence of clonal expansion and somatic mutation. Blood. 2002;100:1388–1398. [PubMed] [Google Scholar]
- 22.Lacroix-Desmazes S, Kaveri SV, Mouthon L, Ayouba A, Malanchere E, Coutinho A, Kazatchkine MD. Self-reactive antibodies (natural autoantibodies) in healthy individuals. J Immunol Methods. 1998;216:117–137. doi: 10.1016/s0022-1759(98)00074-x. [DOI] [PubMed] [Google Scholar]
- 23.Mouthon L, Nobrega A, Nicolas N, Kaveri SV, Barreau C, Coutinho A, Kazatchkine MD. Invariance and restriction toward a limited set of self-antigens characterize neonatal IgM antibody repertoires and prevail in autoreactive repertoires of healthy adults. Proc Natl Acad Sci U S A. 1995;92:3839–3843. doi: 10.1073/pnas.92.9.3839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Avrameas S, Ternynck T, Tsonis IA, Lymberi P. Naturally occurring B-cell autoreactivity: a critical overview. J Autoimmun. 2007;29:213–218. doi: 10.1016/j.jaut.2007.07.010. [DOI] [PubMed] [Google Scholar]
- 25.Wardemann H, Yurasov S, Schaefer A, Young JW, Meffre E, Nussenzweig MC. Predominant autoantibody production by early human B cell precursors. Science. 2003;301:1374–1377. doi: 10.1126/science.1086907. [DOI] [PubMed] [Google Scholar]
- 26.Goodnow CC, Sprent J, Fazekas de St GB, Vinuesa CG. Cellular and genetic mechanisms of self tolerance and autoimmunity. Nature. 2005;435:590–597. doi: 10.1038/nature03724. [DOI] [PubMed] [Google Scholar]
- 27.Julien S, Soulas P, Garaud JC, Martin T, Pasquali JL. B cell positive selection by soluble self-antigen. J Immunol. 2002;169:4198–4204. doi: 10.4049/jimmunol.169.8.4198. [DOI] [PubMed] [Google Scholar]
- 28.Soulas P, Woods A, Jaulhac B, Knapp AM, Pasquali JL, Martin T, Korganow AS. Autoantigen, innate immunity, and T cells cooperate to break B cell tolerance during bacterial infection. J Clin Invest. 2005;115:2257–2267. doi: 10.1172/JCI24646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mo JA, Holmdahl R. The B cell response to autologous type II collagen: biased V gene repertoire with V gene sharing and epitope shift. J Immunol. 1996;157:2440–2448. [PubMed] [Google Scholar]
- 30.Mukherjee J, Nussbaum G, Scharff MD, Casadevall A. Protective and nonprotective monoclonal antibodies to Cryptococcus neoformans originating from one B cell. J Exp Med. 1995;181:405–409. doi: 10.1084/jem.181.1.405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shlomchik MJ, Craft JE, Mamula MJ. From T to B and back again: positive feedback in systemic autoimmune disease. Nat Rev Immunol. 2001;1:147–153. doi: 10.1038/35100573. [DOI] [PubMed] [Google Scholar]
- 32.Panigrahi AK, Goodman NG, Eisenberg RA, Rickels MR, Naji A, Luning Prak ET. RS rearrangement frequency as a marker of receptor editing in lupus and type 1 diabetes. J Exp Med. 2008;205:2985–2994. doi: 10.1084/jem.20082053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Veldman CM, Gebhard KL, Uter W, Wassmuth R, Grotzinger J, Schultz E, Hertl M. T cell recognition of desmoglein 3 peptides in patients with pemphigus vulgaris and healthy individuals. J Immunol. 2004;172:3883–3892. doi: 10.4049/jimmunol.172.6.3883. [DOI] [PubMed] [Google Scholar]
- 34.Hertl M, Eming R, Veldman C. T cell control in autoimmune bullous skin disorders. J Clin Invest. 2006;116:1159–1166. doi: 10.1172/JCI28547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mouquet H, Farci S, Joly P, Maillere B, Leblond J, Drouot L, Leprince J, Tonon MC, Loiseau P, Charron D, Tron F, Gilbert D. A truncated alternative spliced isoform of human desmoglein 1 contains a specific T cell epitope binding to the pemphigus foliaceus-associated HLA class II DRbeta1*0102 molecule. J Immunol. 2006;177:6517–6526. doi: 10.4049/jimmunol.177.9.6517. [DOI] [PubMed] [Google Scholar]