

Abstract
The application of proteomic techniques to neuroscientific research provides an opportunity for a greater understanding of nervous system structure and function. As increasing amounts of neuroproteomic data become available, it is necessary to formulate methods to integrate these data in a meaningful way to obtain a more comprehensive picture of neuronal subcompartments. Furthermore, computational methods can be used to make biologically relevant predictions from large proteomic datasets. Here, we applied an integrated proteomics and systems biology approach to characterize the presynaptic nerve terminal. For this, we carried out proteomic analyses of presynaptically enriched fractions, and generated a presynaptic literature-based protein-protein interaction (PPI) network. We combined these with other proteomic analyses to generate a core list of 117 presynaptic proteins, and used graph theory-inspired algorithms to predict 92 additional components and a presynaptic complex containing 17 proteins. Some of these predictions were validated experimentally, indicating that the computational analyses can identify novel proteins and complexes in a subcellular compartment. We conclude that the combination of techniques (proteomics, data integration, and computational analyses) used in this study are useful in obtaining a comprehensive understanding of functional components, especially low-abundance entities and/or interactions in the presynaptic nerve terminal.
Keywords: computational biology, graph theory, mass spectrometry, presynaptic nerve terminal, signaling networks
Introduction
In recent years, extensive efforts have been made, using subcellular fractionation techniques and large-scale mass spectrometric (MS) analyses, to identify proteins associated with various synaptic preparations [1], including synaptosomes [2-5], synaptic membranes [6-8], the postsynaptic density (PSD) [9-17], synaptic vesicles [18-22], and the presynapse [23, 24]. These neuroproteomic studies have revealed a high degree of complexity in synaptic composition: it is estimated that synapses may contain over 1000 different types of proteins [25]. However, despite a tremendous increase in the rate of discovery of synaptic components, our understanding of synaptic organization has lagged behind, largely because of the lack of understanding of how synaptic proteins interact to form complexes and signaling networks. The question is, once we generate large lists of proteins using proteomics, what else can be learned?
In order to synthesize the data from proteomic studies in a meaningful way, a range of computational techniques can be employed [26]. Information from the biochemical literature, particularly protein-protein interaction (PPI) data, can be applied to increase our understanding of functional pathways within a cellular compartment of interest. Biologically relevant predictions of novel proteins and interactions can be made by applying graph theory-based algorithms to proteomic datasets. Such systems-level approaches have only recently been applied to neuroproteomic studies. A network representation of the postsynaptic NMDA receptor complex has been generated using a combination of proteomics, to identify components of the complex, and literature mining, to identify interactions among these components [27]. A model of signaling networks in hippocampal CA1 neurons has also been generated by manual curation of interaction data from the experimental literature [28]. Analysis of the networks generated in these studies has shown that molecular networks with simple design principles are likely to underlie synaptic signaling.
In this study, we describe an interdisciplinary approach that combines proteomics with graph theory analysis to characterize the protein composition of the presynaptic nerve terminal. First, we carried out proteomic experiments using a fractionation method that allows for the enrichment of rodent presynaptic proteins (and separation from the PSD [29]), and tandem mass spectrometry (LC-MS/MS) for the identification of these proteins. Second, using a computational approach, we merged available presynaptic proteomic lists, extracted presynaptic components and interactions from the literature, and used graph analysis algorithms to evaluate and enrich the knowledge about the presynaptic proteome. These data were used to make predictions of novel presynaptic components and interactions, several of which were validated experimentally. The approach used here is generally applicable to analyzing large datasets from high-throughput proteomic studies.
Materials and Methods
Subcellular Fractionation
Isolation of a presynaptic (PRE) fraction was performed essentially as described in Phillips et al. [29]. Male wild-type C57B6 mice (20-25 g) or Sprague-Dawley rats (200-250 g) were sacrificed by decapitation and the brains rapidly removed. The hippocampi from 4 (for Western blotting) or 10 (for MS/MS analysis) mice, and the striata from 3 (for Western blotting) or 5 (for MS/MS analysis) rats were combined and homogenized in 3 ml of 0.32 M sucrose, 0.1 mM CaCl2, with 30 μl each of protease inhibitor cocktail and phosphatase inhibitor cocktail (Sigma, St. Louis, MO) at 4 °C. All of the following fractionation steps were carried out at 4 °C unless otherwise specified. The homogenate was brought to a final concentration of 1.25 M sucrose by the addition of 2 M sucrose (12 ml) and 0.1 mM CaCl2 (5 ml). The homogenate was then placed in a 40 ml ultracentrifuge tube and overlaid with 10 ml 1 M sucrose, 0.1 mM CaCl2. The gradients were centrifuged at 100,000g for 3 hrs. The synaptosomal fraction (4-5 ml) was collected at the 1.25 M/1 M interface. To obtain synaptic membranes, the synaptosomal fraction was brought to a volume of 35 ml with 20 mM Tris-Cl pH 6, 0.1 mM CaCl2, containing 1% Triton X-100 (TX-100) and 350 μl each of protease and phosphatase inhibitor cocktails, mixed for 20 min, and centrifuged at 40,000g for 20 min. The pellet containing the isolated synaptic membranes was collected. To separate a presynaptic fraction from the PSD, the pellet was resuspended in 20 ml of 20 mM Tris-Cl pH 8, 1% TX-100, 0.1 mM CaCl2. The mixture was again mixed for 20 min, and centrifuged at 40,000g for 20 min. The insoluble pellet containing the PSD fraction was collected and stored at -80 °C until use. The supernatant was removed and concentrated to 1 ml using an Amicon Ultra-15 filter (5,000 MW cut-off, Millipore, Bedford, MA). The concentrate was precipitated with 9 ml of acetone by incubation at −20 °C for 12 hrs, and centrifugation at 15,000g for 30 min. The resulting pellet, containing the PRE fraction, was stored at −80 °C until use.
Western blotting
Total protein concentrations of the different hippocampal fractions (homogenate, synaptosomes, synaptic junctions, PSD, and PRE) were determined using the BCA protein assay (Pierce, Rockford, IL). PRE and PSD pellets were resuspended in 1% or 0.1% SDS. Equal amounts of protein from each fraction were resolved on 7.5% SDS-PAGE gels. Gels were transferred to nitrocellulose membranes (Scheicher & Schuell, Bioscence, Keene, NH) by electroblotting. Membranes were blocked with Odyssey blocking buffer (LI-COR Biosciences, Lincoln, NE) and then incubated with selective primary antibodies: Clathrin heavy chain (1:6000, BD Biosciences, San Jose, CA), Syntaxin 1 (1:2000, Chemicon, Temecula, CA), SNAP25 (1:20,000, Sigma, St. Louis, MO), PSD95 (1:50,000, Upstate, Lake Placid, NY), GluR1 (1:1000, Chemicon, Temecula, CA), CAMKIIα (1:10,000, Upstate, Lake Placid, NY), IQGAP (1:1000, BD Transduction, San Jose, CA), GEF-H1 (1:500, Cell Signaling, Danvers, MA), PCTAIRE 1 (1:250, Cell Signaling, Danvers, MA), or RIN1 (1:250, BD Transduction, San Jose CA). Protein bands were detected using IR800-labeled goat anti-mouse IgG or IR700-labeled goat anti-rabbit IgG secondary antibodies (1:20,000, LI-COR Biosciences, Lincoln, NE) and the Odyssey infrared imaging system.
Proteomics
In-gel digestion
The hippocampal PRE and PSD fractions were resuspended in 200 μl of 1% SDS, and protein concentrations were determined using the BCA protein assay (Pierce, Rockford, IL). 100 μg of protein from each fraction was separated by 7.5% SDS-PAGE. Following electrophoresis, the proteins were visualized by Coomassie blue staining, using 1% PhastGel Blue (Amersham Biosciences, Buckinghamshire, UK). The entire protein lanes were sequentially cut into 26 gel slices and destained with 45% acetonitrile in 100 mM ammonium bicarbonate. The resulting gel slices were incubated with 10 mM tris(2-carboxyethyl)phosphine hydrochloride, alkylated by the addition of 50 mM iodoacetamide, and then digested in situ with trypsin (100 ng per band in 50 mM ammonium bicarbonate). The tryptic peptides were extracted using POROS 20 R2 beads (Applied Biosystems, Foster City, CA) in 0.2% trifluoroacetic acid containing 5% formic acid. The extracted peptides were concentrated by loading the POROS beads onto C18 Zip-tips (Millipore, Bedford, MA), and eluted with 30% and 75% of acetonitrile containing 0.1% trifluoroacetic acid. The eluates were dried under vacuum using a Speed Vac concentrator.
In-solution digestion
The hippocampal PRE and PSD fractions were resuspended in 50 mM Tris-Cl, 0.1% SDS, incubated with 40 mM tris(2-carboxyethyl)phosphine hydrochloride and then digested with trypsin (100 ng in distilled water). The tryptic peptides were loaded onto a cation-exchange cartridge containing POROS 50 HS beads (Applied Biosystems, Foster City, CA) and eluted with 500 mM potassium chloride in 5 mM phosphate buffer and 25% acetonitrile. In-solution digestion was also used to process the striatal PRE fraction. In this case, the tryptic peptides were eluted from the cation-exchange cartridge using a step gradient of increasing potassium chloride concentration (25, 50, 75, 100, 150, 200, 250, 350 mM). The eluates were dried under vacuum using a Speed Vac concentrator.
Mass spectrometry
The resulting peptides were dissolved in 2-25 μl of HPLC sample solvents containing water:methanol:acetic acid:trifluoroacetic acid (70:30:0.5:0.01, v/v/v/v). Capillary-HPLC-MS/MS analysis was conducted on an LCQ ion trap mass spectrometer (Thermo Finnigan, San Jose, CA) coupled with an online MicroPro-HPLC system (Eldex Laboratories, Napa, CA). Two μl of tryptic peptide solution was injected into a Magic C18 column (0.2 × 50 mm for in-gel digests, or 0.2 × 150 mm for in-solution digests, 5 μm, 200 Å, Michrom BioResources, Auburn, CA) which had been equilibrated with 70% solvent A (0.5% acetic acid and 0.01% trifluoroacetic acid in water:methanol (95:5, v/v)) and 30% solvent B (0.5% acetic acid and 0.01% trifluoroacetic acid in methanol:water (95:5,v/v)). Peptides were separated and eluted from the HPLC column with a linear gradient of 30-95% solvent B in 15 min or 30-70% solvent B in 100 min, at a flow rate of 2.0 μl/min for in-gel digests and in-solution digests, respectively. The eluted peptides were sprayed directly into the LCQ mass spectrometer (2.8 kV). The LCQ mass spectrometer was operated in a data-dependent mode for measuring the molecular masses of peptides (parent peptides) and collecting MS/MS peptide fragmentation spectra.
Database search and protein identification
The measured molecular masses of parent peptides and their MS/MS data were used to search the National Center for Biotechnology Information (NCBI) Reference Sequence (RefSeq) database using the program Sonar (Genomic Solutions, Ann Arbor, MI). The same data were searched, using identical parameters, against a random database of NCBI non-redundant mouse sequences generated by the program decoy.pl from Matrix Science, in order to determine the false positive discovery rate. The false positive rate (FPR) = RP/(RP+NP) was calculated, where RP and NP are the number of confirmed matches derived from the randomized and normal database, respectively. By assigning both protein and peptide identification thresholds as < 1, the FPR equals 0.01. By assigning a protein identification threshold of protein score < 0.1 with peptide score < 0.1, the FPR equals 0. Therefore, protein identifications were made based on Sonar expectation values (E-values) of < 0.1 either at the protein or peptide level. BLAST searches were performed for hypothetical and unknown proteins using the NCBI Protein: Protein BLAST web server.
Literature-based presynaptic PPI network
From biochemical research publications reporting direct (binary) interactions between presynaptic proteins and metabolites, we manually extracted and constructed a network of presynaptic PPIs. We abstracted interactions to a mixed graph (directed/undirected), where proteins are represented as nodes and their direct interactions as links. In order to generate a high quality dataset with minimal inherent bias, binary interactions were included only from primary publications describing presynaptic mammalian interactions with no high-throughput data (e.g. yeast two-hybrid or other proteomic methods) included, unless confirmed by other techniques. In order to effectively incorporate data from multiple sources, we used UniProt (http://www.expasy.uniprot.org/) accession numbers (human and mouse) and Entrez Gene (http://www.ncbi.nlm.nih.gov/) gene names (human), the standard for protein identification. In the few cases where protein identifiers could not be mapped to UniProt, their original identifiers were retained. The network was analyzed and visualized using SNAVI [30]. This network contains 127 proteins and small molecules (nodes) and 229 interactions (links), from 145 publications. Self-interactions were not included, whereas interactions involving calcium were included in the network, as calcium plays a central role in neurotransmitter release from the presynaptic nerve terminal. In all, four types of interactions were incorporated – binding, phosphorylation, dephosphorylation, and channel opening. A web-based interface that provides access to this network is provided at http://amp.pharm.mssm.edu/presynaptome.
Generation of a core presynaptic dataset
In order to generate a core presynaptic list, we compiled lists of proteins from our proteomic studies of PRE fractions, our literature-based presynaptic network (converted to list of components), and two published proteomic studies of presynaptic fractions. The first study [23] used the same fractionation protocol applied in our proteomic studies to separate presynaptic and PSD fractions from the rat forebrain, and reported a list of proteins associated with each fraction using multi-dimensional protein identification technology (MudPIT). The second study [19] isolated fractions containing free synaptic vesicles or synaptic vesicles associated with presynaptic plasma membrane from the rat brain using subcellular fractionation, immunoaffinity purification, and sucrose gradient centrifugation, and identified proteins by two-dimensional gel elelectrophoresis followed by matrix-assisted laser desorption-ionization time-of-flight (MALDI TOF) MS. Only the proteins identified in the fraction containing synaptic vesicles associated with presynaptic plasma membrane were considered in our study, since this fraction contained components of the synaptic vesicle trafficking machinery that regulate presynaptic nerve terminal function. In order to compile these lists into a “merged list” and eliminate redundancy, the proteins from all proteomic lists were assigned human accession numbers and gene names using Uniprot and Entrez Gene.
Prediction of Presynaptic Proteins and Complexes
A “background” literature-based protein-protein interaction network was created by merging interactions from BioGrid [31], HPRD [32], PPID [33], and a CA1 neuronal regulatory network [28]. We excluded interactions that originate from research articles reporting more than five interactions to reduce the chance for false positives. This network has 6,442 proteins and 17,879 interactions extracted from 12,462 publications.
A binomial proportions test was used to evaluate the significance of interactions between proteins from the background dataset with proteins from the “merged list” of 306 proteins identified by proteomics. The binomial proportions test provides a good approximation to the Fisher Exact Test, which is used to evaluate the likelihood of a discrete event as compared to what would be expected by chance. For this analysis, the z-score for each protein from the background dataset was computed using the following equation:
| (1) |
Where:
N1 = # of proteins in the merged list (= 306)
N2 = # of proteins in background dataset (= 6442)
p1 = # of direct interactions with proteins in the merged list
p2 = # of direct interactions with proteins in the background network
| (2) |
A higher z-score for a protein would indicate that the number of its interactions with proteins from our experimentally determined seed list is significantly enriched compared with the number of its interactions with other protein partners [34].
Of the 6,442 proteins from the background list, 646 interacted with at least two proteins from the merged list, and 92 of these showed a significant preference (z-score > 3) to interact with proteins from the merged list. A z-score of 3 was chosen since this corresponds to a p-value of ∼ 0.01, which is a standard cutoff for statistical significance. Also, this z-score provided a reasonable number of proteins that could be further analyzed. The 92 proteins with z-score > 3 were evaluated to determine whether they have previously shown to be presynaptic by searching PubMed, SynDB [35] (a database of synaptic proteins) and GO [36].
To predict a presynaptic complex, proteins from the merged proteomics list (306 proteins) were analyzed for the presence of overlapping direct protein interactions (shared neighbors), using interactions from the background dataset (6,442 proteins). 21 pairs of proteins from the merged list were found to have at least four shared direct interacting partners. Other thresholds were tested; four was chosen to produce an acceptable balance between comprehensiveness and stringency. These proteins do not directly interact with each other and do not share sequence homology. The percent of shared neighbors for all pairs of proteins was then calculated as follows and used for ranking the probability that a pair of proteins may exist in a complex:
| (3) |
Where: SN = shared neighbors, ON1 = other neighbors (not shared) of protein 1, and ON2 = other neighbors of protein 2.
The protein interactions from this publication have been submitted to the IMEx (http://imex.sf.net) consortium through IntAct [37] and have been assigned the identifier IM-11648.
Immunocytochemistry
Dissociated neuronal cultures were prepared from the cortices of embryonic day 18 Sprague-Dawley rats, as described [38]. Neurons (16 days in vitro) were fixed with 2% paraformaldeyhyde (PFA) / 2% sucrose for 15 min, permeabilized with 0.25 % TX-100 for 5 min, and incubated for 1 h at room temperature in blocking solution consisting of 2% BSA. Cells were then incubated overnight at 4 °C with antibodies to: RIN1 (1:100, BD Transduction, San Jose, CA), PCTAIRE 1 (1:50, Cell Signaling, Danvers, MA), SV2 (1:20, DSHB, Iowa City, IA) or synaptophysin (1:500, Sigma, St. Louis, MO). The antibodies for GEF-H1 and IQGAP1 (see Western blotting, above) were not of suitable quality for immunohistochemical analysis; the signal for each antibody was too low to be detected, even at very high concentrations (e.g. 1:20). Cells were incubated with Alexa-594 anti-rabbit and Alexa-488 anti-mouse secondary antibodies (1:1000, Molecular Probes, Eugene, OR) for 1 h at room temperature. Coverslips were mounted using Mowiol (Sigma, St. Louis, MO), and visualized using a Leica TCS SP1 confocal microscope equipped with four external lasers (350, 488, 568, and 633 nm, Leica Microsystem). Images were acquired with a ×100/1.32 PL APO objective lens, and analyzed in sequential scanning mode.
Co-Immunoprecipitation
Mouse hippocampal synaptosomal fractions were prepared in the same way as described in the “Subcellular Fractionation” section above. The synaptosomal fraction contains presynaptic membranes, postsynaptic membranes and PSD, and subsynaptic web material [29]. Synaptosomal fractions (200 μg protein) were resuspended in lysis buffer (100 mM NaCl, 5 mM EDTA, 10 mM NaHPO4, pH 7.2) containing 1% TX-100 and protease and phosphatase inhibitor cocktails, and incubated at 4 °C for 20 min. The lysates were subjected to immunoprecipitation at 4 °C overnight with anti-synapsin 1 antibody (4 μg, Stressgen, Victoria, BC) and pre-washed protein A/G agarose beads (40 μl, Pierce, Rockford, IL). As a control, the lysates were incubated with protein A/G agarose beads alone. The immunoprecipitates were washed twice with lysis buffer containing 0.25% TX-100, and once with PBS containing 5 mM EDTA. Bound proteins were eluted with Laemmli loading buffer at 100 °C for 20 min, resolved by 7.5% SDS-PAGE, and immunoblotted with antibodies to dynamin (1:5000, BD Biosciences, San Jose, CA), CAMKIIα (1:10,000, Upstate, Lake Placid, NY), MAP2 (1:2000, Chemicon, Temecula, CA), and synapsin I (1:20,000, Pierce, Rockford, IL). Protein bands were detected using IR800-labeled goat anti-mouse IgG or IR700-labeled goat anti-rabbit IgG secondary antibodies (1:20,000, LI-COR Biosciences, Lincoln, NE) and the Odyssey infrared imaging system.
Results and Discussion
With the advent of high-throughput proteomics, it is now possible to systematically catalogue the components within a subcellular compartment. In this study, we describe an approach to characterize the composition of the presynaptic nerve terminal using subcellular proteomics and systems biology. First, we carried out proteomic studies of proteins enriched in the presynapse. For this, we separated presynaptic (PRE) and postsynaptic (PSD) fractions from rodent hippocampus and striatum by an anionic extraction method, as described in Materials and Methods. To verify the extent of the purification, the various fractions were subjected to Western Blotting, using antibodies to known presynaptic proteins: Syntaxin I and SNAP25, and to known PSD proteins: GluR1 and PSD95. In addition, the fractions were probed with antibodies to clathrin heavy chain, an endocytic protein that was previously shown to be enriched in presynaptic fractions [23], and CAMKII, a major component of the PSD [39] that also associates with presynaptic vesicles [40]. The PRE fraction is enriched in presynaptic proteins and excludes proteins enriched in the PSD fraction (Figure 1).
Figure 1.
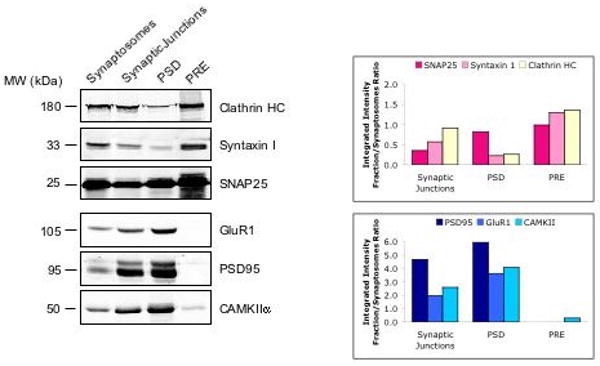
Biochemical validation of the separation protocol used to identify proteins from the mouse hippocampal presynaptic (PRE) fraction. To demonstrate the purity of the fractions, equal amounts (10 to 30 μg) of protein from hippocampal synaptosomes, synaptic junctions, PSD, and PRE fractions were separated by SDS-PAGE and probed with antibodies to presynaptic markers (clathrin heavy chain (HC), syntaxin I, and SNAP25) and postsynaptic markers (GluR1, PSD95, and CAMKIIα). Protein bands were quantified using the Odyssey infrared imaging system. Bar graphs (right) show the integrated intensity of protein bands in each fraction relative to the synaptosomal fraction.
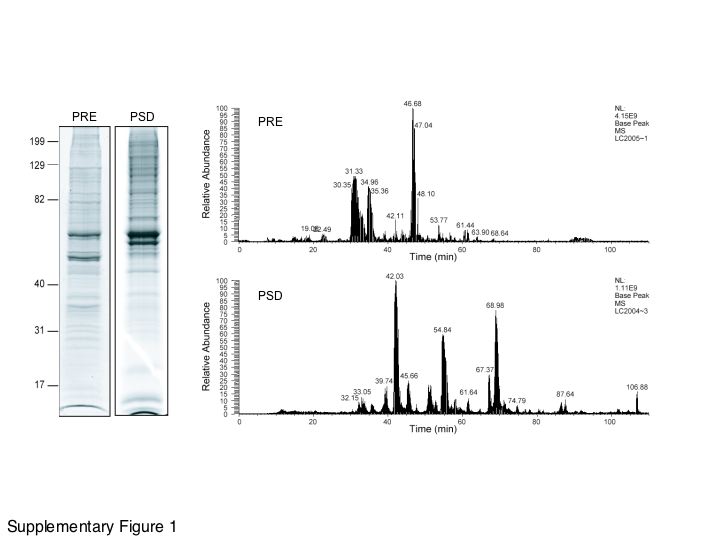
We then identified proteins in the PRE fractions by LC-MS/MS following either in-gel or in-solution digestion (Supplementary Figure 1). Proteins were identified based on highly stringent statistical analysis (in both the quality of the MS/MS peptide fragment ion spectra and the significance of amino acid sequence matches) using the program Sonar, which has recently been reported to be one of the most specific MS/MS database search algorithms [41]. In the hippocampal PRE fraction, we identified a total of 138 proteins (Supplementary Table 1). The profiling of the hippocampal PSD proteins has been previously reported [42]. In the striatal PRE fraction, we identified 121 proteins (Supplementary Table 2). The relatively low number of proteins identified in each of our PRE fractions suggests that these lists are far from comprehensive. The presynaptic proteome likely includes both abundant proteins (e.g. those that are found across different types of synapses and at high levels) and rare proteins (e.g. those that are synapse or brain region-specific). Although subcellular fractionation is the method of choice to reduce the complexity of samples for MS analysis, there remains a large bias in MS data against low-abundant proteins in a sample. In order to address this, we used a graph theory-inspired computational approach to evaluate and enrich the knowledge about proteins identified in presynaptic fractions by us and by others.
In a first step to further analyze the PRE lists produced by proteomics, we manually extracted PPI data from the biochemical and physiological literature to generate an in silico network that represents only presynaptic interactions, as described in Materials and Methods (Supplementary Figure 2, Supplementary Table 3). This network, made of 229 direct (binary) interactions between 127 presynaptic proteins, was generated without considering the results from the proteomics experiments, and is provided as a web-based resource at http://amp.pharm.mssm.edu/presynaptome. Since other studies have reported lists of presynaptic proteins identified by proteomic approaches, we also extracted the data from two recently published proteomic studies of presynaptic fractions [19, 23]. Compilation of these lists with the two lists we developed experimentally, and the list we created from the literature-based network, resulted in a “merged list” containing 393 entries (306 proteins from proteomics, and 87 entries exclusively from the literature) (Supplementary Table 4). A similar strategy focusing solely on proteomic data has been applied to characterize the postsynaptic proteome [25].
In order to readily merge and analyze data from various sources, we extended all protein and interaction data experimentally verified in other mammalian model organisms to orthologous proteins in human (Supplementary Tables 1, 2, and 4). It is a common assumption that PPIs can be inferred through homology transfer from one model organism to another, since functionally linked proteins are likely to evolve together, and therefore should have homologs in evolutionarily related organisms. Although this is not always the case, particularly when comparing prokaryotes and simple eukaryotes with higher eukaryotes [43], PPIs have been shown to be well conserved between protein pairs with at least 80% sequence identity [44]. A recent study examining the evolutionary conservation of proteins, interactions, and complexes showed that mouse and rat show the greatest conservation of human proteins over all, followed by fly, worm, and yeast [45]. The same study found that nearly 70% of human interactions are conserved in mice. Based on these data, it is believed that PPIs from higher eukaryotes such as mouse and rat are highly conserved when compared to human.
Comparison of the lists of proteins derived from the proteomic studies revealed 13-22% overlap (Supplementary Tables 3, 5). Although this is a significant overlap when compared with the expected overlap for randomly generated lists of genes, we would expect the overlap among these lists to be higher. A low degree of overlap could be due to brain regional variation, different strategies of sample preparation, protein separation, and/or run-to-run differences in MS analysis that are routinely observed. In the merged list, 45 proteins (15%) were detected experimentally three or more times, 56 proteins (18%) were detected twice, and the rest (67%) were detected once (Figure 2A, Supplementary Table 6). We designated proteins that were identified two or more times as the “core list” (containing 101 proteins). The intent of the core list is to represent proteins that are likely to be associated with most mammalian presynaptic terminals. By filtering out the proteins that were only identified once experimentally, we limit the number of protein contaminants, as well as proteins that may be specific to a single brain region, species, or even methods of sample preparation and/or protein identification. For example, with the subcellular fractionation technique used in this study, samples may contain contaminant postsynaptic proteins that remain adherent to the presynaptic fraction; however, the identification of such contaminants is less likely with repeated experiments. Thus, while the original proteomic lists and the merged list include valuable data that were used for further computational analyses, the core list represents a highly stringent subgroup of mammalian presynaptic proteins. The contribution of each list to the core list and to the merged list is illustrated in Figure 2B. For example, our hippocampal PRE list contributed 79 proteins to the core list, indicating that these proteins have been validated as being present in the presynapse by one or more of the other lists.
Figure 2.
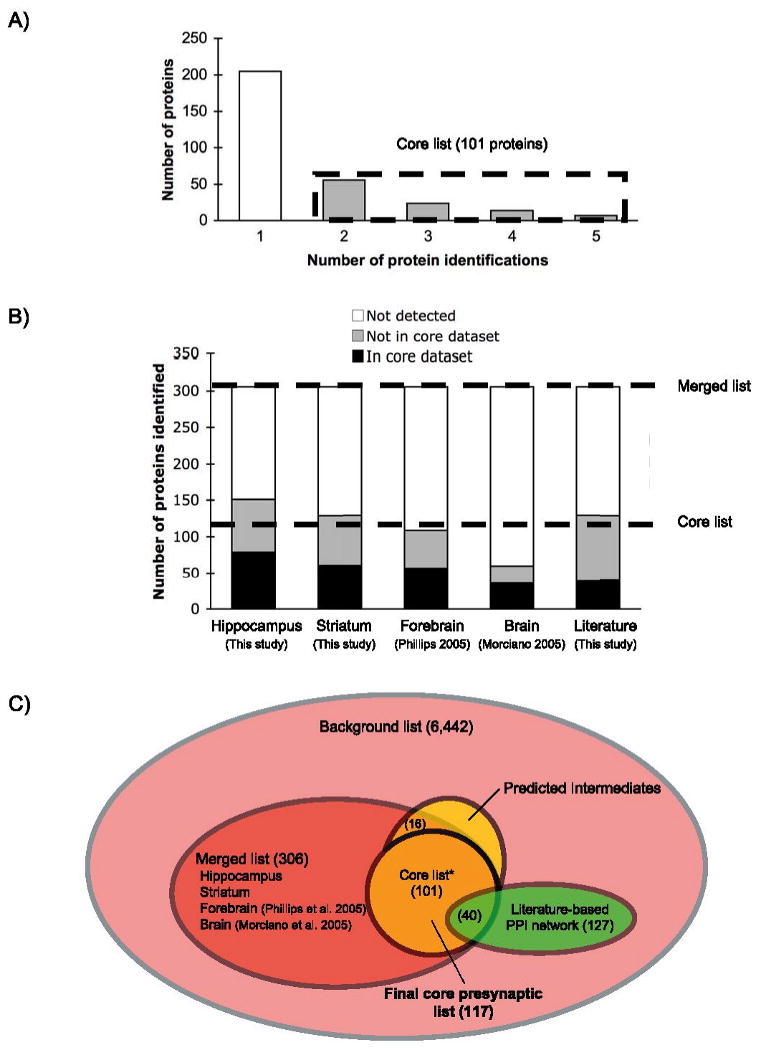
A) To characterize the proteins identified by proteomics, we compiled lists of proteins from our proteomic studies with other studies (Morciano et al., 2005; Phillips et al., 2005) and our literature-based network. The majority (67%) of proteins in the merged list (containing 306 proteins) were identified only once in proteomic studies. The subset of proteins detected in two or more independent lists was taken as the “core list” (101 proteins). B) Contributions of each individual list to the core list and to the merged list. “Not detected” indicates proteins that were not identified in the list. “Not in core list” indicates proteins that were identified in the list but did not contribute to the core list. “In core list” indicates proteins that were identified in the list and contributed to the core list. C) Schematic illustrating the data compilation process used to generate the final core presynaptic list of 117 proteins. Protein lists from our proteomic studies, two other published studies, and our literature-based presynaptic network were combined to form a merged list containing 306 proteins. We placed proteins that were identified two or more times in a core list containing 101 proteins. To enrich this list with additional proteins, we used network analysis to identify 16 intermediates from the merged list that interact directly with proteins from the core list. These proteins were added to the core list to generate the final core presynaptic list of 117 proteins (see Table 1).
Using Gene Ontology, we mapped the “biological process” to proteins from the hippocampal and striatal PRE fractions as well as to proteins in the core list (Supplementary Table 7). The core list is enriched for proteins involved in presynaptic functions, such as neurotransmission. Proteins belonging to transport- or secretion-related biological processes (intracellular transport (18.6%), vesicle-mediated transport (15.3%), protein transport (14.4%), secretion (9.3%), and secretion pathway (9.3%)) are more highly represented in the core dataset. On the other hand, proteins belonging to several metabolic- or catabolic-related processes (cofactor metabolism, macromolecular catabolism, negative regulation of metabolism, carbohydrate metabolism, organic acid metabolism, and electron transport) are under-represented in the core list. Thus, by integrating lists from different sources, we were able to enrich for proteins with established presynaptic functions.
To further analyze the merged list of PRE proteins, we sought to identify literature-based PPIs among the proteins in the merged list. For this we consolidated and filtered several literature-based mammalian PPI networks from BioGrid [31], HPRD [32], PPID [33], and a neuronal signaling network we developed for a prior study [28] (see Materials and Methods for details). We “connected” proteins from the core list by linking pairs of proteins through shared neighbors, using interactions from the consolidated and filtered literature-based mammalian PPI network (“background network”). Between the 101 proteins in the core list (“Top 101”), we found 13 direct interactions, 222 interactions using 1st-level shared neighbors (path length of one extra node and two links), and 1,772 interactions using 2nd –level shared neighbors (path length of two extra nodes and four links). The same analysis was performed using 0, 1, 2, or 3-level shared neighbors to connect the 45 proteins identified 3 or more times (“Top 45”) in the merged list (Supplementary Table 8). A total of 226 intermediate proteins were found to “connect” core list proteins. Among them, 16 consisted of proteins that had been detected once in proteomic studies (Supplementary Table 9). Since these proteins have been shown to interact with proteins from the core list, they are likely to be bona-fide components of the presynaptic nerve terminal proteome, and were therefore upgraded to the core list. This resulted in a “final” core presynaptic list made of 117 proteins (Table 1, Figure 2C).
Table 1.
Core presynaptic list containing 117 proteins.
| Accession # (UniProt) | Protein Name | Gene Name (Entrez Gene) |
|---|---|---|
| Trafficking (32) | ||
| Q10567 | AP-1 complex subunit beta-1 | AP1B1 |
| O95782 | AP-2 complex subunit alpha-1 | AP2A1 |
| O94973 | AP-2 complex subunit alpha-2 | AP2A2 |
| P63010 | AP-2 complex subunit beta-1 | AP2B1 |
| Q96CW1 | AP-2 complex subunit mu-1 | AP2M1 |
| Q93050 | V-ATPase subunit a1 | ATP6V0A1 |
| P61421 | V-ATPase subunit d | ATP6V0D1 |
| P38606 | V-ATPase subunit A1 | ATP6V1A |
| P21281 | V-ATPase subunit B2 | ATP6V1B2 |
| P21283 | V-ATPase subunit C | ATP6V1C1 |
| P36543 | V-ATPase subunit E | ATP6V1E1 |
| P09496 | Clathrin light chain | CLTA |
| Q00610 | Clathrin heavy chain | CLTC |
| Q05193 | Dynamin 1 | DNM1 |
| Q14204 | Dynein heavy chain, cytosolic | DYNC1H1 |
| P54920 | Alpha-soluble NSF attachment protein | NAPA |
| P46459 | N-ethylmaleimide sensitive fusion protein | NSF |
| Q9BY11 | Protein kinase C and casein kinase substrate in neurons protein 1 | PACSIN1 |
| P20336 | Ras-related protein Rab-3A | RAB3A |
| Q86UR5 | Rab3-interacting molecule 1 | RIMS1 |
| Q99962 | SH3-containing GRB2-like protein 2 | SH3GL2 |
| Q9Y6R1 | Synaptosomal-associated protein 25 | SNAP25 |
| Q16623 | Syntaxin 1A | STX1A |
| P61764 | Syntaxin-binding protein 1 | STXBP1 |
| Q15833 | Syntaxin-binding protein 2 | STXBP2 |
| P17600 | Synapsin 1 | SYN1 |
| Q92777 | Synapsin 2 | SYN2 |
| O43426 | Synaptojanin 1 | SYNJ1 |
| P21579 | Synaptotagmin 1 | SYT1 |
| Q8N9I0 | Synaptotagmin 2 | SYT2 |
| Q96QK1 | Vacuolar protein sorting 35 | VPS35 |
| P63027 | Vesicle-associated membrane protein 2 | VAMP2 |
| Signaling (22) | ||
| Q2M2I8 | AP-2 associated kinase 1 | AAK1 |
| P62158 | Calmodulin | CALM1 |
| Q9UQM7 | CAMKII alpha | CAMK2A |
| P09543 | 2′,3′-cyclic-nucleotide 3′-phosphodiesterase | CNP |
| P68400 | Casein kinase II subunit alpha | CSNK2A1 |
| Q16555 | Dihydropyrimidinase-related protein 2 | DPYSL2 |
| P63096 | G protein G(i) alpha 1 | GNAI1 |
| P04899 | G protein G(i) alpha 2 | GNAI2 |
| P08754 | G protein G(i) alpha 3 | GNAI3 |
| P09471 | G protein G(o) alpha 1 | GNAO1 |
| P62873 | G protein beta 1 | GNB1 |
| P62879 | G protein beta 2 | GNB2 |
| P07948 | Tyrosine-protein kinase Lyn | LYN |
| P28482 | Mitogen-activated protein kinase 1 | MAPK1 |
| Q9NQ66 | Phospholipase C beta 1 | PLCB1 |
| Q08209 | Protein phosphatase 2B catalytic subunit alpha isoform | PPP3CA |
| P16298 | Protein phosphatase 2B catalytic subunit beta isoform | PPP3CB |
| P17252 | Protein kinase C alpha type | PRKCA |
| P31946 | 14-3-3 protein beta/alpha | YWHAB |
| P62258 | 14-3-3 protein epsilon | YWHAE |
| Q04917 | 14-3-3 protein eta | YWHAH |
| P63104 | 14-3-3 protein zeta/delta | YWHAZ |
| Receptors/Channels (3) | ||
| P13637 | Na/K ATPase alpha-3 chain | ATP1A3 |
| P21796 | Voltage-dependent anion-selective channel protein 1 | VDAC1 |
| P45880 | Voltage-dependent anion-selective channel protein 2 | VDAC2 |
| Scaffolding/Clustering (2) | ||
| Q01484 | Ankyrin 2 | ANK2 |
| Q9UQF2 | JNK-interacting protein 1 | MAPK8IP1 |
| Cell Adhesion (4) | ||
| Q12860 | Contactin 1 | CNTN1 |
| P32004 | Neural cell adhesion molecule L1 precursor | L1CAM |
| P13591 | Neural cell adhesion molecule 1 | NCAM1 |
| Q92823 | Neuronal cell adhesion molecule precursor | NRCAM |
| Cytoskeletal (23) | ||
| P68133 | Alpha-actin 1 | ACTA1 |
| P60709 | Beta-actin | ACTB |
| P63261 | Gamma-actin | ACTG1 |
| P12814 | Alpha-actinin 1 | ACTN1 |
| P35609 | Alpha-actinin 2 | ACTN2 |
| Q08043 | Alpha-actinin 3 | ACTN3 |
| P35611 | Adducin 1 | ADD1 |
| Q13561 | Dynactin subunit 2 | DCTN2 |
| P11171 | Protein 4.1 | EPB41 |
| P11137 | Microtubule-associated protein 2 | MAP2 |
| P07196 | Neurofilament triplet L protein | NEFL |
| Q9UH03 | Septin 3 | SEPT3 |
| Q99719 | Septin 5 | SEPT5 |
| Q16181 | Septin 7 | SEPT7 |
| Q13813 | Spectrin alpha chain, brain | SPTAN1 |
| Q01082 | Spectrin beta chain, brain 1 | SPTBN1 |
| O15020 | Spectrin beta chain, brain 2 | SPTBN2 |
| P09493 | Tropomyosin 1 alpha chain | TPM1 |
| P68366 | Tubulin alpha 1 chain | TUBA1 |
| Q9BQE3 | Tubulin alpha 6 chain | TUBA6 |
| P07437 | Tubulin beta 2 chain | TUBB2A |
| Q13509 | Tubulin beat 3 chain | TUBB3 |
| P04350 | Tubulin beta 4 chain | TUBB4 |
| Regulatory (7) | ||
| P13639 | Elongation factor 2 | EEF2 |
| P63241 | Eukaryotic translation initiation factor 5A-1 | EIF5A |
| P07900 | Heat shock protein 90 alpha | HSP90AA1 |
| P11142 | Heat shock cognate 71 kDa protein | HSPA8 |
| P49411 | Elongation factor Tu, mitochondrial | TUFM |
| P62937 | Peptidyl-prolyl cis-trans isomerase A | PPIA |
| P35232 | Prohibitin | PHB |
| Metabolic/Mitochondrial (16) | ||
| P04075 | Aldolase A | ALDOA |
| P25705 | ATP synthase subunit alpha | ATP5A1 |
| P06576 | ATP synthase subunit beta | ATP5B |
| P12277 | Creatine kinase B chain | CKB |
| P12532 | Creatine kinase ubiquitous | CKMT1A |
| P08574 | Cytochrome c-1 | CYC1 |
| P10515 | Pyruvate dehydrogenase complext E2 subunit | DLAT |
| P00367 | Glutamate dehydrogenase 1 | GLUD1 |
| P19367 | Hexokinase 1 | HK1 |
| P50213 | Isocitrate dehydrogenase [NAD] subunit alpha | IDH3A |
| O95299 | NADH dehydrogenase [ubiquinone] 1 alpha subcomplex subunit 10 | NDUFA10 |
| O75489 | NADH-ubiquinone oxidoreductase 30 kDa subunit | NDUFS3 |
| P17858 | 6-phosphofructokinase, liver type | PFKL |
| P08237 | 6-phosphofructokinase, muscle type | PFKM |
| P14618 | Pyruvate kinase isozymes M1/M2 | PKM2 |
| P22695 | Ubiquinol-cytochrome-c reductase complex core protein 2 | UQCRC2 |
| Other (8) | ||
| P69905 | Hemoglobin subunit alpha | HBA1 |
| P68871 | Hemoglobin subunit beta | HBB |
| P02686 | Myelin basic protein | MBP |
| O60313 | Dynamin-like 120 kDa protein | OPA1 |
| Q99584 | Protein S100-A13 | S100A13 |
| P04216 | Thy-1 membrane glycoprotein precursor | THY1 |
| P55072 | Valosin-containing protein | VCP |
| P62760 | Visinin-like protein 1 | VSNL1 |
This “final” list represents a core portion of presynaptic proteins but is not comprehensive, since low abundance proteins or proteins associated with a specific brain region are likely to be missing from this list. In the next step, in order to predict novel presynaptic proteins not detected experimentally, we used a binomial proportions test to identify proteins from the background network that preferentially interact with proteins identified experimentally to be presynaptic (Supplementary Table 10). Similar strategies using graph theory have been applied to enrich large-scale datasets in yeast by predicting PPIs [46-48]. The binomial proportions test was used to find proteins from the background network that specifically interact with presynaptic proteins, while pruning out proteins that interact with a large number of other non-presynaptic proteins, and as such could be interacting with some presynaptic proteins but not specifically. We found 92 proteins from the background network that show a significant preference (z-score > 3) to interact with proteins from the merged list, suggesting that these proteins could also exist at presynaptic nerve terminals.
The proteins with z-scores > 3 were compared to those with z-scores < -1, by categorizing them according to Gene Ontology's “biological process”, “cellular component”, and “molecular function” (Supplementary Figure 3). We find that the list of proteins with z-scores > 3 contains a higher proportion of membrane proteins (61%) and transport-related proteins (31%), while the list of proteins with z-scores < -1 contains a higher proportion of nuclear proteins (54%), transcriptional regulators (48%) and metabolism-related proteins (82%). This is consistent with the notion that the statistical test identifies proteins that have a higher chance of being presynaptic, by virtue of their subcellular localization, function, and ability to interact with previously identified presynaptic proteins. Indeed, of the 92 proteins with z-scores > 3, 42 had previously been identified as presynaptic proteins, as indicated by a database search in PubMed, SynDB [35], or GO [36] (Supplementary Table 11). This leaves 50 proteins that preferentially interact with the merged list, which have not been previously identified as presynaptic in any of these databases.
In order to verify the predictions that these proteins are indeed present in the presynapse, we selected five top-ranked proteins that have available antibodies (z-scores in brackets): PCTAIRE-1 (4.8), GEF-H1 (ARHGEF2, 4.2), RIN1 (3.9), NUMB (3.6), and IQGAP1 (3.5). These proteins were also selected because they are known to be involved in signal transduction processes [49-53] and would be of potential interest at the presynapse. We examined the selected proteins in fractions obtained during the purification process. Among them, four could be clearly detected in the PRE fraction by Western blotting (Figure 3A). The protein NUMB could not be detected in any of the fractions, possibly due to the poor quality of the antibody. For RIN1, the size of the protein in the PRE fraction appeared to be of a lower molecular weight as compared to those in the homogenate and synaptosomal fractions; this could indicate selective post-translational processing or the presence of an alternatively spliced variant. To further confirm the subcellular localization of the predicted proteins to be presynaptic, the localization of two of them, RIN1 and PCTAIRE-1, was examined in cultured primary cortical neurons by immunofluoresence. We find that these proteins co-localize with the presynaptic markers SV2 or synaptophysin (Figure 3B), confirming that these predicted proteins are present at the presynapse. PCTAIRE-1 is a kinase that has been shown to phosphorylate NSF in PC12 cells [49]; it is therefore conceivable that PCTAIRE-1 could play a role in regulating vesicle trafficking at presynaptic nerve terminals. RIN1 is a Ras effector that has previously been shown to modulate postsynaptic plasticity in aversive memory formation in the amygdala [50]; a presynaptic form of RIN could play a similar role in regulating signaling pathways at the presynaptic nerve terminal. Overall, these data are consistent with the idea that, using computational methods, it is possible to enrich proteomic data by including low abundance proteins such as signaling molecules.
Figure 3.
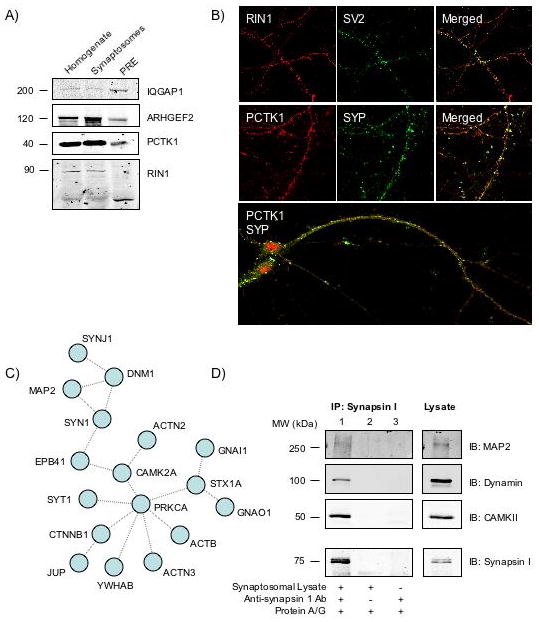
Prediction and experimental validation of novel presynaptic components and complexes. A) To validate the presence of some of the predicted proteins in the PRE fraction, 50 μg of protein from homogenate, synaptosomes, and PRE fractions were separated by SDS-PAGE and probed with selective antibodies to IQGAP, GEF-H1, RIN1, and PCTK1 by Western Blotting. B) To further confirm the presence of these proteins at the presynapse, immunofluorescence studies were performed using cultured primary cortical neurons. The cells were fixed with PFA, permeabilized, and probed for the localization of RIN1 and PCTK1, and their co-localization with the presynaptic markers SV2 or synaptophysin (SYP) using immunocytochemistry. C) To predict a novel presynaptic complex, proteins from the merged list were analyzed for their ability to interact indirectly (via shared neighbors). A schematic of the predicted complex (containing 17 proteins) is shown. Proteins are indicated by their gene names and links represent indirect interactions. D) Validation of the predicted presynaptic protein complex by co-immunoprecipitation. Left panels: Mouse hippocampal synaptosomal fractions were immunoprecipitated using anti-synapsin I antibody. MAP2, dynamin, and CAMKIIα were detected in the immunoprecipitate by Western Blotting (lane 1). Lane 2 represents immunoprecipitation without synapsin antibody, and lane 3 represents immunoprecipitation without synaptosomal lysate. Right panels: Western Blotting was carried out on the synaptosomal lysate as a control. Bottom panels: Blots were reprobed with synapsin I antibody as a control.
Finally, a “shared neighbor” analysis was applied to identify potential presynaptic complexes. Previous studies in yeast have shown that two proteins sharing a significantly large number of common interaction partners have close functional associations and are likely to exist in a complex [54, 55]. We hypothesized that non-homologous presynaptic proteins that share many interacting partners, but have not been shown to interact directly, may be present in a complex. We computed the percent of shared direct interacting partners (shared neighbors) between proteins from the merged list of 306 proteins identified by proteomics. We found that 21 pairs of non-homologous proteins have at least four shared neighbors, but have not been previously described to directly interact with each other (Supplementary Table 12). Using this information, we generated a hypothetical protein complex containing 17 proteins (Figure 3C). This complex included synapsin I and dynamin, which have been shown to co-precipitate with Src in PC12 cells [56], and MAP2, which has been shown to co-localize with synapsin I in the olfactory bulb glomerulus [57]. Using co-immunoprecipitation experiments, we biochemically validated some of the predictions made by this analysis. In mouse hippocampal synaptosomes, synapsin I co-immunoprecipitates with three other proteins from the predicted complex: dynamin, CAMKII, and MAP2 (Figure 3D), supporting the presence of the predicted interacting proteins in this complex. These results validate the presence of some parts of the computationally predicted complex at presynaptic nerve terminals, and show that this method of identifying proteins with shared interactors can be used to successfully identify novel interacting complexes.
Concluding Remarks
Subcellular fractionation is frequently used in neuroproteomic studies to concentrate and enrich proteins associated with a specific subcompartment of the nervous system [1]. This approach has the advantage of simplifying the complexity of whole tissue extracts, and maximizing the probability of detecting low abundance proteins by MS [58, 59]. In addition, the fractionation of cells into specialized subcompartments provides the possibility to link proteomic data with functional units [60]. However, protein lists generated by MS are by no means comprehensive; low abundance proteins, such as signaling molecules, and membrane-bound proteins, such as receptors and channels, remain notoriously difficult to identify in high-throughput studies [59], and contaminants remain in the sample preparations. An important next step is to develop tools to sieve the data obtained from high-throughput studies, and integrate it with data from the biochemical literature, in order to obtain a clearer and fuller picture of the compartment of interest. In this study, we used a combination of proteomics and computational biology approaches to characterize components in the presynaptic nerve terminal. The core list of presynaptic proteins serves as a useful resource for future functional studies that will attempt to characterize mammalian presynaptic cell signaling pathways and presynaptic organization under physiological and perturbed states.
An exciting feature of this study is that we have been able to make biologically relevant predictions that can be tested experimentally. In addition to generating a core presynaptic list of proteins, we used computational approaches to evaluate PPIs within this compartment and predict novel presynaptic components and complexes. Several computational predictions were validated using biochemical methods, indicating that the network analyses used can accurately predict proteins and interactions within a subcellular compartment. This process allowed us to increase the number of presynaptic components that were identified by MS, and helped in predicting a potential presynaptic complex comprising a number of important signaling molecules; future studies evaluating presynaptic function could target the modulation of such a complex rather than focus on individual proteins. Importantly, the computational analyses that we applied can readily be used to study other subcellular compartments [61].
Supplementary Material
Supplementary Figure 1. Two experimental approaches used in the identification of hippocampal PRE and PSD proteins. To achieve highly comprehensive protein identification, both in-gel digestion and in-solution digestion were used. (A) SDS-PAGE separation of PRE and PSD proteins prepared for in-gel trypsin digestion and LC-MS/MS protein identification (details are provided in the Materials and Methods). (B) Reverse-phase HPLC profile of tryptic peptides of PRE and PSD proteins from in-solution digestion. PRE and PSD proteins were digested by trypsin directly, and the tryptic peptides were analyzed by 1-dimensional reverse-phase HPLC-MS/MS with a two-hour elution gradient. The elution time for each of the major peaks is indicated.
{kind=link}
Supplementary Figure 2. Literature-based presynaptic protein-protein interaction (PPI) network, containing 127 proteins and 229 interactions. The network was generated by manually extracting mammalian presynaptic interactions from the literature. In this graphical representation of the network, proteins are represented as nodes (and designated by human gene names), and their direct interactions are represented as links. A web-based interface providing access to this network can be found at http://amp.pharm.mssm.edu/presynaptome.
{kind=link}
Supplementary Figure 3. Functional annotation of presynaptic datasets. Proteins were assigned to Gene Ontology biological processes, as described in Materials and Methods (see Table S7). The percentage of proteins in the hippocampal PRE fraction, striatal PRE fraction, and final core presynaptic dataset that belonged to each of the categories is indicated. The total number of proteins in each dataset was taken as 100%.
{kind=link}
Acknowledgments
We would like to thank E. Sobie, L. Fricker, and I. Gomes for critical reading of the manuscript, and K. Gagnidze, I. Carcea, and D. Benson for help with immunocytochemistry, and support from NIH grants DA08863 and DA019521 (to LAD), GM54508 and DK 38761 (to RI), CA88325 and RR017802 (to RW), 1P50GM071558-01A27398 (SBCNY), and R24 CA095823 (MSSM Microscopy Shared Facility).
Abbreviations
- PPI
protein-protein interaction
- PRE
presynaptic
- PSD
postsynaptic density
Footnotes
The authors have no conflicts of interest to declare.
Contributor Information
Noura S. Abul-Husn, Department of Pharmacology & Systems Therapeutics, Mount Sinai School of Medicine One Gustave L. Levy Place, New York, NY 10029
Ittai Bushlin, Department of Pharmacology & Systems Therapeutics, Mount Sinai School of Medicine One Gustave L. Levy Place, New York, NY 10029.
José A. Morón, Department of Pharmacology & Systems Therapeutics, Mount Sinai School of Medicine One Gustave L. Levy Place, New York, NY 10029
Sherry L. Jenkins, Department of Pharmacology & Systems Therapeutics, Mount Sinai School of Medicine One Gustave L. Levy Place, New York, NY 10029
Georgia Dolios, Department of Genetics & Genomic Sciences, Mount Sinai School of Medicine One Gustave L. Levy Place, New York, NY 10029.
Rong Wang, Department of Genetics & Genomic Sciences, Mount Sinai School of Medicine One Gustave L. Levy Place, New York, NY 10029.
Ravi Iyengar, Department of Pharmacology & Systems Therapeutics, Mount Sinai School of Medicine One Gustave L. Levy Place, New York, NY 10029.
Avi Ma'ayan, Department of Pharmacology & Systems Therapeutics, Mount Sinai School of Medicine One Gustave L. Levy Place, New York, NY 10029.
Lakshmi A. Devi, Department of Pharmacology & Systems Therapeutics, Mount Sinai School of Medicine One Gustave L. Levy Place, New York, NY 10029.
References
- 1.Abul-Husn NS, Devi LA. Neuroproteomics of the synapse and drug addiction. J Pharmacol Exp Ther. 2006;318:461–468. doi: 10.1124/jpet.105.091520. [DOI] [PubMed] [Google Scholar]
- 2.Schrimpf SP, Meskenaite V, Brunner E, Rutishauser D, et al. Proteomic analysis of synaptosomes using isotope-coded affinity tags and mass spectrometry. Proteomics. 2005;5:2531–2541. doi: 10.1002/pmic.200401198. [DOI] [PubMed] [Google Scholar]
- 3.Witzmann FA, Arnold RJ, Bai F, Hrncirova P, et al. A proteomic survey of rat cerebral cortical synaptosomes. Proteomics. 2005;5:2177–2201. doi: 10.1002/pmic.200401102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Corti V, Sanchez-Ruiz Y, Piccoli G, Bergamaschi A, et al. Protein fingerprints of cultured CA3-CA1 hippocampal neurons: comparative analysis of the distribution of synaptosomal and cytosolic proteins. BMC Neurosci. 2008;9:36. doi: 10.1186/1471-2202-9-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.McClatchy DB, Liao L, Park SK, Venable JD, Yates JR. Quantification of the synaptosomal proteome of the rat cerebellum during post-natal development. Genome Res. 2007;17:1378–1388. doi: 10.1101/gr.6375007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Stevens SM, Jr, Zharikova AD, Prokai L. Proteomic analysis of the synaptic plasma membrane fraction isolated from rat forebrain. Brain Res Mol Brain Res. 2003;117:116–128. doi: 10.1016/s0169-328x(03)00282-1. [DOI] [PubMed] [Google Scholar]
- 7.Jia JY, Lamer S, Schumann M, Schmidt MR, et al. Quantitative proteomics analysis of detergent-resistant membranes from chemical synapses: evidence for cholesterol as spatial organizer of synaptic vesicle cycling. Mol Cell Proteomics. 2006;5:2060–2071. doi: 10.1074/mcp.M600161-MCP200. [DOI] [PubMed] [Google Scholar]
- 8.Li KW, Miller S, Klychnikov O, Loos M, et al. Quantitative proteomics and protein network analysis of hippocampal synapses of CaMKIIalpha mutant mice. J Proteome Res. 2007;6:3127–3133. doi: 10.1021/pr070086w. [DOI] [PubMed] [Google Scholar]
- 9.Jordan BA, Fernholz BD, Boussac M, Xu C, et al. Identification and verification of novel rodent postsynaptic density proteins. Mol Cell Proteomics. 2004;3:857–871. doi: 10.1074/mcp.M400045-MCP200. [DOI] [PubMed] [Google Scholar]
- 10.Li KW, Hornshaw MP, Van Der Schors RC, Watson R, et al. Proteomics analysis of rat brain postsynaptic density. Implications of the diverse protein functional groups for the integration of synaptic physiology. J Biol Chem. 2004;279:987–1002. doi: 10.1074/jbc.M303116200. [DOI] [PubMed] [Google Scholar]
- 11.Peng J, Kim MJ, Cheng D, Duong DM, et al. Semiquantitative proteomic analysis of rat forebrain postsynaptic density fractions by mass spectrometry. J Biol Chem. 2004;279:21003–21011. doi: 10.1074/jbc.M400103200. [DOI] [PubMed] [Google Scholar]
- 12.Yoshimura Y, Yamauchi Y, Shinkawa T, Taoka M, et al. Molecular constituents of the postsynaptic density fraction revealed by proteomic analysis using multidimensional liquid chromatography-tandem mass spectrometry. J Neurochem. 2004;88:759–768. doi: 10.1046/j.1471-4159.2003.02136.x. [DOI] [PubMed] [Google Scholar]
- 13.Trinidad JC, Specht CG, Thalhammer A, Schoepfer R, Burlingame AL. Comprehensive identification of phosphorylation sites in postsynaptic density preparations. Mol Cell Proteomics. 2006;5:914–922. doi: 10.1074/mcp.T500041-MCP200. [DOI] [PubMed] [Google Scholar]
- 14.Cheng D, Hoogenraad CC, Rush J, Ramm E, et al. Relative and absolute quantification of postsynaptic density proteome isolated from rat forebrain and cerebellum. Mol Cell Proteomics. 2006;5:1158–1170. doi: 10.1074/mcp.D500009-MCP200. [DOI] [PubMed] [Google Scholar]
- 15.Dosemeci A, Tao-Cheng JH, Vinade L, Jaffe H. Preparation of postsynaptic density fraction from hippocampal slices and proteomic analysis. Biochem Biophys Res Commun. 2006;339:687–694. doi: 10.1016/j.bbrc.2005.11.069. [DOI] [PubMed] [Google Scholar]
- 16.Emes RD, Pocklington AJ, Anderson CN, Bayes A, et al. Evolutionary expansion and anatomical specialization of synapse proteome complexity. Nat Neurosci. 2008;11:799–806. doi: 10.1038/nn.2135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Trinidad JC, Thalhammer A, Specht CG, Lynn AJ, et al. Quantitative analysis of synaptic phosphorylation and protein expression. Mol Cell Proteomics. 2008;7:684–696. doi: 10.1074/mcp.M700170-MCP200. [DOI] [PubMed] [Google Scholar]
- 18.Coughenour HD, Spaulding RS, Thompson CM. The synaptic vesicle proteome: a comparative study in membrane protein identification. Proteomics. 2004;4:3141–3155. doi: 10.1002/pmic.200300817. [DOI] [PubMed] [Google Scholar]
- 19.Morciano M, Burre J, Corvey C, Karas M, et al. Immunoisolation of two synaptic vesicle pools from synaptosomes: a proteomics analysis. J Neurochem. 2005;95:1732–1745. doi: 10.1111/j.1471-4159.2005.03506.x. [DOI] [PubMed] [Google Scholar]
- 20.Takamori S, Holt M, Stenius K, Lemke EA, et al. Molecular anatomy of a trafficking organelle. Cell. 2006;127:831–846. doi: 10.1016/j.cell.2006.10.030. [DOI] [PubMed] [Google Scholar]
- 21.Burre J, Beckhaus T, Schagger H, Corvey C, et al. Analysis of the synaptic vesicle proteome using three gel-based protein separation techniques. Proteomics. 2006;6:6250–6262. doi: 10.1002/pmic.200600357. [DOI] [PubMed] [Google Scholar]
- 22.Burre J, Zimmermann H, Volknandt W. Immunoisolation and subfractionation of synaptic vesicle proteins. Anal Biochem. 2007;362:172–181. doi: 10.1016/j.ab.2006.12.045. [DOI] [PubMed] [Google Scholar]
- 23.Phillips GR, Florens L, Tanaka H, Khaing ZZ, et al. Proteomic comparison of two fractions derived from the transsynaptic scaffold. J Neurosci Res. 2005;81:762–775. doi: 10.1002/jnr.20614. [DOI] [PubMed] [Google Scholar]
- 24.Khanna R, Zougman A, Stanley EF. A proteomic screen for presynaptic terminal N-type calcium channel (CaV2.2) binding partners. J Biochem Mol Biol. 2007;40:302–314. doi: 10.5483/bmbrep.2007.40.3.302. [DOI] [PubMed] [Google Scholar]
- 25.Collins MO, Husi H, Yu L, Brandon JM, et al. Molecular characterization and comparison of the components and multiprotein complexes in the postsynaptic proteome. J Neurochem. 2005 doi: 10.1111/j.1471-4159.2005.03507.x. [DOI] [PubMed] [Google Scholar]
- 26.Armstrong JD, Pocklington AJ, Cumiskey MA, Grant SG. Reconstructing protein complexes: from proteomics to systems biology. Proteomics. 2006;6:4724–4731. doi: 10.1002/pmic.200500895. [DOI] [PubMed] [Google Scholar]
- 27.Pocklington AJ, Cumiskey M, Armstrong JD, Grant SG. The proteomes of neurotransmitter receptor complexes form modular networks with distributed functionality underlying plasticity and behaviour. Mol Syst Biol. 2006;2:2006 0023. doi: 10.1038/msb4100041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ma'ayan A, Jenkins SL, Neves S, Hasseldine A, et al. Formation of regulatory patterns during signal propagation in a Mammalian cellular network. Science. 2005;309:1078–1083. doi: 10.1126/science.1108876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Phillips GR, Huang JK, Wang Y, Tanaka H, et al. The presynaptic particle web: ultrastructure, composition, dissolution, and reconstitution. Neuron. 2001;32:63–77. doi: 10.1016/s0896-6273(01)00450-0. [DOI] [PubMed] [Google Scholar]
- 30.Ma'ayan A, Jenkins SL, Webb RL, Berger SI, et al. SNAVI: Desktop application for analysis and visualization of large-scale signaling networks. BMC Syst Biol. 2009;3:10. doi: 10.1186/1752-0509-3-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Stark C, Breitkreutz BJ, Reguly T, Boucher L, et al. BioGRID: a general repository for interaction datasets. Nucleic Acids Res. 2006;34:D535–539. doi: 10.1093/nar/gkj109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mishra GR, Suresh M, Kumaran K, Kannabiran N, et al. Human protein reference database--2006 update. Nucleic Acids Res. 2006;34:D411–414. doi: 10.1093/nar/gkj141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Husi H, Grant SG. In: Neuroscience Databases: A Practical Guide. Kotter R, editor. Kluwer Academic Publishers; Boston/Dordrecht/London: 2002. pp. 51–62. [Google Scholar]
- 34.Berger SI, Posner JM, Ma'ayan A. Genes2Networks: connecting lists of gene symbols using mammalian protein interactions databases. BMC Bioinformatics. 2007;8:372. doi: 10.1186/1471-2105-8-372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang W, Zhang Y, Zheng H, Zhang C, et al. SynDB: a Synapse protein DataBase based on synapse ontology. Nucleic Acids Res. 2007;35:D737–741. doi: 10.1093/nar/gkl876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gene Ontology Consortium, Creating the gene ontology resource: design and implementation. Genome Res. 2001;11:1425–1433. doi: 10.1101/gr.180801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kerrien S, Alam-Faruque Y, Aranda B, Bancarz I, et al. IntAct--open source resource for molecular interaction data. Nucleic Acids Res. 2007;35:D561–565. doi: 10.1093/nar/gkl958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Anderson TR, Shah PA, Benson DL. Maturation of glutamatergic and GABAergic synapse composition in hippocampal neurons. Neuropharmacology. 2004;47:694–705. doi: 10.1016/j.neuropharm.2004.07.023. [DOI] [PubMed] [Google Scholar]
- 39.Ziff EB. Enlightening the postsynaptic density. Neuron. 1997;19:1163–1174. doi: 10.1016/s0896-6273(00)80409-2. [DOI] [PubMed] [Google Scholar]
- 40.Benfenati F, Onofri F, Czernik AJ, Valtorta F. Biochemical and functional characterization of the synaptic vesicle-associated form of CA2+/calmodulin-dependent protein kinase II. Brain Res Mol Brain Res. 1996;40:297–309. doi: 10.1016/0169-328x(96)00053-8. [DOI] [PubMed] [Google Scholar]
- 41.Kapp EA, Schutz F, Connolly LM, Chakel JA, et al. An evaluation, comparison, and accurate benchmarking of several publicly available MS/MS search algorithms: sensitivity and specificity analysis. Proteomics. 2005;5:3475–3490. doi: 10.1002/pmic.200500126. [DOI] [PubMed] [Google Scholar]
- 42.Moron JA, Abul-Husn NS, Rozenfeld R, Dolios G, et al. Morphine administration alters the profile of hippocampal postsynaptic density-associated proteins: A proteomic study focusing on endocytic proteins. Mol Cell Proteomics. 2006 doi: 10.1074/mcp.M600184-MCP200. [DOI] [PubMed] [Google Scholar]
- 43.Mika S, Rost B. Protein-protein interactions more conserved within species than across species. PLoS Comput Biol. 2006;2:e79. doi: 10.1371/journal.pcbi.0020079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yu H, Luscombe NM, Lu HX, Zhu X, et al. Annotation transfer between genomes: protein-protein interologs and protein-DNA regulogs. Genome Res. 2004;14:1107–1118. doi: 10.1101/gr.1774904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Brown KR, Jurisica I. Unequal evolutionary conservation of human protein interactions in interologous networks. Genome Biol. 2007;8:R95. doi: 10.1186/gb-2007-8-5-r95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bader GD, Hogue CW. Analyzing yeast protein-protein interaction data obtained from different sources. Nat Biotechnol. 2002;20:991–997. doi: 10.1038/nbt1002-991. [DOI] [PubMed] [Google Scholar]
- 47.Yu H, Paccanaro A, Trifonov V, Gerstein M. Predicting interactions in protein networks by completing defective cliques. Bioinformatics. 2006;22:823–829. doi: 10.1093/bioinformatics/btl014. [DOI] [PubMed] [Google Scholar]
- 48.Albert I, Albert R. Conserved network motifs allow protein-protein interaction prediction. Bioinformatics. 2004;20:3346–3352. doi: 10.1093/bioinformatics/bth402. [DOI] [PubMed] [Google Scholar]
- 49.Liu Y, Cheng K, Gong K, Fu AK, Ip NY. Pctaire1 phosphorylates N-ethylmaleimide-sensitive fusion protein: implications in the regulation of its hexamerization and exocytosis. J Biol Chem. 2006;281:9852–9858. doi: 10.1074/jbc.M513496200. [DOI] [PubMed] [Google Scholar]
- 50.Dhaka A, Costa RM, Hu H, Irvin DK, et al. The RAS effector RIN1 modulates the formation of aversive memories. J Neurosci. 2003;23:748–757. doi: 10.1523/JNEUROSCI.23-03-00748.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Krendel M, Zenke FT, Bokoch GM. Nucleotide exchange factor GEF-H1 mediates cross-talk between microtubules and the actin cytoskeleton. Nat Cell Biol. 2002;4:294–301. doi: 10.1038/ncb773. [DOI] [PubMed] [Google Scholar]
- 52.Li Z, McNulty DE, Marler KJ, Lim L, et al. IQGAP1 promotes neurite outgrowth in a phosphorylation-dependent manner. J Biol Chem. 2005;280:13871–13878. doi: 10.1074/jbc.M413482200. [DOI] [PubMed] [Google Scholar]
- 53.Chan SL, Pedersen WA, Zhu H, Mattson MP. Numb modifies neuronal vulnerability to amyloid beta-peptide in an isoform-specific manner by a mechanism involving altered calcium homeostasis: implications for neuronal death in Alzheimer's disease. Neuromolecular Med. 2002;1:55–67. doi: 10.1385/NMM:1:1:55. [DOI] [PubMed] [Google Scholar]
- 54.Okada K, Kanaya S, Asai K. Accurate extraction of functional associations between proteins based on common interaction partners and common domains. Bioinformatics. 2005;21:2043–2048. doi: 10.1093/bioinformatics/bti305. [DOI] [PubMed] [Google Scholar]
- 55.Samanta MP, Liang S. Predicting protein functions from redundancies in large-scale protein interaction networks. Proc Natl Acad Sci U S A. 2003;100:12579–12583. doi: 10.1073/pnas.2132527100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Foster-Barber A, Bishop JM. Src interacts with dynamin and synapsin in neuronal cells. Proc Natl Acad Sci U S A. 1998;95:4673–4677. doi: 10.1073/pnas.95.8.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kasowski HJ, Kim H, Greer CA. Compartmental organization of the olfactory bulb glomerulus. J Comp Neurol. 1999;407:261–274. [PubMed] [Google Scholar]
- 58.Stasyk T, Huber LA. Zooming in: fractionation strategies in proteomics. Proteomics. 2004;4:3704–3716. doi: 10.1002/pmic.200401048. [DOI] [PubMed] [Google Scholar]
- 59.Righetti PG, Castagna A, Antonioli P, Boschetti E. Prefractionation techniques in proteome analysis: the mining tools of the third millennium. Electrophoresis. 2005;26:297–319. doi: 10.1002/elps.200406189. [DOI] [PubMed] [Google Scholar]
- 60.Brunet S, Thibault P, Gagnon E, Kearney P, et al. Organelle proteomics: looking at less to see more. Trends Cell Biol. 2003;13:629–638. doi: 10.1016/j.tcb.2003.10.006. [DOI] [PubMed] [Google Scholar]
- 61.Bromberg KD, Ma'ayan A, Neves SR, Iyengar R. Design logic of a cannabinoid receptor signaling network that triggers neurite outgrowth. Science. 2008;320:903–909. doi: 10.1126/science.1152662. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1. Two experimental approaches used in the identification of hippocampal PRE and PSD proteins. To achieve highly comprehensive protein identification, both in-gel digestion and in-solution digestion were used. (A) SDS-PAGE separation of PRE and PSD proteins prepared for in-gel trypsin digestion and LC-MS/MS protein identification (details are provided in the Materials and Methods). (B) Reverse-phase HPLC profile of tryptic peptides of PRE and PSD proteins from in-solution digestion. PRE and PSD proteins were digested by trypsin directly, and the tryptic peptides were analyzed by 1-dimensional reverse-phase HPLC-MS/MS with a two-hour elution gradient. The elution time for each of the major peaks is indicated.
Supplementary Figure 2. Literature-based presynaptic protein-protein interaction (PPI) network, containing 127 proteins and 229 interactions. The network was generated by manually extracting mammalian presynaptic interactions from the literature. In this graphical representation of the network, proteins are represented as nodes (and designated by human gene names), and their direct interactions are represented as links. A web-based interface providing access to this network can be found at http://amp.pharm.mssm.edu/presynaptome.
Supplementary Figure 3. Functional annotation of presynaptic datasets. Proteins were assigned to Gene Ontology biological processes, as described in Materials and Methods (see Table S7). The percentage of proteins in the hippocampal PRE fraction, striatal PRE fraction, and final core presynaptic dataset that belonged to each of the categories is indicated. The total number of proteins in each dataset was taken as 100%.