

Abstract
We assume that quantitative measurements on a considered trait and unphased genotype data at certain marker loci are available on a sample of individuals from a background population. Our goal is to map quantitative trait loci by using a Bayesian model that performs, and makes use of, probabilistic reconstructions of the recent unobserved genealogical history (a pedigree and a gene flow at the marker loci) of the sampled individuals. This work extends variance component-based linkage analysis to settings where the unobserved pedigrees are considered as latent variables. In addition to the measured trait values and unphased genotype data at the marker loci, the method requires as an input estimates of the population allele frequencies and of a marker map, as well as some parameters related to the population size and the mating behavior. Given such data, the posterior distribution of the trait parameters (the number, the locations, and the relative variance contributions of the trait loci) is studied by using the reversible-jump Markov chain Monte Carlo methodology. We also introduce two shortcuts related to the trait parameters that allow us to do analytic integration, instead of stochastic sampling, in some parts of the algorithm. The method is tested on two simulated data sets. Comparisons with traditional variance component linkage analysis and association analysis demonstrate the benefits of our approach in a gene mapping context.
ASSUME that quantitative measurements on certain trait and unphased genotype data at certain marker loci are available on a sample of individuals from a population. In quantitative trait locus (QTL) mapping the goal is to find positions in the genome in which the genetic variation between the individuals explains the observed differences in the considered quantitative trait. A central task in QTL mapping is to estimate how the sampled individuals are related to each other in different parts of the genome, since such information provides a means to identify regions where the estimated inheritance pattern is in accordance with the observed similarities in the trait values. Important pieces of information for estimating the locus-specific relatedness structures would be given by pedigree records and genotyped ancestors. However, for example, in wild animal and plant populations there are situations where no data on the genetic history are available or where such data are available only on a very recent history of the sampled individuals. In view of such situations, we have earlier introduced a Bayesian model that estimates the unobserved recent history of the sampled individuals by performing reconstructions of pedigrees and gene flows at the linked marker loci (Gasbarra et al. 2007a). Here we extend that work to map QTL.
Many QTL mapping methods adopt a two-step strategy where one first estimates the relatedness structure from marker data, either locuswise or at the level of individuals (Bink et al. 2008), and then incorporates the estimates into subsequent QTL or association analyses as if they were observed quantities (e.g., Meuwissen and Goddard 2004, 2007; Yu et al. 2006). A special feature of our approach is that both of these tasks are carried out simultaneously within a single Bayesian model. Similar ideas have been used in some coalescent-based gene mapping methods that model jointly the relatedness structure of a trait locus and the phenotype (e.g., Larribe et al. 2002; Morris et al. 2002; Zöllner and Pritchard 2005; Minichiello and Durbin 2006). Our method differs from these in that we are working with the recent genetic history of the sample (individuals) and not using continuous time approximations of coalescent trees and recombination graphs.
Our method can be seen as an extension of pedigree-based linkage analysis to situations where no pedigrees are observed. Thus, even though the relationships between the sampled individuals may be unknown, there should still be close relatives in the sample so that linkage information can be extracted. These kinds of data may be mostly available in wild plant or animal species (Frentiu et al. 2008), but may also be available for humans. We also consider similar marker maps that are used in pedigree-based linkage analysis. In our examples we consider ∼100 loci with polymorphic markers (say, with six alleles) that are moderately spaced along the chromosomes (say, 4 cM apart from each other).
In this article our phenotype model considers only additive genetic effects for QTL and for the polygene, but extensions to environmental covariates and genetic interaction effects would be technically straightforward. We analyze the phenotype model by estimating the genetic components of phenotypic variance. An advantage of a variance component model is that the numbers of QTL alleles and their effect sizes need not be specified (see Yi and Xu 2000).
Computationally, we use the reversible-jump Markov chain Monte Carlo methodology, which allows us to treat the number of QTL, their positions, and their variance contributions as random variables. Because the method is computationally intensive, we have introduced two shortcuts in the algorithm that allow for an analytic integration over the allelic paths at the QTL and over the residual variance. As a result, the central parameters of interest are the relative phenotypic variance contributions of the QTL with respect to the residual variance.
We illustrate the usefulness of the method with two examples. Comparisons of our results with the fixed-pedigree variance component linkage analysis program SOLAR (Almasy and Blangero 1998) and with the association analysis package TASSEL (Bradbury et al. 2007) clearly show the advantage that is gained from being able to model the unobserved part of the recent genetic history of the sampled individuals.
MODEL FOR GENETIC HISTORY
Consider a sample of n individuals belonging to the current generation of the population. We build a probability model for their joint genetic history, up to T generations backward in time, at certain marker loci whose relative positions (with respect to each other) are assumed to be known.
The configuration space of possible ancestral histories has three components: the pedigree specifying the relationships between the individuals, the paths of alleles of these individuals at the marker loci, and the types of the alleles of the founder individuals at generation T. We described the same probability model on the configuration space earlier (Gasbarra et al. 2007a) and therefore provide here only a brief summary of this model.
Pedigree model:
For pedigrees we use the probability model introduced by Gasbarra et al. (2005). The model considers an isolated population with nonoverlapping generations indexed backward in time by t = 0, 1,…, T, with t = 0 referring to the present and t = T to the founder generation. The population is characterized by four sets of parameters: N′t, N″t, αt, and βt, for t = 1, … , T. The parameters N′t and N″t describe, respectively, the number of males and females belonging to generation t of the population. Parameter αt controls the differences of reproductive success between males in generation t: large values of αt imply nearly equal numbers of children for each male, whereas for small values of αt there will be a few dominant males who are mainly responsible for the reproduction. Parameter βt tunes the degree of monogamy (of males) in generation t: large values of βt lead to random mating and small values of βt introduce more permanent family structures into the pedigree. Naturally the roles of males and females can be changed in the model. We denote this probability measure on pedigree graphs by 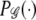 .
.
Flow of alleles through the pedigree:
We assume a fixed marker map with L loci and denote the recombination fractions between loci by ρ = (ρ(l, l′): 1 ≤ l < l′ ≤ L). Note that several chromosomes can be modeled simultaneously by using the recombination fraction 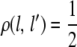 to indicate that markers l and l′ lie in different linkage groups.
to indicate that markers l and l′ lie in different linkage groups.
By definition, the genome of each individual in the pedigree consists of a pair of paternal and maternal haplotypes. The flow of alleles through the pedigree is determined by the grandparental origins that for haplotype i are denoted by 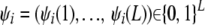 . The convention used here is that ψi(l) = 0 if the allele at locus l of haplotype i is of grandmaternal origin, and ψi(l) = 1 in the case of grandpaternal origin.
. The convention used here is that ψi(l) = 0 if the allele at locus l of haplotype i is of grandmaternal origin, and ψi(l) = 1 in the case of grandpaternal origin.
If an allele carried by an individual in generation t > 0 is transmitted to some individual in the present generation, we say that the allele is ancestral and otherwise that it is censored. Since we are actually interested only in the paths of the ancestral alleles, we set ψi(l) = Ø if the allele at locus l of haplotype i is censored.
The probability of a set 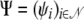 of grandparental origins of nonfounder haplotypes on the pedigree is given by
of grandparental origins of nonfounder haplotypes on the pedigree is given by
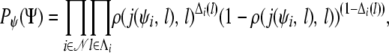 |
where Λi = {l : ψi(l) ≠ Ø}, ρ(l, l′) is the recombination fraction between loci l and l′, with the convention that 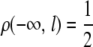 , j(ψi, l) denotes the last uncensored locus of haplotype i before l, with the convention that j(ψi, l) = −∞ if l is the first uncensored locus of its chromosome in the haplotype i, and Δi(l) = |ψi(l) − ψi(j(ψi, l))| with the convention that ψi(−∞) = 0.
, j(ψi, l) denotes the last uncensored locus of haplotype i before l, with the convention that j(ψi, l) = −∞ if l is the first uncensored locus of its chromosome in the haplotype i, and Δi(l) = |ψi(l) − ψi(j(ψi, l))| with the convention that ψi(−∞) = 0.
Types of founder alleles:
Denote by 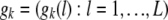 the ordered genotype of individual k and let
the ordered genotype of individual k and let 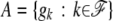 be the set of founder genotypes. Assuming linkage equilibrium at the founder generation, the probability of the founder alleles is given by
be the set of founder genotypes. Assuming linkage equilibrium at the founder generation, the probability of the founder alleles is given by
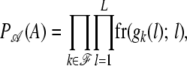 |
where the population genotype frequencies fr(·; l) at each marker locus l are assumed given. (If Hardy–Weinberg equilibrium is assumed, we can use the population allele frequencies instead.) The genotype frequencies are extended to partially or totally censored genotypes in the obvious way. Note that the ordered founder alleles together with the grandparental origins of the nonfounder haplotypes determine the flow of alleles in the pedigree.
Prior distribution for genetic history:
Given the pedigree parameters (N′t, N″t, βt, αt, T), the population genotype frequencies and the recombination fractions between the marker loci, a configuration ω consisting of a pedigree G and a gene flow with founder alleles A and grandparental origins Ψ is assigned the (prior) probability
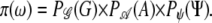 |
Our earlier work (Gasbarra et al. 2007a) studied this distribution conditionally on the observed marker data and in this article we further add to the model a contribution from a quantitative phenotype.
Variance component model for the phenotypes:
We use a variance component model that is similar to, for example, those by Almasy and Blangero (1998) and Yi and Xu (2000). A slight difference between the standard model development (e.g., Equation 1 in Yi and Xu 2000) and the one given below is that here the covariances at the QTL are derived explicitly using the corresponding incidence matrices (Xq). This formulation is chosen because our state space can be augmented to contain the exact paths at the putative QTL, and thus we could work with the exact identity-by-descent (IBD) matrices at the QTL (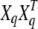 ). However, in this article we use the conditional expectations of the IBD matrices at QTL, given the allelic paths at the flanking markers. We formulate the model at the level of single alleles, and therefore certain variance parameters in our model equal half of the corresponding quantities when they are considered at the level of genotypes.
). However, in this article we use the conditional expectations of the IBD matrices at QTL, given the allelic paths at the flanking markers. We formulate the model at the level of single alleles, and therefore certain variance parameters in our model equal half of the corresponding quantities when they are considered at the level of genotypes.
To derive the model, we suppose that a quantitative phenotype is measured from each of the n sampled individuals from the current generation of the population. In the following we explain the phenotype model conditionally on genetic history ω. Note, however, that both structures (genetic history and phenotype parameters) are random variables in our model and we are studying their joint posterior distribution conditionally on the observed marker data and the observed phenotype values. Conditionally on genetic history ω we adopt a simple regression model for the phenotypes,
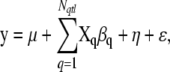 |
(1) |
where y is the n-dimensional vector of phenotypes, μ is the population mean, Xq is the n × 2f matrix describing which of the 2f founder alleles each individual carries at QTL q, βq is the 2f-dimensional vector of founder allele effects at QTL q, η is the n-dimensional vector of polygenic contributions (sum of the effects of QTL located outside of the marker map), and ɛ is the n-dimensional vector of residual errors. Possible extensions of the model, which would contain also covariates and genetic effects of higher degrees, are considered in the discussion.
We assume that 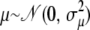 (univariate normal distribution),
(univariate normal distribution), 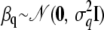 (multivariate normal distribution),
(multivariate normal distribution), 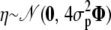 , where Φ is the kinship matrix calculated from the pedigree structure, and
, where Φ is the kinship matrix calculated from the pedigree structure, and 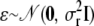 . Here
. Here 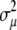 ,
, 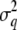 ,
, 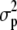 , and
, and 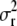 are the variance components related to the population mean, a single allele at QTL q, the polygenic contribution, and the residual effect, respectively. The element
are the variance components related to the population mean, a single allele at QTL q, the polygenic contribution, and the residual effect, respectively. The element 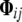 of the kinship matrix is the conditional probability, given the pedigree structure, that a randomly sampled allele at a random locus from individual i is IBD with a randomly sampled allele from the same locus from individual j (see, e.g., Lange 2002). In this model the number of QTL (Nqtl) is considered as a random variable, and each QTL has an exact position specified in terms of the genetic distance to the nearest marker locus.
of the kinship matrix is the conditional probability, given the pedigree structure, that a randomly sampled allele at a random locus from individual i is IBD with a randomly sampled allele from the same locus from individual j (see, e.g., Lange 2002). In this model the number of QTL (Nqtl) is considered as a random variable, and each QTL has an exact position specified in terms of the genetic distance to the nearest marker locus.
For a given genetic history (pedigree, allele paths, and founder alleles at the marker loci), the phenotype model (1) is simply
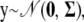 |
(2) |
where the covariance matrix has the form
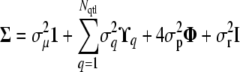 |
(3) |
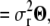 |
(4) |
with
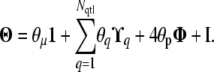 |
(5) |
Here 1 is the n × n matrix full of ones, 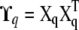 is the (unscaled) covariance matrix at QTL q, and
is the (unscaled) covariance matrix at QTL q, and 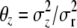 for z = μ, q, p. The representation of Σ in terms of Θ becomes useful in the practical computations that we describe below.
for z = μ, q, p. The representation of Σ in terms of Θ becomes useful in the practical computations that we describe below.
Priors:
We assume that a priori 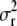 ∼ IG(a, d) (inverse-gamma distribution) and that each θz ∼ Exp(1) (exponential distribution). A priori, each marker interval (between two adjacent markers or between the extreme markers and the endpoints of the chromosome) may contain at most one QTL and this happens with probability 1 − exp(−λΔ), where Δ is the genetic length of the interval and λ > 0 is a hyperparameter chosen according to our prior guess on the total number of QTL.
∼ IG(a, d) (inverse-gamma distribution) and that each θz ∼ Exp(1) (exponential distribution). A priori, each marker interval (between two adjacent markers or between the extreme markers and the endpoints of the chromosome) may contain at most one QTL and this happens with probability 1 − exp(−λΔ), where Δ is the genetic length of the interval and λ > 0 is a hyperparameter chosen according to our prior guess on the total number of QTL.
Posteriors:
In our earlier works we estimated the posterior distribution of the genetic histories of the sampled individuals, conditionally on the genotype observations at the marker loci. In this article our focus is on the posterior distribution of the QTL parameters, namely the number, the positions, and the relative variances of the QTL, as well as the relative variance of the polygenic component with respect to the residual variance. The posterior is calculated conditionally on the marker and phenotype data and on fixed population parameters. We described earlier how to apply Markov chain Monte Carlo (MCMC) methods to estimate the posterior distribution of the genetic histories. Here we add new parts to our MCMC algorithm that also update the QTL parameters. Before introducing the MCMC algorithm we consider briefly two simplifying steps by which we are able to integrate out analytically several variables of the phenotype model during the MCMC run. These analytic integrations decrease the number of variables that need to be updated in the MCMC algorithm and thereby shorten its running time.
Integrating σr2 out:
Instead of updating parameter 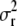 in the MCMC algorithm, we combine the likelihood function from (2), an inverse-gamma prior for
in the MCMC algorithm, we combine the likelihood function from (2), an inverse-gamma prior for 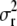 and a representation
and a representation 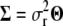 to analytically integrate
to analytically integrate 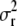 out from the likelihood formula. (See appendix b for details.) As a result we get the likelihood function
out from the likelihood formula. (See appendix b for details.) As a result we get the likelihood function
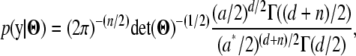 |
where 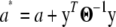 , and a and d are the parameters of the inverse-gamma prior for
, and a and d are the parameters of the inverse-gamma prior for 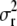 .
.
Integrating over allelic paths at QTL:
It would be possible to include in the configuration the complete information of the allelic paths at the QTL and thus use the exact 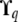 matrices in the model. However, using the exact recursive formulas given in appendix a we are able to compute
matrices in the model. However, using the exact recursive formulas given in appendix a we are able to compute 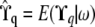 at each QTL q, i.e., the conditional expectation of
at each QTL q, i.e., the conditional expectation of 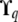 given the available allelic paths at the (nearest informative) marker loci. In this article we have approximated the exact model (2) by replacing
given the available allelic paths at the (nearest informative) marker loci. In this article we have approximated the exact model (2) by replacing 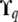 with
with 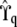 in Equation 5 when calculating matrix Θ.
in Equation 5 when calculating matrix Θ.
MCMC ALGORITHM
In our earlier articles (Gasbarra et al. 2007a,b) the MCMC algorithms explored the space of possible genetic histories of the sampled individuals using several different Metropolis–Hastings updating schemes. Here we add to the algorithm further Metropolis–Hastings proposal steps involving QTL parameters. As the number of QTL (Nqtl) is a random variable that affects the dimensionality of the model, we update it using the reversible-jump MCMC methodology (Green 1995). Earlier, such ideas were used for QTL mapping, for example, by Heath (1997) and Sillanpää and Arjas (1998). Next we describe the Metropolis–Hastings schemes that are used to update the variables (in each iteration of the MCMC run).
Updating genetic history:
We use the proposal distributions explained in Gasbarra et al. (2007a) to update the pedigree and allelic paths. The only modification is that in the current algorithm the phenotype-likelihood contribution is taken into account when calculating the acceptance probability of the proposed configuration.
Adding and removing QTL:
In each iteration with probability  we propose a deletion of an existing QTL. The candidate QTL for deletion is sampled with a probability proportional to the inverses of the effect variance. When we propose to delete a QTL, we also propose to transfer its variance contribution to the polygenic variance. Note that the variance related to a QTL q is
we propose a deletion of an existing QTL. The candidate QTL for deletion is sampled with a probability proportional to the inverses of the effect variance. When we propose to delete a QTL, we also propose to transfer its variance contribution to the polygenic variance. Note that the variance related to a QTL q is 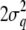 , i.e., twice the variance contribution of a single QTL allele, since each individual carries two QTL alleles.
, i.e., twice the variance contribution of a single QTL allele, since each individual carries two QTL alleles.
In the case that no deletion is proposed, we attempt to add a new QTL at a location sampled uniformly on the available genetic map. If the sampled interval already contains a QTL, we propose to replace the existing one with a new one. The variance of the proposed QTL is sampled as a uniformly random proportion of the current polygenic variance that is simultaneously proposed to decrease accordingly.
Both of these updates propose a change in the number of QTL and since there are continuous variables involved in the QTL model, the theory of reversible-jump MCMC (Green 1995) is adapted in computing acceptance probabilities of the moves.
Updating current QTL:
We choose randomly a QTL from Nqtl possibilities and propose a modification to its relative variance. In addition, with probability 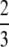 we simultaneously attempt to modify its location.
we simultaneously attempt to modify its location.
The new position is sampled from a normal distribution centered at the current position (with fixed standard deviation given as a tuning parameter to the algorithm). If the new position is outside of the chromosome or if there is already a QTL in the proposed interval, we refrain from modifying the location.
Parameter θq for the chosen QTL is perturbed by multiplying it with a random variable from a lognormal distribution (whose parameters are fixed for the whole MCMC run).
Simultaneously with the perturbation of θq we also propose to change the parameters θp and θμ with similar lognormal proposals.
Updating θp and θμ:
We also have separate updates for θp and θμ that propose no modifications to any other variable. These are implemented with lognormal proposals (see above).
RESULTS
There are three main points that we aim to illustrate by the following examples. First, our method produces good results in comparison with a widely used variance component QTL mapping software SOLAR (Almasy and Blangero, 1998), even in cases where we are using less information than SOLAR. More specifically, our method is able to handle also data that do not include records on pedigrees, whereas traditional linkage analysis packages like SOLAR work only on the known parts of a considered pedigree. Second, we show that if the data include close relatives, then our method is able to separate the true signals from false positives more efficiently than an association analysis method TASSEL (Bradbury et al. 2007) that utilizes an estimated relatedness matrix as a part of the mixed linear model for the phenotype. Our third point is that, in some genetic mapping situations, it is important to be able to explicitly model the genetic relatedness between the sampled individuals beyond only one or two generations backward in time to find strong enough QTL signals. And this is exactly what our method is designed to do.
Linkage analysis on close relatives:
When sampling from natural populations, one frequently encounters situations where the pedigree relationships between individuals are not known, but where it is possible that even close relatives are included among the sampled individuals. When such samples are analyzed for the associations between genetic markers and phenotypic values, it is necessary to account for the relatedness structure to avoid false positives. In this example we show how our method is able to simultaneously account for the unknown relatedness and to estimate the locations on the genome that are partly responsible for the phenotypic variation.
Data simulation:
We consider 50 nuclear families each with three children, whose parents are interconnected via a pedigree structure that is simulated, 20 generations backward in time, using the pedigree model of Gasbarra et al. (2005). (The population parameters are αt = 5, βt = 10−3, N′t = N″t = 1500 − 50t, for generations t = 1, 2, … , 19, where 1 is the parents' generation and 19 is the founder generation.) Given the simulated pedigree structure, the genetic marker data are simulated on four chromosomes by first sampling the alleles for the founders (who are assumed to be in linkage and Hardy–Weinberg equilibria) and then dropping the genes through the pedigree according to Mendelian rules and the recombination model. Each chromosome contains 30 microsatellite markers, each with six alleles that are equally frequent in the population at the founder generation. The recombination fractions between adjacent markers of the same chromosome are 0.04. Conditionally on the marker data, two QTL were simulated on the genome, q1 between markers 38 and 39, and q2 between markers 96 and 97, on chromosomes 2 and 4, respectively. For the founder individuals, five equally frequent alleles are assumed to exist in the population at both QTL. The effects of the five alleles are sampled from 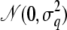 , with σ1 = 0.5 and σ2 = 0.7 for QTL q1 and q2, respectively. Finally, the phenotypes for the sampled individuals at generation 0 are simulated as yi = Qi + ɛi, where Qi is the sum of the effects of the four QTL alleles that i carries and
, with σ1 = 0.5 and σ2 = 0.7 for QTL q1 and q2, respectively. Finally, the phenotypes for the sampled individuals at generation 0 are simulated as yi = Qi + ɛi, where Qi is the sum of the effects of the four QTL alleles that i carries and 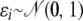 are sampled as independent residual effects. The resulting empirical sample variance of the QTL allele effects among the sampled individuals was 0.36 for q1 and 0.48 for q2, and the residual variance was 0.79.
are sampled as independent residual effects. The resulting empirical sample variance of the QTL allele effects among the sampled individuals was 0.36 for q1 and 0.48 for q2, and the residual variance was 0.79.
Results:
We applied our method to analyze the phenotypic values of 150 individuals belonging to the youngest generation of the above-explained simulated genetic history combined with their unphased marker genotype data. We carried out a pedigree reconstruction for only a single generation backward, which turned out to be enough in this case, as the nuclear families were large enough to contain linkage information (three children in each family). The prior distribution for the existence of QTL was chosen in such a way that the expected number of QTL over all four chromosomes was about five 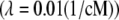 . By simulations the population parameters for the parents' generation were adjusted to correspond to a population where monogamy is common and very large numbers of offspring in the same family are rare (N′ = N″ = 625, β = 10−4, and α = 0.625).
. By simulations the population parameters for the parents' generation were adjusted to correspond to a population where monogamy is common and very large numbers of offspring in the same family are rare (N′ = N″ = 625, β = 10−4, and α = 0.625).
The allele frequencies among parents were assumed to be uniform and the recombination fractions were assumed known exactly (i.e., 0.04 between adjacent markers).
We executed four independent runs of the algorithm, each running for 55,000 MCMC iterations, and then discarded the first 5000 iterations as a burn-in part. The results from different runs were similar, suggesting that the chains had converged. The elapsed time of the runs was ∼5 days but a running time of a single day would have already been sufficient to achieve qualitatively the same results. The results (averaged over all four runs) describing the expected value of the ratio of the QTL variance to the residual variance, as a function of chromosomal location, are shown in the top panel of Figure 1. The measurement unit of location is an interval between two adjacent markers. The positions of the simulated QTL are marked with asterisks on chromosomes 2 and 4. Both QTL can be identified from the posterior curve of QTL variance, and no considerable additional signals are present elsewhere on the marker map. The top panel of Figure 1 also shows two intervals surrounding the QTL positions and the corresponding posterior probabilities (≈0.68 and ≈0.74) that these intervals contain at least one QTL. These intervals were chosen manually to cover the regions that were estimated to have relatively high contributions to the phenotypic variance.
Figure 1.—

Results of example I. Rows from top to bottom correspond to our method, SOLAR, and TASSEL, respectively. The two true QTL are marked with “*” and the posterior probabilities that the marked intervals contain at least one QTL are shown for our method.
For comparison we also run the data with SOLAR (Almasy and Blangero 1998), which is a widely used variance component linkage analysis package for quantitative traits. It requires pedigree(s), marker data, and the marker map as input and estimates the IBD distribution between members of the same pedigree at several locations between the markers. The resulting IBD information is then utilized in a sequential testing procedure for the existence of a QTL between the markers. SOLAR is able to compute the IBD estimates using the multipoint method unless the pedigree structures are too complicated.
In this example we gave SOLAR the correct family structures (50 nuclear families with three children each) accompanied with the correct marker map and the genetic marker data on the children. The resulting multipoint LOD score curves are displayed in the middle panel of Figure 1. It can be seen that also SOLAR gives signals for the two real QTL, but it also assigns nonzero scores to some other regions. The reason for this may be that SOLAR has a model for only a single QTL, and since these data contain two QTL, this may result in some additional noise in SOLAR's tests. The overall shapes of the curves in the top and middle panels of Figure 1 are quite similar and both concentrate strongly in the vicinity of the true QTL. The fact that we have used less information in our analysis than in the SOLAR run does not seem to result in less accurate signals for the true QTL. Indeed, with these data our method was able to reconstruct the nuclear families with three children very accurately.
The bottom panel of Figure 1 displays marker related P-values from the association analysis program TASSEL (Bradbury et al. 2007). TASSEL applies a mixed linear model (MLM) to explain the phenotypes by using markers (each marker separately) and can also include covariates, population structure, and relatedness structure in the analysis. Here we applied a model that included the relatedness structure between individuals calculated using the moment estimator of Lynch and Ritland (1999). This approach is close to our own in the sense that both try to accommodate for the relatedness between the sampled individuals and also allow for a polygenic component in the model.
For these data it turned out that this combination of an association analysis with relatedness estimates was not able to separate the true QTL signals from false positives. However, one must keep in mind that this data set is more suitable for linkage analysis than for association analysis as the marker distances are relatively long and there are close relatives in the data. The association analysis results indicate, however, that these data are not too obvious in the sense that one could have established the QTL positions directly from the correlations between the marker data and phenotypes without modeling the linkage within the families.
Advantage from modeling several generations:
In the above example the nuclear families were large enough (with respect to the QTL effects, sample size, and marker allele distribution) so that the QTL could be found already by a model that included only a single generation of the ancestors of the sampled individuals. We now consider a more difficult situation where we have to model several additional generations backward in time before clear signals of the QTL can be identified.
Data simulation:
We use a similar procedure to generate the data set as in the previous example, considering 50 nuclear families but now with only two children in each. The parents of those families are connected by a simulated 18-generation pedigree, which has a bottleneck in its recent history as shown in Figure 2. Only the first 7 generations of the complete pedigree are actually shown in Figure 2, and the remaining generations were simulated as in the previous example. We consider marker data on a single chromosome containing 100 microsatellite markers, each with six alleles. The recombination fractions between the adjacent markers are again 0.04. Conditionally on the marker data, two QTL were simulated on the chromosome, with q1 between markers 13 and 14 and q2 between markers 86 and 87. For the founder individuals, five equally frequent alleles were assumed to exist in the population at both QTL. The effects for the five types of founder alleles were sampled from 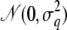 , with σ1 = 0.5 for QTL q1 and σ2 = 0.3 for q2. The final phenotypes for the sampled individuals at generation 0 were created by adding independent standard normal random variables to the sum of the QTL effects of each individual. The resulting sample variance of the QTL allele effects among the sampled individuals was 0.26 for q1, and 0.12 for q2, with residual variance equal to 0.96.
, with σ1 = 0.5 for QTL q1 and σ2 = 0.3 for q2. The final phenotypes for the sampled individuals at generation 0 were created by adding independent standard normal random variables to the sum of the QTL effects of each individual. The resulting sample variance of the QTL allele effects among the sampled individuals was 0.26 for q1, and 0.12 for q2, with residual variance equal to 0.96.
Figure 2.—

The 7 youngest generations from the 20-generation pedigree that was used in the data simulation of example II, drawn with Pedfiddler (J. C. Loredo-Osti and K. Morgan).
Results:
We applied our method to analyze the phenotypic values of 100 individuals belonging to the youngest generation of the above-explained simulated genetic history combined with their unphased marker genotype data. Separate reconstructions were carried out for T = 1, 2, 3, 4 generations backward in time. For the values of the population parameters we used αt = 10, βt = 0.001, N′t = N″t = 1000 − 25t, for generations t = 1, 2, … , 4. They were chosen on the basis of a similar simulation experiment as in the previous example.
For each value of T, four separate MCMC runs were executed and their average values are reported here. The elapsed time of the runs was ∼6 days but qualitatively similar results were already achieved within a single day. Figure 3 shows how the posterior of relative QTL effects, as a function of location, develops as more generations are included. In particular the step from T = 3 to T = 4 seems to bring the QTL effects to levels that correspond well to those calculated in the data simulation. Thus it seems evident that in these data the power for detecting QTL comes from a more distant past than the parents' or grandparents' generations. Each panel of Figure 3 also shows two intervals surrounding the QTL positions and the corresponding posterior probabilities that these intervals contain at least one QTL. These intervals were chosen manually to cover the regions that were estimated to have relatively high contributions to the phenotypic variance. For both intervals the corresponding QTL probabilities consistently increase as more generations are included in the model. This phenomenon further confirms that we are able to capture stronger QTL signals from these data by modeling several generations simultaneously.
Figure 3.—

Results of example II with varying numbers of reconstructed generations. The numerical values are the posterior probabilities that the marked intervals contain at least one QTL.
The same phenomenon can also be seen in Figure 4, where we have also included results from a SOLAR analysis based on the correct nuclear family structure (middle panel) and from a TASSEL analysis with the relatedness matrix estimated as in the previous example (bottom panel). The top panel contains the data from our method with T = 4. Since the families are smaller and QTL weaker than in the previous example, SOLAR does not seem to be able to catch the QTL q1. However, SOLAR might perform better if one first identifies q2 and then fixes it as a covariate when searching for a second QTL. Note again that we have used more information in the SOLAR run than with our own method, since even though we have modeled the history four generations backward in time, it is still based only on the genotype data at the youngest generation and no part of the pedigree was considered known to us. We also attempted to analyze these data with SOLAR by giving it the correct pedigree structure up to the grandparents' generation, but SOLAR was not able to handle that due to the complexity of the pedigree.
Figure 4.—

Results of example II. Rows from top to bottom correspond to our method, SOLAR, and TASSEL, respectively. The two true QTL are marked with “*” and the posterior probabilities that the marked intervals contain at least one QTL are shown for our method.
The association analysis with relatedness estimates incorporated into the linear model was not able to catch the true QTL positions either. Moreover, since the data include close relatives, association analyses that test a single marker at a time can be expected to produce some amount of false positives regardless of the correction.
DISCUSSION
In this article we have extended our earlier model for marker data-based pedigree and gene flow estimation to also account for a quantitative phenotype via a variance component approach. In the model, the phenotypic variance is decomposed into a random number of QTL effects, a polygenic effect, and a residual. The structures of the covariance matrices for QTL effects are determined by the expected IBD sharing at the QTL positions, given the IBD sharing at the flanking marker loci, and the polygenic covariance structure is dictated by the pedigree configuration. This approach is targeted at the settings where the sampled individuals are likely to be related to each other within a few of the most recent generations, but where their exact relationships are not known. If more specific knowledge of the relationships were available, it would also be possible to fix the known parts of the pedigree. Thus the traditional variance component linkage analysis that operates only on the fixed pedigree becomes a special case of our framework.
Our approach is applicable to diploid species and can adapt to several (nonrandom) mating scenarios as well as to user-specified marker maps. The practical usefulness of our method depends especially on the amount of available marker data and on the degree of relatedness between the sampled individuals.
Marker data:
In recent years the development of genotyping technologies has been extremely rapid, and therefore the gene mapping studies in humans are currently carried out with hundreds of thousands of single-nucleotide polymorphisms (SNPs) distributed over the genome. It is not computationally possible to handle such data at once by the approach taken in this article. However, a stepwise strategy, where linkage analysis is carried out first, and then association analysis is employed conditionally on the results of the preliminary linkage analysis, is possible, especially in population isolates. Such a strategy is also the motivation behind the methods that detect associations in the presence of linkage signals (Cantor et al. 2005). It could also be possible to treat several tightly linked SNPs as a single multiallelic locus, if credible haplotype information were available. In that case the linkage between the combined SNPs should be so tight that it would be feasible to assume that no recombinations have occurred within the SNP blocks during the most recent generations of the history of the sample. This would allow us to pick a sparser and computationally more tractable marker map from the original large SNP panel, while still maintaining some of the information carried by the polymorphic haplotype blocks.
Relatedness:
In our approach the detection of QTL is based on the correlations between the allele sharing and phenotypic similarities among the sampled individuals. Thus, to identify the QTL, it is necessary that the sampled individuals share ancestors within the estimated pedigree. Furthermore, close relatives such as siblings and cousins provide a valuable source of information concerning the transmission of the alleles between the generations. In contrast, the genetic material of the sampled individuals who are isolated from the rest of the pedigree may spread out arbitrarily among the ancestors, because there is no haplotype information coming from genotyped relatives. Hence, our approach is likely to be most useful in settings where the sampled individuals are closely related but the exact pedigree records are not available or not reliable. Such cases could be encountered, for example, in some wild animal populations. Indeed, a recent topic of general interest in such a context is the estimation of the relatedness structure or of the pedigree of the sampled individuals (Frentiu et al. 2008; Pemberton 2008). The approach introduced here not only provides a means to estimate the relatedness structure, but also offers a simultaneous analysis of a quantitative trait.
Phenotype model:
In this article we considered only additive genetic effects, but if required, our phenotype model could be easily extended to include environmental covariates as well as genetic interaction effects. For example, inclusion of dominance effects would require estimating whether each pair of the sampled individuals shares two alleles IBD at the QTL or a polygene and would be a technically straightforward addition to the current MCMC algorithm. However, identifiability problems (for the polygene) due to small sample sizes are common already with known pedigrees (Miztal 1997; Waldmann et al. 2008), and when also a pedigree is estimated, such problems are likely to increase. The lack of a known pedigree structure together with relatively small sample sizes may also be a reason for the relatively large estimates for the additive polygenic component in our examples, [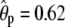 and
and 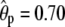 in examples I and II (T = 4), respectively]. It is likely that the polygenic component has here captured some of the variation due to the QTL and to the residual variance. To estimate it more accurately, we would need larger sample and family sizes. For example, Yi and Xu (2000) assumed in their examples that the pedigrees of 500 full-sib families with six siblings in each were known, compared to our settings of 50 full-sib families with two to three siblings in each and without a known pedigree structure. The important thing in our examples was that we were able to get good estimates of the relative effects of the QTL.
in examples I and II (T = 4), respectively]. It is likely that the polygenic component has here captured some of the variation due to the QTL and to the residual variance. To estimate it more accurately, we would need larger sample and family sizes. For example, Yi and Xu (2000) assumed in their examples that the pedigrees of 500 full-sib families with six siblings in each were known, compared to our settings of 50 full-sib families with two to three siblings in each and without a known pedigree structure. The important thing in our examples was that we were able to get good estimates of the relative effects of the QTL.
MCMC algorithm:
The proposal distributions that modify the pedigree and the gene flow at the marker loci are the same as in our previous article (Gasbarra et al. 2007a), while the corresponding acceptance ratios have been updated to take into account the phenotype model. The main addition was the updates for the phenotype parameters, implemented with the reversible-jump MCMC (RJMCMC) methodology. In recent QTL literature, RJMCMC algorithms have been accused of being complicated and slowly mixing, and other approaches to model selection have been considered (e.g., Wang et al. 2005). On the other hand, in a recent comparison by O'Hara and Sillanpää (2009) RJMCMC was found to provide a competent alternative to other Bayesian model selection methods. Here updating of the phenotype parameters did not notably slow down the sampler, as most of the time is still spent on the computationally demanding block updates for the pedigree and the gene flow at the marker loci. Furthermore, our phenotype updates were speeded up by integrating out the residual variance and the exact inheritance paths at the QTL loci. We also note that in our variance component model the effect of each QTL is characterized by a single (relative) variance value. This may further facilitate the mixing of our algorithm compared to the models that estimate the absolute effects of all possible QTL alleles/genotypes (e.g., Wang et al. 2005).
During the example analyses we found that a good initial state for the multigeneration pedigree model can be created sequentially, one generation at a time. The idea is that first the algorithm is run for a certain number of iterations on the state space of t-generation pedigrees, and then the final configuration of that run is augmented to a (t + 1)-generation configuration. By repeating this procedure for t = 1, … , T − 1, an initial state containing T ancestral generations can be created. This strategy seemed to yield more realistic initial states than our previous practice (Gasbarra et al. 2007a), which sampled a pedigree and allelic paths from the youngest generation to the founder generation in one go. A natural explanation for this improvement is that a multigeneration pedigree with a gene flow imposes strong dependencies between the variables, and therefore it is better to resample and improve the current configuration locally before it is extended to the next generation.
Another way to improve the mixing of the sampler could be an application of parallel computation, e.g., in the form of Metropolis-coupled Markov chain Monte Carlo (MCMCMC) (Geyer 1991), where a number of processors running in parallel would execute separate MCMC algorithms, of which only one would correspond exactly to the target distribution. The recombination likelihoods of the other MCMC samplers would then have different degrees of relaxation, represented by a temperature parameter. The chains with higher temperature values would pay less attention to the recombination likelihoods and as a result would explore the configuration space more freely. At certain points of time the chains at the adjacent temperature values would communicate and possibly switch their temperature values according to the Metropolis–Hastings rule. The final results would be collected only from the coldest chain, i.e., from the chain with the original target distribution.
Conclusion:
Our experiences with the method reported here suggest that a joint estimation of the recent relatedness structure and of the locations of quantitative trait loci is not only feasible but also advantageous in certain situations compared to other approaches that are not able to model the inheritance process using estimated pedigrees. The practical usefulness of this method depends, in a complex way, on several factors such as the degree of relatedness of the sampled individuals, the genetic architecture of the trait, and the effect sizes of the QTL and needs to be considered separately in any particular situation. An ability to analyze these kinds of complex models helps in filling the gap between the frameworks of linkage and association analyses and as such deserves further development.
Acknowledgments
We are thankful to two anonymous reviewers for their comments that helped us to improve the manuscript. This work was supported by grant nos. 50178, 202324, and 53297 (Centre of Population Genetic Analyses) from the Academy of Finland and by the ComBi Graduate School (M.P.).
APPENDIX A: EXACT RECURSIVE COMPUTATION OF THE CONDITIONAL COVARIANCES AT THE QTL GIVEN THE ALLELIC PATHS AT THE FLANKING MARKER LOCI
As usual, we denote by ω a configuration that describes the pedigree together with the paths and types of the (ancestral) alleles at marker loci. We numerate the haplotypes in the pedigree, starting from the founder generation. Let ψi(l) ∈ {0, 1} be the grandparental origin of the allele at marker locus l on haplotype i, with the convention that ψi(l) = 0 when the allele is inherited from the grandmother and ψi(l) = 1 if from the grandfather.
Let 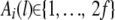 be the founder allele of haplotype i; that is, Ai(l) = k if the allele at position l of haplotype i is inherited from the kth founder haplotype. Suppose that a candidate QTL is located at position l + Δ between markers l and l′ (l < l + Δ < l′).
be the founder allele of haplotype i; that is, Ai(l) = k if the allele at position l of haplotype i is inherited from the kth founder haplotype. Suppose that a candidate QTL is located at position l + Δ between markers l and l′ (l < l + Δ < l′).
If the ith haplotype belongs to the founder generation,
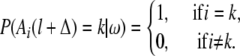 |
Otherwise, haplotype i is formed by recombining the haplotypes gf and gm transmitted from a grandfather and a grandmother, respectively. Then the conditional distribution of the grandparental origin at the locus (l + Δ), given the flanking markers, is
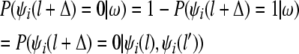 |
and according to the Haldane recombination model
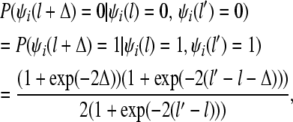 |
and
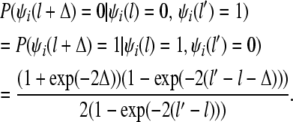 |
Using these conditional recombination probabilities we find the recursive formulas for the conditional IBD probabilities of nonfounder haplotypes:
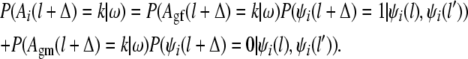 |
Since we follow only the paths of the ancestral alleles, it is possible that the grandparental origins of the ith haplotype are not known at the flanking markers of the QTL. Then in the formulas above, l and l′ are the closest markers on the left and on the right of the candidate QTL at which the configuration ω determines the grandparental origins. In the case that there are not any ancestral alleles on the left (or right) side of the QTL, the corresponding grandparental origin probabilities are set equal to  .
.
Since we want to compute conditional genetic covariances between individuals, we use the same idea to compute the joint conditional distribution of the IBD indicators on two haplotypes. Again the recursion starts from the founder generation, where the IBD indicators are fully determined.
If i and i′ are two separate haplotypes obtained by recombining the haplotypes gf, gm and gf′, gm′, respectively, then
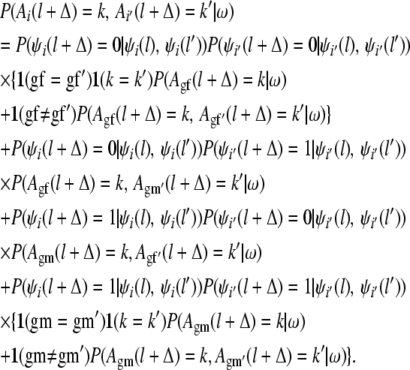 |
Here 1(x = y) is the indicator that equals 1, if x = y, and 0 otherwise.
Using the above formulas, the expectation of the conditional covariance between individuals I and J at QTL q given the allele paths is calculated as
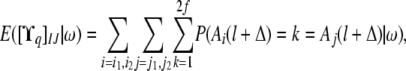 |
where i1 and i2 are the haplotypes of I and j1 and j2 are the haplotypes of J.
APPENDIX B: ANALYTIC INTEGRATION OF σr2 FROM THE LIKELIHOOD
With the notation introduced in the variance component model for the phenotypes section we assume that 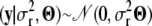 and that
and that 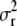 ∼ IG(a, d). Then
∼ IG(a, d). Then
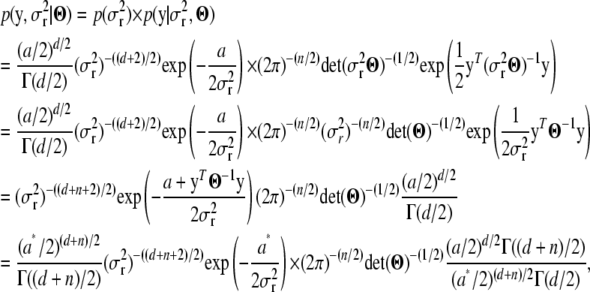 |
where 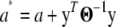 . Now the term left of the multiplication symbol is the density of the IG(a*, d + n) distribution (as the function of
. Now the term left of the multiplication symbol is the density of the IG(a*, d + n) distribution (as the function of 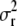 ), whence its integral (with respect to
), whence its integral (with respect to 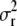 ) is 1, and only the term on the right side remains. That is,
) is 1, and only the term on the right side remains. That is,
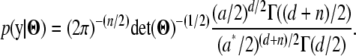 |
References
- Almasy, L., and J. Blangero, 1998. Multipoint quantitative trait linkage analysis in general pedigrees. Am. J. Hum. Genet. 62: 1198–1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bink, M. C. A. M., A. D. Anderson, W. E. van de Weg and E. A. Thompson, 2008. Comparison of marker-based pairwise relatedness estimators on a pedigreed plant population. Theor. Appl. Genet. 117: 843–855. [DOI] [PubMed] [Google Scholar]
- Bradbury, P. J., Z. Zhang, D. E. Kroon, T. M. Casstevens, Y. Ramdoss et al., 2007. TASSEL: software for association mapping of complex traits in diverse samples. Bioinformatics 23: 2633–2635. [DOI] [PubMed] [Google Scholar]
- Cantor, R. M., G. K. Chen, P. Pajukanta and K. Lange, 2005. Association testing in a linked region using large pedigrees. Am. J. Hum. Genet. 76: 538–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frentiu, F. D., S. M. Clegg, J. Chittock, T. Burke, M. W. Blows et al., 2008. Pedigree-free animal models: the relatedness matrix reloaded. Proc. R. Soc. Lond. Ser. B Biol. Sci. 275: 639–647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasbarra, D., M. J. Sillanpää and E. Arjas, 2005. Backward simulation of ancestors of sampled individuals. Theor. Popul. Biol. 67: 75–83. [DOI] [PubMed] [Google Scholar]
- Gasbarra, D., M. Pirinen, M. J. Sillanpää and E. Arjas, 2007. a Estimating genealogies from linked marker data: a Bayesian approach. BMC Bioinformatics 8: 411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasbarra, D., M. Pirinen, M. J. Sillanpää, E. Salmela and E. Arjas, 2007. b Estimating genealogies from unlinked marker data: a Bayesian approach. Theor. Popul. Biol. 72: 305–322. [DOI] [PubMed] [Google Scholar]
- Geyer, C. J., 1991. Markov chain Monte Carlo maximum likelihood, pp. 156–163 in Computing Science and Statistics: Proceedings of the 23rd Symposium on the Interface. Interface Foundation, Fairfax, VA.
- Green, P. J., 1995. Reversible jump Markov chain Monte Carlo computation and Bayesian model determination. Biometrika 82: 711–732. [Google Scholar]
- Heath, S. C., 1997. Markov chain Monte Carlo segregation and linkage analysis for oligogenic models. Am. J. Hum. Genet. 61: 748–760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lange, K., 2002. Mathematical and Statistical Methods for Genetic Analysis. Springer, New York.
- Larribe, F., S. Lessard and N. J. Schork, 2002. Gene mapping via the ancestral recombination graph. Theor. Popul. Biol. 62: 215–229. [DOI] [PubMed] [Google Scholar]
- Lynch, M., and K. Ritland, 1999. Estimation of pairwise relatedness with molecular markers. Genetics 152: 1753–1766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meuwissen, T. H. E., and M. E. Goddard, 2004. Mapping multiple QTL using linkage disequilibrium and linkage analysis information and multitrait data. Genet. Sel. Evol. 36: 261–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meuwissen, T. H. E., and M. E. Goddard, 2007. Multipoint identity-by-descent prediction using dense markers to map quantitative trait loci and estimate effective population size. Genetics 176: 2551–2560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minichiello, M. J., and R. Durbin, 2006. Mapping trait loci by use of inferred ancestral recombination graphs. Am. J. Hum. Genet. 79: 910–922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miztal, I., 1997. Estimation of variance components with large-scale dominant models. J. Dairy Sci. 80: 965–974. [Google Scholar]
- Morris, A. P., J. C. Whittaker and D. J. Balding, 2002. Fine-scale mapping of disease loci via shattered coalescent modeling of genealogies. Am. J. Hum. Genet. 70: 686–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Hara, R. B., and M. J. Sillanpää, 2009. A review of Bayesian variable selection methods: what, how and which. Bayesian Anal. 4: 85–118. [Google Scholar]
- Pemberton, J. M., 2008. Wild pedigrees: the way forward. Proc. R. Soc. Lond. Ser. B Biol. Sci. 275: 613–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sillanpää, M. J., and E. Arjas, 1998. Bayesian mapping of multiple quantitative trait loci from incomplete inbred line cross data. Genetics 148: 1373–1388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waldmann, P., J. Hallander, F. Hoti and M. J. Sillanpää, 2008. Efficient Markov chain Monte Carlo implementation of Bayesian analysis of additive and dominance genetic variances in noninbred pedigrees. Genetics 179: 1101–1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, H., Y. M. Zhang, X. Li, G. L. Masinde, S. Mohan et al., 2005. Bayesian shrinkage estimation of quantitative trait loci parameters. Genetics 170: 465–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi, N., and S. Xu, 2000. Bayesian mapping of quantitative trait loci under the identity-by-descent-based variance component model. Genetics 156: 411–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu, J., G. Pressoir, W. H. Briggs, I. Vroh Bi, M. Yamasaki et al., 2006. A unified mixed-model method for association mapping that accounts for multiple levels of relatedness. Nat. Genet. 38: 203–208. [DOI] [PubMed] [Google Scholar]
- Zöllner, S., and J. K. Pritchard, 2005. Coalescent-based association mapping and fine mapping of complex trait loci. Genetics 169: 1071–1092. [DOI] [PMC free article] [PubMed] [Google Scholar]