

Abstract
The p62/sequestosome 1 protein has been identified as a component of pathological protein inclusions in neurodegenerative diseases including amyotrophic lateral sclerosis (ALS). P62 has also been implicated in autophagy, a process of mass degradation of intracellular proteins and organelles. Autophagy is a critical pathway for degrading misfolded and/or damaged proteins, including the copper-zinc superoxide dismutase (SOD1) mutants linked to familial ALS. We previously reported that p62 interacted with ALS mutants of SOD1 and that the ubiquitin-association (UBA) domain of p62 was dispensable for the interaction. In this study, we identified two distinct regions of p62 that were essential to its binding to mutant SOD1: the N-terminal PB1 domain (residues 1-104) and a separate internal region (residues 178–224) termed here as SOD1 mutant interaction region (SMIR). The PB1 domain is required for appropriate oligomeric status of p62 and the SMIR is the actual region interacting with mutant SOD1. Within the SMIR, the conserved W184, H190 and positively charged R183, R186, K187 and K189 residues are critical to the p62-mutant SOD1 interaction since substitution of these residues with alanine resulted in significantly abolished binding. In addition, SMIR and the p62 sequence responsible for the interaction with LC3, a protein essential for autophagy activation, are independent of each other. In cells lacking p62, the existence of mutant SOD1 in acidic autolysosomes decreased, suggesting that p62 can function as an adaptor between mutant SOD1 and the autophagy machinery. This study provides a novel molecular mechanism by which mutant SOD1 can be recognized by p62 in an ubiquitin-independent fashion and targeted for the autophagy-lysosome degradation pathway.
Keywords: Sequestosome 1/p62, familial ALS, mutant SOD1, autophagy
Introduction
Sequestosome 1/p62 is a conserved multifunctional protein that is mainly involved in either cellular signaling or protein degradation and aggregation (Moscat et al. 2007, Seibenhener et al. 2007). The diverse functions of p62 are reflected by its domain structure. The N-terminal PB1 domain heterodimerizes with other PB1 domains and can also form homodimers and homooligomers (Wilson et al. 2003, Lamark et al. 2003). The ZZ-type zinc finger mediates the interaction with RIPK1 (Sanz et al. 1999). The LC3 interaction region (LIR) can directly interact with LC3, a protein essential to autophagosome formation (Pankiv et al. 2007). The C-terminal ubiquitin association (UBA) domain is responsible for ubiquitin binding (Vadlamudi et al. 1996, Ciani et al. 2003, Seibenhener et al. 2004).
The p62 protein was identified as a common component of cytoplasmic inclusions in protein aggregation diseases. It was found in the intracellular protein inclusions in ALS (Mizuno et al. 2006, Parkinson et al. 2006, Gal et al. 2007), other neurodegenerative disorders (Kuusisto et al. 2001, Zatloukal et al. 2002), as well as liver diseases (Denk et al. 2006, Zatloukal et al. 2002) and muscle disorders (Janue et al. 2007). It was recently reported that the Drosophila p62 ortholog was required for forming protein inclusions in the adult fly brain (Nezis et al. 2008).
The p62 protein was implicated in both major protein degradation pathways: autophagy and the ubiquitin-proteasome system. It can act as a shuttling factor to the proteasome (Babu et al. 2005, Seibenhener et al. 2004). The p62 protein also interacts directly with LC3 and facilitates the degradation of ubiquitinated protein aggregates by autophagy (Pankiv et al. 2007). Depletion of p62 inhibited the recruitment of LC3 to autophagosomes (Bjorkoy et al. 2005). Whereas it is generally supposed that the interaction between polyubiquitinated protein aggregates and p62 is mediated by the polyubiquitin-UBA domain interaction, the autophagic substrate recognition mechanism is not fully understood.
A subset of the familial form of ALS is caused by inherited mutations in the Cu/Zn superoxide dismutase 1 (SOD1) gene (Rosen et al. 1993, Valentine et al. 2005). The SOD1 protein can be degraded by both proteasome and autophagy (Di Noto et al. 2005, Kabuta et al. 2006). Several ALS mutants of SOD1 are more heavily polyubiquitinated than the WT protein (Urushitani et al. 2002). While the degradation of mutant SOD1 by the ubiquitin-proteasome system has been studied more thoroughly, the mechanism of targeting of mutant SOD1 to the autophagy machinery is largely unexplored.
We have previously reported that p62 preferentially interacted with the A4V and G93A ALS SOD1 mutants (Gal et al. 2007). Surprisingly, the truncated p62 lacking the UBA domain was still able to bind mutant SOD1, suggesting that an alternative pathway for autophagic substrate recognition may exist. We strive to investigate the nature of the UBA-independent interaction in this study. The results hereby demonstrate that two non-contiguous regions of p62 are necessary for an efficient p62-mutant SOD1 interaction. The N-terminal PB1 domain is needed to ensure oligomerization of p62, whereas an internal region spanning residues 178 to 224 forms the actual SOD1 mutant interaction region hereby termed as “SMIR”. The interactions between p62 and mutant SOD1, LC3 and polyubiquitin chains are mediated by the SMIR, LIR and UBA domains, respectively. In addition to facilitating the sequestration of mutant SOD1 into inclusions (Gal et al. 2007, Strom et al. 2008), the results suggest that p62 could target mutant SOD1 to autophagy and proteasome pathways in both ubiquitin-dependent and independent fashions.
Materials and methods
Plasmid construction
The p62 constructs were based on the MGC-5968 mouse p62 clone (ATCC). The SOD1 plasmids were constructed as described in (Zhang & Zhu 2006). The mCherry-GFP vector was a gift from Dr. Geir Bjorkoy at University of Tromso (Pankiv et al. 2007). More details on plasmid construction and a list of all constructs used in this study (Table S1) can be found in Supplemental Data.
Cell culture and transfection
The HEK293 and NSC34 cells were cultured in DMEM medium supplemented with 10% FBS and penicillin/streptomycin in a 37°C incubator in 5% CO2. The wild-type and p62 knockout mouse embryonic fibroblast (MEF) cells were a gift from Dr. Masaaki Komatsu at Tokyo Metropolitan Institute of Medical Science and were cultured as described in (Komatsu et al. 2007). Cells were transfected in a 6-well format using the Lipofectamine transfection reagent.
Primary mouse motor neuron culture
Primary mouse motor neurons were purified and cultured following a published protocol (Gingras et al. 2007). Briefly, spinal cords were dissected from embryonic day 13 C57B6 mice, and dissociated by trituration. Motor neurons were purified by differential centrifugation using a step gradient of NycoPrep (Accurate Chemical) solutions. The motor neurons were then plated on poly-lysine treated glass coverslips and incubated at 37°C,5% CO2 in growth-factor supplemented medium. The cells were transfected 4 days after plating using Lipofectamine LTX.
Immunoprecipitation and Western blotting
The cells were harvested 2 days after transfection, washed with 1X PBS, and lysed in 1X RIPA buffer supplemented with protease inhibitor cocktail (P-8340, Sigma), 0.625 mg/ml N-ethylmaleimide (Sigma), 1 mM sodium orthovanadate (Sigma), 10μM MG132 (Calbiochem) and 0.2 mM PMSF (Sigma). The WT SOD1 and G93A mutant SOD1 transgenic mice were sacrificed and the spinal cords were dissected as previously described (Zhang et al. 2007). All animal procedures were approved by the university IACUC committee. The mouse spinal cord extracts were prepared with the above buffer using a dounce homogenizer.
The HA immunoprecipitations were performed in 500μl final volume containing 1 mg protein extract using 2μg mouse monoclonal anti-HA antibody (Santa Cruz, sc-7392) and Protein-G Sepharose (GE Healthcare, 17-0618-01). The p62 immunoprecipitations were performed using 2 μg mouse monoclonal anti-p62 antibody (Abnova, H00008878-M01) and Protein-G Sepharose. The FLAG immunoprecipitations were performed using anti-FLAG M2 Affinity Gel (Sigma, F2426). The IP samples and the corresponding extracts were subjected to SDS-PAGE followed by Western blotting using the following antibodies: anti-HA (Santa Cruz, sc-7392 and sc-805), anti-FLAG (Sigma, F3165, F7425 and A8592), anti-actin (Santa Cruz, sc-1616), anti-DsRed (Clontech, 632496), anti-GFP (Santa Cruz, sc-8334), anti-LC3 (MBL, PM036), and anti-p62 (Santa Cruz, sc-25575 and Abnova, H00008878-M01). The bands were visualized using ECL reagents (Pierce).
Native gel electrophoresis
Cellular extracts were prepared in RIPA buffer supplemented with protease inhibitors as above. The extracts were mixed with native sample buffer (Biorad) and 35 μg total protein of each sample was electrophoresed on 4–20% ReadyGel Tris-HCl gradient gels using non-denaturing TRIS-glycine running buffer (Biorad). The gels were incubated in the transfer buffer supplemented with 0.25% SDS at 70oC for 10 min before transfer.
Fluorescence microscopy
For the scoring of SOD1-p62 co-aggregation, NSC34 cells were transfected with SOD1-GFP and DsRedM-p62 constructs in 6-well plates. The cells were inspected 24 hours after transfection using a Zeiss Axiovert 100 microscope. The number of doubly transfected cells and cells with DsRed-positive and/or GFP-positive inclusions were counted in ten random viewfields with approximately 30 transfected cells per viewfield. Statistical analysis was carried out using Student’s t-test.
For confocal microscopy, primary mouse motor neurons and NSC34 cells were cultured on sterile coverslips and transfected with SOD1-GFP and DsRedM-p62 constructs. Three days (primary motor neurons) or 24 hours (NSC34 cells) after transfection, the cells were fixed using 4% (w/v) paraformaldehyde in PBS at room temperature. After washing with PBS, the coverslips were mounted on slides with Vectashield mounting medium (Vector Laboratories) containing DAPI (Invitrogen) to visualize the cell nuclei. Fluorescence microscopy was carried out using a Leica DM IRBE confocal microscope with a 100x objective.
Live cell imaging of WT and p62 knockout MEF cells was performed using a Leica SP5 inverted confocal microscope equipped with an environmental chamber. The mCherry-GFP-SOD1 plasmids were transfected into MEF cells in 35mm poly-D-lysine coated glass-bottom culture dishes (MatTek, P35GC-1.5-14-C). Cells were imaged 24 hours after transfection using a 100x objective and images in both green and red channels were acquired. Image analysis was performed using the ImageJ program (http://rsb.info.nih.gov/ij/). Details regarding quantification of overlapping mCherry and GFP signals are described in the Supplemental Data.
Results
Interaction between p62 and ALS-linked SOD1 mutants
We previously reported the accumulation of p62 and co-localization of p62 with mutant SOD1 in G93A mutant transgenic mouse spinal cord motor neurons (Gal et al. 2007). We hereby tested the interaction of endogenous p62 with G93A mutant SOD1 in the ALS mouse model. As shown in Figure 1A, G93A mutant SOD1 was co-immunoprecipitated with endogenous p62 from the spinal cord extract of G93A SOD1 transgenic mouse (lane 2). In contrast, WT SOD1 was not co-precipitated with endogenous p62 from spinal cord extract of WT SOD1 transgenic mouse (lane 1). In addition, we tested the interaction between p62 and multiple ALS-linked SOD1 mutants in cultured cells. As shown in Figure S1, four different SOD1 mutants A4V, G37R, G85R and G93A were all co-immunoprecipitated with p62 (lanes 2–5) whereas WT SOD1 was not (lane 1). No co-precipitation was detected in four controls (lanes 6–9). All SOD1 proteins were abundant at similar levels in the post-immunoprecipitation supernatants and cell extracts, eliminating the possibility of artifacts arising from different protein levels. Consistent with the published co-localization data obtained from ALS mice (Gal et al. 2007), the tight co-localization of p62 and mutant SOD1 in intracellular inclusions was also observed in primary mouse motor neurons transfected with DsRedM-p62 and GFP-tagged A4V SOD1 (Figure 1B). In primary motor neurons transfected with WT SOD1, no SOD1 inclusions were observed while p62 self-aggregates were evident. Low magnification images showing the morphology of the primary motor neurons are shown in Figure S2. The interaction and co-localization results from transgenic animals, primary motor neurons and cultured cells consistently support the interaction between p62 and the ALS-linked SOD1 mutants.
Figure 1. The interaction and co-localization between p62 and the ALS-linked SOD1 mutants.

(A) The interaction between endogenous p62 and mutant SOD1 in G93A SOD1 transgenic mouse spinal cord. Protein extracts were prepared from spinal cords dissected from 125-days old WT and G93A SOD1 transgenic mice and subjected to p62 immunoprecipitation followed by Western blotting using specified antibodies. (B). Co-localization of p62 and mutant SOD1 in primary mouse motor neurons. Primary motor neurons were prepared, cultured, transfected with DsRedM-p62 and WT or A4V mutant SOD1-EGFP, and subjected to confocal microscopic analysis. The scale bars are 10 μm.
An internal region of p62 is responsible for ALS mutant SOD1 binding
Since the UBA domain within p62 was not required for the p62-mutant SOD1 interaction, we started searching for the sequences responsible for the interaction. The structure of known functional domains of the p62 protein and the exon structure of the corresponding mouse p62 gene are shown in Figure 2A. The full-length (FL) p62 and several domain deletion constructs were generated to map the region(s) of p62 necessary for the binding of SOD1 mutants associated with familial ALS. The deletion constructs were co-transfected with HA-tagged A4V SOD1 into HEK293 cells and HA-SOD1 immunoprecipitations were performed. As shown in Figure 2B, deletion of the ZZ-type zinc finger (ΔZZ, lane 3), UBA domain (ΔUBA, lane 8), or LIR (ΔLIR, lane 7) had no effect on the p62-mutant SOD1 interaction. However, deletion of the PB1 domain (ΔPB1, lane 2), the sequences encoded by exon 4 (residues S178-S224) (Δexon4, lane 4), exon 5 (residues A225-G252) (Δexon5, lane 5), or exons 4 and 5 (Δexon4-5, lane 6) all resulted in abolished or significantly reduced p62 co-precipitated with A4V SOD1. These data suggest that the PB1 domain and the sequences encoded by exons 4 and 5 are critical to the p62-mutant SOD1 interaction.
Figure 2. Mapping the p62 domains necessary for the interaction with A4V SOD1.

(A) The domain structure of the p62 protein and the exon structure of the mouse p62 gene. (B). The DsRedM-tagged full-length and domain deletion p62 mutants were co-transfected with HA-tagged A4V mutant SOD1. HA immunoprecipitation followed by Western blotting was performed to test which domains were required for the interaction. Deletion of the PB1 domain, exon 4 or exon 5 resulted in impaired binding of A4V SOD1 to p62. * denotes the antibody light chain in the immunoprecipitation samples. (C). HA-SOD1 immunoprecipitation was similarly carried out as in (B). The PB1 domain alone (lane 1) or PB1 and ZZ-type zinc finger together (lane 2) were not co-precipitated with A4V SOD1. The construct containing both PB1 domain and exon 4 (lane 3) was the minimal sequence required for interaction. (D). The interaction was tested by reciprocal FLAG-p62 immunoprecipitation. The protein expression levels in the cell lysates were examined in all experiments. The antibody used is specified beside each panel.
A successive series of C-terminal deletion constructs, all containing the PB1 domain, was tested (Figure 2C). The PB1 domain alone (lane 1) or PB1 along with the ZZ-type zinc finger (exons 1–3, lane 2) did not interact with A4V SOD1. Inclusion of exon 4 (exons 1-4, lane 3) and exon 5 (exons 1–5, lane 4) resulted in regained interaction with A4V SOD1. The ΔUBA (lane 5) and FL (lane 6) p62 were positive controls. The results from Figure 2B and 2C suggest that the PB1 domain is necessary but not sufficient for the p62-A4V interaction. Along with the PB1 domain, the sequence encoded by exons 4 and 5 is likely to be the domain responsible for the interaction. The role of the PB1 domain will be addressed in the next section.
In the reciprocal p62 immunoprecipitation experiments (Figure 2D), A4V SOD1 was co-precipitated with FL p62 (lane 5) while WT SOD1 was not co-precipitated with any p62 constructs tested (lanes 1–4). It was verified that the p62 construct lacking exons 4 and 5 ( exons4–5, lane 6) lost its ability to interact with A4V SOD1. Consistent with the earlier results, deletion of LIR (ΔLIR, lane 7) or the UBA domain (ΔUBA, lane 8) did not impair the interaction.
The role of the PB1 domain in the p62-A4V SOD1 interaction
The PB1 domain has been shown to be responsible for homooligomerization of the p62 protein (Lamark et al. 2003, Wilson et al. 2003). We hypothesized that the PB1 domain might be necessary to ensure the oligomerization of p62 to support its interaction with mutant SOD1. We have utilized two tools to test this hypothesis. First, the K7A/D69A double mutation in the PB1 domain of p62, which was shown to suppress the ability of p62 to oligomerize significantly (Lamark et al. 2003), was used to test if it would impair the p62-mutant SOD1 interaction. Second, we used monomeric (pDsRed-Monomer-C1, Clontech) and tetrameric (pDsRed2-C1, Clontech) versions of the DsRed fluorescent protein tag (designated as DsRedM and DsRedT) (Strongin et al. 2007) to manipulate the oligomeric status of the DsRed-tagged fusion proteins. As shown in Figure 3A, monomeric DsRedM-tagged FL p62 (lane 1) was co-precipitated with A4V SOD1 whereas DsRedM-tagged K7A/D69A double mutant p62 (lane 2) was barely co-precipitated with A4V SOD1. Consistent with results shown in Figure 2, the DsRedM tagged ΔPB1 (lane 3) and ΔPB1ΔUBA (lane 4) deletion mutants were not co-precipitated with A4VSOD1. In contrast, when the ΔPB1 (lane 7) and ΔPB1ΔUBA (lane 8) deletion mutants were tagged with tetrameric DsRedT, they were co-precipitated with A4V SOD1. A very recent study reported an additional PB1 domain mutant R21A/D69A that impaired the oligomerization of p62 (Kirkin et al. 2009). The DsRedM-tagged R21A/D69A mutant p62 was also unable to bind A4V SOD1 (Figure 3B). The results suggest that the PB1 domain facilitates oligomerization of p62 to enable the interaction with mutant SOD1.
Figure 3. The role of the PB1 domain in the interaction.

(A) The indicated p62 constructs, tagged with either the monomeric (DsRedM) or the tetrameric (DsRedT) DsRed were co-transfected with HA-tagged A4V SOD1. The cell extracts were subjected to HA immunoprecipitation followed by Western blotting. (B). The DsRedM tagged WT, K7A/D69A or R21A/D69A p62 construct was co-transfected with HA-tagged A4V SOD1. The cell extracts were subjected to HA immunoprecipitation followed by Western blotting. (C). Demonstration of the oligomerization status of selected DsRed-p62 constructs by native gel electrophoresis followed by Western blotting. A slower band (*) and a faster band (●) in native gel electrophoresis indicate an oligomeric and a monomeric form, respectively. The panels of the DsRed blot of the native gel were obtained with 5 sec (short) and 40 sec (long) exposition times of the same membrane. The protein expression levels in extracts were assessed in all experiments. The antibody used in each experiment is specified beside each panel.
To assess the effect of the K7A/D69A and R21A/D69A mutations on the oligomerization of p62 and the effect of the DsRedM and DsRedT tags on the oligomerization of the PB1-deleted p62 constructs, native polyacrylamide gel electrophoresis analysis was performed (Figure 3C). The DsRedM-tagged FL p62 migrated largely as a slower band (*) in native gel electrophoresis, indicating that DsRedM-FL-p62 mainly existed as an oligomeric form. In contrast, DsRedM-tagged K7A/D69A and R21A/D69A mutants largely migrated as the faster-migrating band (bull;), suggesting the mutants were dominantly monomeric. DsRedM-tagged ΔPB1 also migrated clearly as a faster monomeric form, whereas DsRedT-tagged ΔPB1 migrated as a slower oligomeric form. The protein expression levels were examined by SDS-PAGE followed by Western blotting. A longer exposure time was used to visualize the DsRedT-tagged ΔPB1-p62 in the native gel electrophoresis analysis since its expression level was lower. It is known that deletion of the PB1 domain results in monomeric p62, thus the results support that the DsRedM-and DsRedT-tagging strategy can effectively manipulate the oligomeric status of ΔPB1-p62.
Identification of the SOD1 Mutant Interaction Region
To validate that the sequence encoded by exons 4 and 5 in p62 is the region that interacts with A4V SOD1, DsRedM- and DsRedT-tagged exon(4–5) were generated. As shown in Figure 4A, DsRedT-exon(4-5) was co-precipitated with A4V SOD1 (lane 4) whereas DsRedM-exon(4–5) was not(lane 1), although the expression level of DsRedT-exon(4-5) was much lower than that of DsRedM-exon(4-5). The DsRedM-tagged FL p62 (lane 2) was included as a positive control. Native gel electrophoresis showed that DsRedT-exon(4-5) migrated as a slower band whereas DsRedM-exon(4-5) migrated as a faster band (Figure S3), consistent with the earlier results in Figure 3C. The results support that the sequence encoded by exons 4 and 5 along with the tetrameric DsRedT was sufficient to interact with mutant SOD1.
Figure 4. Identification of the SOD1 mutant interaction region (SMIR).

(A) The HA-tagged A4V SOD1 interacted with the segment of p62 encoded by exons 4 and 5 only when it was tagged with the tetrameric DsRedT, but not the monomeric DsRedM. The indicated DsRed-tagged p62 constructs were co-transfected with HA-tagged A4V SOD1, and cellular extracts were subjected to HA immunoprecipitation. (B). The HA-tagged A4V SOD1 interacted with the DsRedT-exon 4, but not DsRedT-exon 5. The sequence encoded by exon 4 was termed as the SOD1 mutant interaction region (SMIR). (C). The DsRedT-tagged SMIR was co-immunoprecipitated with multiple ALS-linked SOD1 mutants (A4V, G37R, G85R and G93A) tagged with HA, but not with WT SOD1.
We further tested whether both exons 4 and 5 are required for interaction with mutant SOD1 (Figure 4B). The DsRedT-tagged exon(4-5) interacted with A4V SOD1 (lane 2) but not with WT SOD1 (lane 1). When the exon 4 and exon 5 was individually tagged with DsRedT, DsRedT-exon4 (lane 3) co-precipitated with A4V SOD1 but DsRedT-exon5 (lane 4) did not. In the domain deletion experiments (Figure 2B), deletion of exon 4 completely abolished the p62-mutant SOD1 interaction whereas deletion of exon 5 significantly suppressed but did not completely abolish the interaction. The results suggest that the sequence encoded by exon 4 is the true region that is essential to the p62-mutant SOD1 interaction. Thus, the region encoded by exon 4 is designated as the SOD1 mutant interaction region (SMIR).
To confirm that the SMIR can interact with multiple ALS-linked SOD1 mutants, co-immunoprecipitation of DsRedT-SMIR and HA-tagged WT and mutant SOD1 was performed. DsRedT-SMIR was co-precipitated with all SOD1 mutants tested (A4V, G37R, G85R and G93A) but not with WT SOD1 (Figure 4C). The results are consistent with Figure S1 that shows the interaction between full-length p62 and the same set of SOD1 mutants.
SMIR residues crucial for the mutant SOD1 interaction
We tested which residues in SMIR are crucial for interaction with mutant SOD1. The alignment of the SMIR sequence from different organisms shows that the SMIR sequence is highly conserved among mammalians but shows more variety in lower vertebrates (Figure 5A). A conserved cluster of basic residues are in the beginning of SMIR: R183, R186, K187 and K189 (positions based on mouse and human p62). There are six tryptophan residues in mouse p62, four of which are clustered in SMIR, with W184 being the most conserved. A nearby histidine residue H190 is also conserved in all organisms.
Figure 5. Critical residues in SMIR crucial for p62-mutant SOD1 interaction.

(A) The alignment of the SMIR sequence of p62 from different organisms. The conserved cluster of positively charged residues (R183, R186, K187, K189) were indicated by “+” and the conserved W184 and H190 residues were also noted. (B). DsRedM-tagged p62 (WT, W184A, H190A or R183A/R186A/K187A/K189A) was co-transfected with HA-tagged A4V SOD1, and the extracts were subjected to HA immunoprecipitation.
Two single amino acid mutants (W184A and H190A) and a quadruple mutant (R183A/R186A/K187A/K189A, AAAA) were generated to evaluate the significance of the conserved residues in the binding of mutant SOD1. Immunoprecipitation studies (Figure 5B) showed that W184A (lane 1) and H190A (lane 2) significantly suppressed the p62-mutant SOD1 interaction, whereas the AAAA mutant nearly abolished the interaction completely (lane 3). WT p62 (lane 5) and Δexon4-p62 (lane 4) were included as positive and negative controls, respectively. The results demonstrated the importance of the conserved residues in SMIR to the p62-mutant SOD1 interaction.
The effect of the SMIR deletion on the inclusion forming properties of p62
We previously reported that p62 sequestered mutant, but not WT SOD1 into inclusions (Gal et al. 2007). It has been shown that the PB1 and UBA domains are responsible for the formation of the p62-positive inclusions (Bjorkoy et al. 2005) and that the UBA domain is responsible for recognizing polyubiquitinated proteins (Vadlamudi et al. 1996, Ciani et al. 2003, Seibenhener et al. 2004). In light of the discovery of the SMIR in this study, we examined how the SMIR deletion would impact the inclusion formation of p62 and the sequestration of A4V SOD1-GFP into the inclusions in NSC34 cells. As previously published (Gal et al. 2007), the percentages of cells containing DsRed positive p62 inclusions, GFP positive SOD1 inclusions and both inclusions were counted simultaneously using separate filters. As shown in Figure 6, the SMIR deletion did not impair the ability of p62 to form inclusions itself. Full-length p62 significantly enhanced A4V SOD1-GFP inclusion formation compared to the DsRedM vector control (p = 1.73x10−4). However, ΔSMIR-p62 had no effect on A4V SOD1 inclusion formation compared to the vector control (p = 0.333). Moreover, compared to FL p62, significantly less SOD1 inclusions formed when ΔSMIR-p62 was co-expressed with A4V SOD1 (p = 3.78x10−4).
Figure 6. The effect of the SMIR and LIR deletion on mutant SOD1 sequestration into p62 inclusions.

DsRedM-p62, DsRedM-p62-ΔSMIR, DsRedM-p62-ΔLIR or DsRedM vector and GFP-tagged A4V mutant SOD1 were co-transfected into NSC34 cells. The inclusion formation was measured by simultaneously monitoring the GFP-positive SOD1 inclusions and DsRed-positive p62 inclusions under fluorescence microscope. Cells containing DsRed, GFP, and both inclusions were counted in 10 random viewfields. The percentage of cells containing inclusions out of the transfected cells was calculated and the average values from three independent experiments are shown.
The expression levels of p62 and ΔSMIR-p62 were comparable (Figure S4A). Confocal microscopic analysis (Figure S4B) showed that DsRedM-FL p62 and A4V SOD1 almost always formed tight co-inclusions, but the DsRedM-ΔSMIR p62 inclusions and A4V SOD1 were often clearly separate. The results support that the deletion of SMIR significantly impaired the ability of p62 to sequester A4V SOD1 into inclusions.
p62 as an adaptor between mutant SOD1 and the autophagy machinery
Autophagy is critical for the degradation of inclusions of aggregated proteins. The LC3 protein is essential to autophagy activation, and p62 was reported to directly bind LC3 (Pankiv et al. 2007). The SMIR responsible for binding with mutant SOD1 is distinct from the reported LIR (Pankiv et al. 2007), thus it is likely that the binding of A4V SOD1 and LC3 by p62 are independent. To confirm this, HA-tagged A4V SOD1, GFP-tagged LC3 and FLAG-tagged p62 (FL, ΔSMIR orΔLIR) were co-transfected into HEK293 cells. Upon FLAG-p62 immunoprecipitation, both A4V SOD1 and LC3 were co-precipitated with FL p62 (Figure 7A, lane 1). However, LC3 was co-precipitated with ΔSMIR-p62, but A4V SOD1 was not (lane 2). Conversely, A4V SOD1 was co-precipitated with ΔLIR-p62, but LC3 was not (lane 3). The results demonstrate that binding of p62 to mutant SOD1 and LC3 are independent of each other and that SMIR and LIR are responsible for interaction with SOD1 and LC3, respectively.
Figure 7. p62 as an adaptor between mutant SOD1 and autophagy.

(A) The p62-LC3 and p62-mutant SOD1 interactions are independent. HA-A4V SOD1, GFP-LC3 and FLAG-tagged FL p62, ΔSMIR-p62, ΔLIR-p62 or FLAG vector were co-transfected, and cellular extracts were subjected to FLAG-p62 immunoprecipitation followed by Western blotting using specified antibodies. (B). The acidification of mCherry-GFP-A4V SOD1 inclusions is impaired in p62 KO MEF cells. The mCherry-GFP-A4V SOD1 plasmid was transfected into WT or p62 KO MEF cells. Live cell imaging was performed 24 hours post transfection. The boxed areas of the entire cell images are enlarged to the right. Scare bars are 10 μm. (C). Quantification of the overlap rate of green and red puncta in the transfected WT and p62 KO MEF cells. A higher overlap rate in the p62 KO cells (p < 0.05) indicated less amount of mutant SOD1 in acidic autolysosomes.
The effect of LIR domain deletion on inclusion formation was examined. As shown in Figure 6, the percentage of cells containing SOD1 inclusions in the presence of ΔLIR-p62 was the same as in the presence of FL p62. The confocal images also showed that DsRedM-ΔLIR-p62 and A4V SOD1-GFP almost always formed tight co-inclusions (Figure S4B). The results support that, unlike SMIR, LIR has little effect on mutant SOD1 suquestration. Thus, LIR and SMIR are two regions in p62 that have distinct and independent functions.
Furthermore, we tested the hypothesis that p62 is a potential adaptor between mutant SOD1 and LC3. When DsRedM-p62 was overexpressed in NSC34 cells whose endogenous p62 level is relatively low, compared to the DsRedM vector control, greater amount of LC3 was co-precipitated by mutant SOD1 (Figure S5). The data support that p62 can be an adaptor between mutant SOD1 and LC3, linking mutant SOD1 to the autophagy machinery.
Moreover, we tested whether the loss of p62 would reduce the existence of mutant SOD1 in acidic autophagic vesicles and lysosomes. The mCherry-GFP double fluorescent tag enables the in vivo monitoring of pH of subcellular compartments since mCherry is acid stable whereas GFP is acid sensitive (Pankiv et al. 2007). We constructed an mCherry-GFP-A4V SOD1 fusion protein and expected that the A4V SOD1 puncta were observed in both green (GFP) and red (mCherry) channels unless they existed in the low pH environment of autolysosomes. Representative confocal microscopic images of mCherry-GFP-A4V SOD1 are shown in Figure 7B. In WT MEF cells, in addition to yellow puncta, a number of red puncta were evidently within “holes” where the green signal was quenched. These red A4V SOD1 aggregates were likely to exist in acidic autolysosomes. In contrast, the puncta in the p62 KO MEF cells were largely yellow, suggesting that they were outside acidic autolysosomes. We quantified the percentage of overlapping green and red puncta in WT and p62 KO MEF cells using the ImageJ program (Figure 7C). The total numbers of pixels of A4V SOD1 puncta observed in the mCherry channel were comparable in WT and p62 KO cells. However, the percentage of overlapping pixels in mCherry and GFP channels increased in p62 KO MEF cells compared to the WT cells. The results suggest that less mCherry-GFP-A4V SOD1 existed in the acidic environment of autolysosomes in the p62 KO cells, supporting that p62 can function as an adaptor between mutant SOD1 and the autophagy-lysosome degradation pathway.
Discussion
The p62 protein has been reported to be present in protein inclusions in a wide range of pathological conditions including neurodegenerative diseases (Kuusisto et al. 2001, Mizuno et al. 2006, Parkinson et al. 2006, Zatloukal et al. 2002). In addition, p62 has been linked to both of the two major protein degradation mechanisms, the ubiquitin-proteasome system and autophagy. The UBA domain at the C-terminus of p62 has been reported to recognize polyubiquitinated proteins and shuttle them to the proteasome (Ciani et al. 2003, Seibenhener et al. 2004, Babu et al. 2005). The p62 protein is a prototype autophagic receptor and facilitates autophagic degradation of aggregated proteins since it can directly interact with LC3 (Pankiv et al. 2007). We previously reported the co-localization of p62 and G93A mutant SOD1 in ALS mouse spinal cord motor neurons (Gal et al. 2007). In this study, we demonstrated the interaction between endogenous p62 and G93A mutant SOD1, but not WT SOD1, in transgenic mouse spinal cord homogenates (Figure 1A). The interaction was also consistently observed between p62 and four SOD1 mutants in cultured cells (Figure S1). Moreover, p62 was co-localized with A4V SOD1 inclusions in primary motor neurons (Figure 1B). The results support the significance of p62 and its interaction with mutant SOD1 in ALS. Deletion of the UBA domain in p62 did not abolish the p62-mutant SOD1 interaction (Gal et al. 2007), suggesting the possibility of a novel ubiquitin-independent mechanism for the interaction. This study is to elucidate the ubiquitin-independent mechanism and the significance of p62 in the degradation and aggregation of mutant SOD1.
The systematic p62 domain deletion analysis led to the identification of two independent, non-contiguous regions involved in mutant SOD1 binding: the N-terminal PB1 domain and an internal segment encoded by exon 4 that we termed as SOD1 mutant interaction region (SMIR). The PB1 domain alone did not bind A4V SOD1, suggesting that the PB1 domain is not where mutant SOD1 interacts with p62. Instead, the PB1 domain is needed to ensure the oligomerization of p62, which appears to be required for the p62-mutant SOD1 interaction. This is consistent with previous reports that the PB1 domain is responsible for the formation of p62 homo-oligomers (Lamark et al. 2003, Wilson et al. 2003). Replacement of the PB1 domain with the monomeric DsRedM tag led to the loss of A4V SOD1 binding. Likewise, K7A/D69A or R21A/D69A double mutation of p62, which have been shown to impair PB1 oligomerization (Lamark et al. 2003, Kirkin et al. 2009), significantly weakened the binding of A4V SOD1 (Figures 3A and 3B). However, chimaeric proteins in which the PB1 domain was replaced with the tetrameric DsRedT retained A4V SOD1 binding (Figure 3A).
The DsRedT-tagged SMIR was co-precipitated with four different ALS-linked SOD1 mutants (A4V, G37R, G85R and G93A, see Figure 4C) as the full-length p62 did (Figure S1), supporting that SMIR is the region within p62 that mutant SOD1 interacts with. Within the SMIR sequence, mutations introduced at conserved residues (W184A and H190A) and removal of a conserved cluster of basic residues (R183A/R186A/K187A/K189A) all significantly impaired the p62-A4V SOD1 interaction, supporting that the region is indeed pivotal for the association (Figure 5B). It is however unknown whether these residues are directly implicated in the interaction, or if they are needed to maintain the proper structure to support the association with mutant SOD1. It also remains to be further elucidated whether p62 directly interacts with mutant SOD1, or yet unknown protein(s) may function as adaptor(s) between p62 and mutant SOD1. Nevertheless, SMIR is the region within p62 that either mutant SOD1 or the yet-unknown adaptor will directly interact with. In addition, it is unclear what molecular trait of mutant SOD1 (for example, misfolding or oxidative damage) leads to its association with p62. The questions will be addressed with direct binding assays using purified proteins in future studies.
The domains flanking the SMIR, the ZZ-type zinc finger and the domain encoded by exon 5 of p62 were also examined. The ZZ-type zinc finger was not needed for the interaction asΔZZ-p62 was co-precipitated with A4V SOD1. Exon 5 appeared to play a role in the interactionsince Δexon5-p62 showed reduced co-precipitation with A4V SOD1 (Figure 2B). However, DsRedT-tagged exon 5 was not co-precipitated with A4V SOD1 (Figure 4B). It is possible that the exon 5 domain might: (a) be necessary for the proper folding of the exon 4 segment, or (b) play a role as a spacer between different p62 domains in the context of the full-length protein.
We reported that p62 facilitated the sequestration of the familial ALS-related SOD1 mutants into inclusions (Gal et al. 2007). In this study, we showed that deletion of the SMIR did not impair the formation of p62 inclusions (Figure 6). The results are consistent with the report that the PB1 UBA domains are the two major determinants of p62 inclusion formation (Bjorkoy et al. 2005). Moreover, due to the lack of the site of mutant SOD1 interaction, ΔSMIR-p62 lost the capability of sequestering mutant SOD1 into inclusions (Figures 6 and S4). The results demonstrate that the SMIR is functionally separate from the PB1 and UBA domains that are directly involved in p62 inclusion formation.
Autophagy enables the large-scale degradation of cytoplasmic proteins and organelles (Mizushima et al. 2008), but the autophagy targeting mechanisms are far from being fully understood in eukaryotic cells,. Here we report that the binding sites for mutant SOD1 (SMIR) and LC3 (LIR) are clearly distinct regions within p62. The co-immunoprecipitation studies show that the binding of mutant SOD1 and LC3 by p62 are independent (Figure 7A). Over-expression of exogenous p62 in NSC34 cells enhanced the association between mutant SOD1 and LC3 (Figure S5). The results establish a link between mutant SOD1 and LC3 with p62 as a potential adaptor. In fact, the p62 protein level was found to be higher in the G93A transgenic mice in our previously published study (Gal et al. 2007). In addition, SOD1 has been reported to be degraded by both the proteasome and autophagy (Kabuta et al. 2006). The live cell image analysis of WT and p62 KO MEF cells using double fluorescently labeled mCherry-GFP-A4V SOD1 showed that lower percentage of A4V SOD1 puncta existed in autolysosomes in the p62 KO cells (Figure 7B and 7C). The results support the hypothesis that p62 can be the protein to recognize mutant SOD1 and target it to the autophagy machinery as illustrated in Figure 8.
Figure 8. Proposed model of how p62 may target mutant SOD1 aggregates to autophagy.
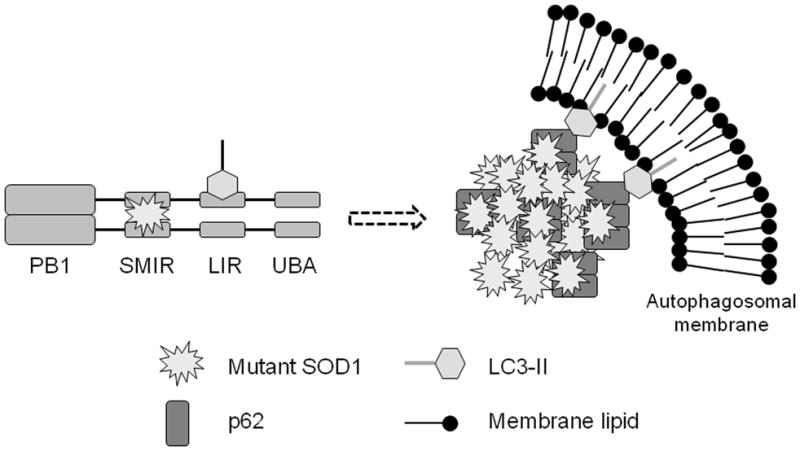
LC3-II represents the phosphatidylethanolamine-conjugated form of LC3 and is associated with the autophagosomal membrane. Misfolded mutant SOD1 is recognized by p62, sequestered to form inclusions, and targeted to autophagy.
As discussed earlier, p62 over-expression can facilitate sequestering mutant SOD1 to inclusions (Figure 6). On the other hand, p62 can also target mutant SOD1 inclusions to autophagosomes for degradation. Thus, p62 appears to play a role in both the formation and degradation of mutant SOD1 inclusions. Previous studies have also suggested that p62 was required for both the formation of polyubiquitin-positive inclusions and the degradation of such inclusions by the autophagy-lysosome pathway (Pankiv et al. 2007, Wooten et al. 2008, Komatsu et al. 2007, Tan et al. 2008). Thus, deletion of p62 may decrease both formation and degradation of mutant SOD1 inclusions, which does not necessarily result in more or less accumulation of mutant SOD1 inclusions. It is noted that the accumulation of A4V SOD1 puncta in the mCherry channel was comparable in WT and p62 KO cells in Figure 7B. However, it is evident that less mutant SOD1 puncta were observed in the acidic autolysosomes in the p62 KO cells compared to p62 WT cells. The results are consistent with the notion that p62 can contribute to both formation and degradation of mutant SOD1 inclusions.
The ubiquitin-proteasome system is also a major mechanism for degrading misfolded proteins. ALS mutant SOD1 has been reported to cause the suppression of proteasome activity (Kabashi et al. 2008). In cultured HEK293 cells overexpressing mutant SOD1, the catalytic activity of proteasomes was slightly higher than that in cells overexpressing WT SOD1 (data not shown). It is conceivable that misfolding-prone mutant SOD1 may induce proteasome activity in the short term, thus proteasome inhibition is less likely in the experimental system in this study. It is noted that measuring the catalytic activity without considering regulatory subunits of proteasome likely has little relevance to the potential role of proteasome in the disease. Transgenic UbG76V-GFP proteasome activity reporter mouse is a valuable tool to assess the overall functionality of the ubiquitin-proteasome system (including contributions of all catalytic and regulatory subunits of proteasomes) in vivo (Lindsten et al. 2003). A recent study using the UbG76V-GFP proteasome activity reporter mice found only mild proteasome impairment in motor neurons in the symptomatic ALS mice (Cheroni et al. 2009). The findings support the significance of studying autophagy in degradation and aggregation of mutant SOD1.
The relationship between mutant SOD1 polyubiquitination, aggregation and autophagy is unclear. The findings from this study provide clear evidence that the p62-mutant SOD1 interaction is mediated by the SMIR sequence in p62. This interaction may serve as a novel mechanism by which mutant SOD1 is recognized by p62 without the UBA domain or polyubiquitin chains. The significance of the findings is substantiated by the evidence that this interaction can contribute to both the sequestration of mutant SOD1 to inclusions (Figure 6) and the targeting of mutant SOD1 to the autophagy-lysosome pathway (Figure 7). It has been reported that certain inclusions can be degraded by autophagy whereas others are autophagy-resistant (Wong et al. 2008). Future studies will address remaining critical questions including how p62 would influence the mutant SOD1 protein degradation and aggregation, how autophagy would modulate the toxicity of mutant SOD1 and motor neuron degeneration in ALS.
Supplementary Material
Acknowledgments
We thank Dr. Geir Bjorkoy for the pDest-mCherry-GFP plasmid and Dr. Masaaki Komatsu for the WT and p62 KO MEF cells. This study was in part supported by the NIH grants R01NS049126 and R21AG032567 to HZ. The support from NIH/NCRR COBRE program (P20RR020171) and NIH/NIEHS Superfund Basic Research Program (P42ES007380) are acknowledged.
Abbreviations
- UBA
ubiquitin association domain
- LIR
LC3 interaction region
- SMIR
SOD1 mutant interaction region
- SOD1
Cu/Zn superoxide dismutase 1
- ALS
amyotrophic lateral sclerosis
- MEF
mouse embryonic fibroblast
- WT
wild-type
- FL
full-length
- KO
knockout
- SDS
sodium dodecyl sulfate
- PAGE
polyacrylamide gel electrophoresis
- GFP
green fluorescent protein
- DsRedM and DsRedT
monomeric and tetrameric red fluorescent protein, respectively
References
- Babu JR, Geetha T, Wooten MW. Sequestosome 1/p62 shuttles polyubiquitinated tau for proteasomal degradation. J Neurochem. 2005;94:192–203. doi: 10.1111/j.1471-4159.2005.03181.x. [DOI] [PubMed] [Google Scholar]
- Bjorkoy G, Lamark T, Brech A, Outzen H, Perander M, Overvatn A, Stenmark H, Johansen T. p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J Cell Biol. 2005;171:603–614. doi: 10.1083/jcb.200507002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheroni C, Marino M, Tortarolo M, et al. Functional alterations of the ubiquitin proteasome system in motor neurons of a mouse model of familial Amyotrophic Lateral Sclerosis. Hum Mol Genet. 2009;18:82–96. doi: 10.1093/hmg/ddn319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciani B, Layfield R, Cavey JR, Sheppard PW, Searle MS. Structure of the ubiquitin-associated domain of p62 (SQSTM1) and implications for mutations that cause Paget's disease of bone. J Biol Chem. 2003;278:37409–37412. doi: 10.1074/jbc.M307416200. [DOI] [PubMed] [Google Scholar]
- Denk H, Stumptner C, Fuchsbichler A, Muller T, Farr G, Muller W, Terracciano L, Zatloukal K. Are the Mallory bodies and intracellular hyaline bodies in neoplastic and non-neoplastic hepatocytes related? J Pathol. 2006;208:653–661. doi: 10.1002/path.1946. [DOI] [PubMed] [Google Scholar]
- Di Noto L, Whitson LJ, Cao X, Hart PJ, Levine RL. Proteasomal degradation of mutant superoxide dismutases linked to amyotrophic lateral sclerosis. J Biol Chem. 2005;280:39907–39913. doi: 10.1074/jbc.M506247200. [DOI] [PubMed] [Google Scholar]
- Gal J, Strom AL, Kilty R, Zhang F, Zhu H. p62 accumulates and enhances aggregate formation in model systems of familial amyotrophic lateral sclerosis. J Biol Chem. 2007;282:11068–11077. doi: 10.1074/jbc.M608787200. [DOI] [PubMed] [Google Scholar]
- Gingras M, Gagnon V, Minotti S, Durham HD, Berthod F. Optimized protocols for isolation of primary motor neurons, astrocytes and microglia from embryonic mouse spinal cord. J Neurosci Methods. 2007;163:111–118. doi: 10.1016/j.jneumeth.2007.02.024. [DOI] [PubMed] [Google Scholar]
- Janue A, Olive M, Ferrer I. Oxidative stress in desminopathies and myotilinopathies: a link between oxidative damage and abnormal protein aggregation. Brain Pathol. 2007;17:377–388. doi: 10.1111/j.1750-3639.2007.00087.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabashi E, Agar JN, Hong Y, Taylor DM, Minotti S, Figlewicz DA, Durham HD. Proteasomes remain intact, but show early focal alteration in their composition in a mouse model of amyotrophic lateral sclerosis. J Neurochem. 2008 doi: 10.1111/j.1471-4159.2008.05317.x. in press. [DOI] [PubMed] [Google Scholar]
- Kabuta T, Suzuki Y, Wada K. Degradation of amyotrophic lateral sclerosis-linked mutant Cu, Zn-superoxide dismutase proteins by macroautophagy and the proteasome. J Biol Chem. 2006;281:30524–30533. doi: 10.1074/jbc.M603337200. [DOI] [PubMed] [Google Scholar]
- Kirkin V, Lamark T, Sou YS, et al. A role for NBR1 in autophagosomal degradation of ubiquitinated substrates. Mol Cell. 2009;33:505–516. doi: 10.1016/j.molcel.2009.01.020. [DOI] [PubMed] [Google Scholar]
- Komatsu M, Waguri S, Koike M, et al. Homeostatic levels of p62 control cytoplasmic inclusion body formation in autophagy-deficient mice. Cell. 2007;131:1149–1163. doi: 10.1016/j.cell.2007.10.035. [DOI] [PubMed] [Google Scholar]
- Kuusisto E, Salminen A, Alafuzoff I. Ubiquitin-binding protein p62 is present in neuronal and glial inclusions in human tauopathies and synucleinopathies. Neuroreport. 2001;12:2085–2090. doi: 10.1097/00001756-200107200-00009. [DOI] [PubMed] [Google Scholar]
- Lamark T, Perander M, Outzen H, Kristiansen K, Overvatn A, Michaelsen E, Bjorkoy G, Johansen T. Interaction codes within the family of mammalian Phox and Bem1p domain-containing proteins. J Biol Chem. 2003;278:34568–34581. doi: 10.1074/jbc.M303221200. [DOI] [PubMed] [Google Scholar]
- Lindsten K, Menendez-Benito V, Masucci MG, Dantuma NP. A transgenic mouse model of the ubiquitin/proteasome system. Nat Biotechnol. 2003;21:897–902. doi: 10.1038/nbt851. [DOI] [PubMed] [Google Scholar]
- Mizuno Y, Amari M, Takatama M, Aizawa H, Mihara B, Okamoto K. Immunoreactivities of p62, an ubiqutin-binding protein, in the spinal anterior horn cells of patients with amyotrophic lateral sclerosis. J Neurol Sci. 2006;249:13–18. doi: 10.1016/j.jns.2006.05.060. [DOI] [PubMed] [Google Scholar]
- Mizushima N, Levine B, Cuervo AM, Klionsky DJ. Autophagy fights disease through cellular self-digestion. Nature. 2008;451:1069–1075. doi: 10.1038/nature06639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moscat J, Diaz-Meco MT, Wooten MW. Signal integration and diversification through the p62 scaffold protein. Trends Biochem Sci. 2007;32:95–100. doi: 10.1016/j.tibs.2006.12.002. [DOI] [PubMed] [Google Scholar]
- Nezis IP, Simonsen A, Sagona AP, Finley K, Gaumer S, Contamine D, Rusten TE, Stenmark H, Brech A. Ref(2)P, the Drosophila melanogaster homologue of mammalian p62, is required for the formation of protein aggregates in adult brain. J Cell Biol. 2008;180:1065–1071. doi: 10.1083/jcb.200711108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pankiv S, Clausen TH, Lamark T, Brech A, Bruun JA, Outzen H, Overvatn A, Bjorkoy G, Johansen T. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem. 2007;282:24131–24145. doi: 10.1074/jbc.M702824200. [DOI] [PubMed] [Google Scholar]
- Parkinson N, Ince PG, Smith MO, et al. ALS phenotypes with mutations in CHMP2B (charged multivesicular body protein 2B) Neurology. 2006;67:1074–1077. doi: 10.1212/01.wnl.0000231510.89311.8b. [DOI] [PubMed] [Google Scholar]
- Rosen DR, Siddique T, Patterson D, et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature. 1993;362:59–62. doi: 10.1038/362059a0. [DOI] [PubMed] [Google Scholar]
- Sanz L, Sanchez P, Lallena MJ, Diaz-Meco MT, Moscat J. The interaction of p62 with RIP links the atypical PKCs to NF-kappaB activation. Embo J. 1999;18:3044–3053. doi: 10.1093/emboj/18.11.3044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seibenhener ML, Babu JR, Geetha T, Wong HC, Krishna NR, Wooten MW. Sequestosome 1/p62 is a polyubiquitin chain binding protein involved in ubiquitin proteasome degradation. Mol Cell Biol. 2004;24:8055–8068. doi: 10.1128/MCB.24.18.8055-8068.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seibenhener ML, Geetha T, Wooten MW. Sequestosome 1/p62--more than just a scaffold. FEBS Lett. 2007;581:175–179. doi: 10.1016/j.febslet.2006.12.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strom AL, Shi P, Zhang F, Gal J, Kilty R, Hayward LJ, Zhu H. Interaction of ALS-related mutant copper-zinc superoxide dismutase with the dynein-dynactin complex contributes to inclusion formation. J Biol Chem. 2008 doi: 10.1074/jbc.M800276200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strongin DE, Bevis B, Khuong N, Downing ME, Strack RL, Sundaram K, Glick BS, Keenan RJ. Structural rearrangements near the chromophore influence the maturation speed and brightness of DsRed variants. Protein Eng Des Sel. 2007;20:525–534. doi: 10.1093/protein/gzm046. [DOI] [PubMed] [Google Scholar]
- Tan JM, Wong ES, Kirkpatrick DS, et al. Lysine 63-linked ubiquitination promotes the formation and autophagic clearance of protein inclusions associated with neurodegenerative diseases. Hum Mol Genet. 2008;17:431–439. doi: 10.1093/hmg/ddm320. [DOI] [PubMed] [Google Scholar]
- Urushitani M, Kurisu J, Tsukita K, Takahashi R. Proteasomal inhibition by misfolded mutant superoxide dismutase 1 induces selective motor neuron death in familial amyotrophic lateral sclerosis. J Neurochem. 2002;83:1030–1042. doi: 10.1046/j.1471-4159.2002.01211.x. [DOI] [PubMed] [Google Scholar]
- Vadlamudi RK, Joung I, Strominger JL, Shin J. p62, a phosphotyrosine-independent ligand of the SH2 domain of p56lck, belongs to a new class of ubiquitin-binding proteins. J Biol Chem. 1996;271:20235–20237. doi: 10.1074/jbc.271.34.20235. [DOI] [PubMed] [Google Scholar]
- Valentine JS, Doucette PA, Zittin Potter S. Copper-zinc superoxide dismutase and amyotrophic lateral sclerosis. Annu Rev Biochem. 2005;74:563–593. doi: 10.1146/annurev.biochem.72.121801.161647. [DOI] [PubMed] [Google Scholar]
- Wilson MI, Gill DJ, Perisic O, Quinn MT, Williams RL. PB1 domain-mediated heterodimerization in NADPH oxidase and signaling complexes of atypical protein kinase C with Par6 and p62. Mol Cell. 2003;12:39–50. doi: 10.1016/s1097-2765(03)00246-6. [DOI] [PubMed] [Google Scholar]
- Wong ESP, Tan JMM, Soong WE, Hussein K, Nukina N, Dawson VL, Dawson TM, Cuervo AM, Lim KL. Autophagy-mediated clearance of aggresomes is not a universal phenomenon. Hum Mol Genet. 2008;17:2570–2582. doi: 10.1093/hmg/ddn157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wooten MW, Geetha T, Babu JR, Seibenhener ML, Peng J, Cox N, Diaz-Meco MT, Moscat J. Essential role of sequestosome 1/p62 in regulating accumulation of Lys63-ubiquitinated proteins. J Biol Chem. 2008;283:6783–6789. doi: 10.1074/jbc.M709496200. [DOI] [PubMed] [Google Scholar]
- Zatloukal K, Stumptner C, Fuchsbichler A, et al. p62 Is a common component of cytoplasmic inclusions in protein aggregation diseases. Am J Pathol. 2002;160:255–263. doi: 10.1016/S0002-9440(10)64369-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang F, Strom AL, Fukada K, Lee S, Hayward LJ, Zhu H. Interaction between familial amyotrophic lateral sclerosis (ALS)-linked SOD1 mutants and the dynein complex. J Biol Chem. 2007;282:16691–16699. doi: 10.1074/jbc.M609743200. [DOI] [PubMed] [Google Scholar]
- Zhang F, Zhu H. Intracellular conformational alterations of mutant SOD1 and the implications for fALS-associated SOD1 mutant induced motor neuron cell death. Biochim Biophys Acta. 2006;1760:404–414. doi: 10.1016/j.bbagen.2005.11.024. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.