

Abstract
AIM
Experimental pain models may help to evaluate the mechanisms of analgesics and target the clinical indications for their use. This review, the second in a series of two, addresses how the efficacy of non-opioid analgesics have been assessed in human volunteers using experimental pain models.
METHODS
A literature search was completed for randomized controlled studies that included human experimental pain models, healthy volunteers and non-opioid analgesics.
RESULTS
Nonsteroidal anti-inflammatory drugs worked against various types of acute pain as well as in hyperalgesia. Analgesia from paracetamol was difficult to detect in experimental pain and the pain needed to be assessed with very sensitive methods like evoked brain potentials. The N-methyl-D-aspartate antagonists exemplified by ketamine generally needed strong, long-lasting or repeated pain in the skin for detectable analgesia, whereas pain in muscle and viscera generally was more easily attenuated. Gabapentin worked well in several models, particularly those inducing hyperalgesia, whereas lamotrigine was weak in modulation of experimental pain. Imipramine attenuated pain in most experimental models, whereas amitriptyline had weaker effects. Delta-9-tetrahydrocannabinol attenuated pain in only a few models.
CONCLUSIONS
Pain induction and assessment are very important for the sensitivity of the pain models. Generally, experimental pain models need to be designed with careful consideration of the pharmacological mechanisms and pharmacokinetics of analgesics. The drawback with the different study designs is also discussed. This knowledge can aid the decisions that need to be taken when designing experimental pain studies for compounds entering Phase I and II trials.
Keywords: clinical trials, experimental pain, non-opioid analgesics
Introduction
The use of various analgesics is the prevailing treatment of pain. The clinical effects typically guide the selection of the analgesics and titration of the dose. However, when treating clinical pain analgesic effects are difficult to evaluate due to a number of confounding factors. These confounders may include variable baseline pain, complaints relating to psychological factors related to the illness, as well as systemic reactions such as fever and general malaise [1]. In assessing the efficacy of analgesics in clinical trials these confounders can bias the outcome.
Experimental pain models are without many of the above confounders and therefore a valuable tool for characterizing analgesics [2]. Using such models, the investigator can control the experimentally induced pain (including the nature, localization, intensity, frequency and duration of the stimulus), and provide quantitative measures of the psychophysical, behavioural or neurophysiological responses [1, 3]. Since such pain models activate only part of the pain system, application of these experimental pain models offers a unique opportunity for exploring the pain system. Reviews of the various models and analgesics exists, but this review presents a more thorough approach and contains updated information [4].
Hence, the aim was to characterize how various experimental models of acute pain and evoked hyperalgesia (see definitions below) detect analgesia of clinically used non-opioid analgesics. This can be divided into the following: (i) to investigate the sensitivity of various experimental models to test clinically used non-opioid analgesics and (ii) to investigate how the dose and dosing regimen can affect the findings. As the physiology of deep and superficial pain differs, the results will furthermore be divided into the tissue (skin, muscle or viscera) in where the pain was induced, and into modalities used for pain stimulation. For a more thorough introduction to human experimental pain models, see the first review in this series involving opioid analgesics [5].
Method
PubMed searches were conducted for articles and abstracts. MeSH and free-text terms for analgesics were combined with the terms ‘experimental pain’, ‘human’, and ‘randomized’. Analgesics included ‘opioids’, ‘nonsteroidal anti-inflammatory drugs’, ‘antidepressants’, ‘anticonvulsants’ and ‘NMDA antagonists’. Only manuscripts published in English were included. There was no limit to the time of publication. Furthermore, we did not feel that the level of evidence could be graded due to the exploratory nature of many of the studies. Some trials test combinations of analgesics, but to avoid too complex results we included only trials where the analgesic in question was tested alone in one of the treatment arms.
To be able to illustrate the importance of various experimental designs, we included only drugs that have been tested in at least five different trials. Trials involving experimental pain often use very small sample sizes because the variation of the outcome measures is less than in traditional clinical trials. Trials with fewer than 10–12 subjects are hard to test statistically and the findings therefore questionable. However, it has been shown that experimental models with a high reproducibility sample size <10 are powered to show the effect of analgesics [6]. Accordingly, we found a well-designed study with a sample size of seven, and this was the minimum sample size for the studies included in this review [7].
Results are summarized and discussed on a pharmacological mechanistic basis at the end of each drug class section.
Nonsteroidal anti-inflammatory drugs and paracetamol in experimental pain
Acetylsalicylic acid (aspirin)
Acetylsalicylic acid has been tested extensively in the past. Topical as well as systemic preparations have been tested (Table 1).
Table 1.
Schematic overview of studies involving acetysalicylic acid in human experimental pain models
| Reference | Dose | Model (pain assessments) | Main findings | |
|---|---|---|---|---|
| Acute models | [10] (n= 30) | Topical application (0.03–5%) | Intradermal continuous infusion of phosphate buffered solution (pH 5.2) VAS | Doses > 1.5% VAS↓ |
| [8] (n= 32) | 1000 mg p.o. | Intracutaneous electrical stimulation VAS, evoked brain potentials | VAS ↓, amplitudes of evoked potentials ↓ latencies of evoked potentials ↔ | |
| [13] (n= 14) | 1000 mg i.v. | Stimulation of nasal mucosa by CO2 VAS, evoked brain potentials, power spectrum | PDT ↔ | |
| Amplitudes and latencies of evoked potentials ↓, power spectrum changed | ||||
| [9] (n= 9) | 750 mg p.o. | Laser skin stimulation, evoked brain potentials | Amplitude ↓ | |
| [14] (n= 24) | 600 and 900 mg p.o. | Ischaemic pain AUCVAS, PTT | No parameters were affected | |
| [12] (n= 10) | 1200 mg p.o. | Electrical stimulation of dental pulp PDT | PDT ↑ | |
| Models inducing hyperalgesia | [16] (n= 60) | 900 mg p.o. | Delayed-onset muscle soreness VAS, McGill questionnaire, deep pressure PDT | No parameters were affected |
| [15] (n= 10) | Topical application (0.25 g ml−1) | Topically applied capsaicin, evoked pain, area of secondary HA to pinprick and heat and allodynia to light touch | Area of HA to pinprick and allodynia to light touch↓ | |
| Area of HA to heat ↔ | ||||
| [11] (n= 12) | 100 and 1500 mg p.o. | Long-lasting pinching of the interdigital web (two different intensities) PDT | PDT ↑ | |
| Only the highest intensity discriminated between doses |
In the column ‘Model’ the method for pain assessment is normal font, and the method for pain induction is bold. PDT, pain detection threshold; PTT, pain tolerance threshold; AUC, area under curve; VAS, visual analogue scale; HA, hyperalgesia.
Acute models
Skin/nasal and dental mucosa: Acetylsalicylic acid has been tested against pain from radiant heat, electrical stimulation, nasal gaseous CO2 stimulation and infusion of low-pH solutions [8–10]. Pain from repeated mechanical impact of the interdigital web is a classical model for detecting analgesia from nonsteroidal anti-inflammatory drugs (NSAIDs), and this model is also sensitive to analgesia from acetylsalicylic acid [11]. Topically applied acetylsalicylic acid worked against the tonic pain from cutaneous infusion of a low-pH solution [10]. Electrical pain evoked in skin or dental pulp was decreased by aspirin [8, 12]. Kobal et al. found only significant modulation of the evoked potentials, not the subjective pain ratings after nasal gaseous CO2 stimulation [13]. Supporting this, Schaffler et al. found good effect of acetylsalicylic acid on the evoked vertex potential from radiant heat stimulation [9].
Muscle: Since acetylsalicylic acid is used frequently for treatment of musculoskeletal pain, trials in muscle pain are very relevant. However, tonic muscle pain, such as ischaemic muscle pain in one study, was unaffected by acetylsalicylic acid when measured with various subjective ratings [14]. An explanation for this could be that the model did not induce prostaglandin production.
Models of hyperalgesia
Skin: Acetylsalicylic acid has not been tested extensively in hyperalgesic models. Allodynia and hyperalgesia from pinprick after capsaicin were diminished by acetylsalicylic acid, whereas heat hyperalgesia was unaffected [15].
Muscle: Acetylsalicylic acid did not affect delayed-onset muscle soreness when both intensity and unpleasantness were rated [16].
Dosing regimes
The therapeutic dose for weak pain is normally set to 100–500 mg, and in most of the experimental trials reviewed here doses within this range were used, although at the higher end of the dose range.
Ibuprofen
Ibuprofen has been extensively tested and is often used as a representative for weak NSAIDs. Topical as well as systemic preparations have been tested (Table 2).
Table 2.
Schematic overview of studies involving ibuprofen in human experimental pain models
| Reference | Dose | Model (pain assessments) | Main findings | |
|---|---|---|---|---|
| Acute models | [18] (n= 20) | 800 mg p.o. | Electrical stimulation (earlobe) PDT, PTT | PDT ↔, PTT ↑ (only for men) |
| [22] (n= 20) | 600 mg p.o. | Static pressure pain (interdigital web) VAS | VAS ↓ | |
| [10] (n= 30) | Topical application (5%) | Intradermal infusion of phosphate buffered solution (pH 5.2) AUCVAS | AUCVAS↓ | |
| [21] (n= 24) | 3 × 400 and 3 × 800 mg p.o. | Repeated pinching (interdigital web) VAS | VAS ↔ | |
| [20] (n= 18) | 400 and 800 mg p.o. | Stimulation of nasal mucosa by CO2 (phasic pain) and dry air (tonic pain), VAS, evoked brain potentials (only phasic pain) | Pain ratings to CO2 stimulation and dry air ↔ | |
| Evoked potentials ↓ (dose related manner) | ||||
| [17] (n= 8) | 600 mg p.o. | Electrical skin stimulation nociceptive reflex | Reflex threshold ↑ | |
| [23] (n= 10) | 400 and 800 mg p.o | Laser skin stimulation PDT | PDT↑ for both doses, no dose–response relation was seen | |
| [24] (n= 12) | 600 mg p.o. | Cold pressor test PDT, peak pain (VAS), AUCVAS | No parameters were affected | |
| [32] (n= 24) | 400 mg p.o. and topical 2 mg cm−2 | Pinprick PDT | PDT ↔ | |
| Models inducing hyperalgesia | [32] (n= 24) | 400 mg p.o. and topical 2 mg cm−2 | Freeze lesion: | p.o. treatment: PDT in primary HA area and secondary HA area ↑ |
| Area of primary and secondary HA: PDT to punctuate stimulation | Topical treatment: PDT in primary HA area ↑, PDT in secondary HA area ↔ | |||
| [34] (n= 34) | 800 mg p.o. | UVB radiation | Heat PDT and PTT ↑ | |
| Primary HA: | ||||
| Heat PDT and PTT | ||||
| [33] (n= 21) | 600 mg p.o. | HA to repetitive pinching to interdigital web VAS | Decrease of VAS (HA) to repetitive pinching was reduced | |
| UVB radiation | Heat PDT ↑ | |||
| HA evaluated by: heat PDT, nonpainful sensations and PDT to mechanical stimulation | Mechanical PDT ↑ | |||
| Nonpainful sensations unaffected | ||||
| [22] (n= 20) | 600 mg p.o. | Burn injury spontaneous pain (VAS during burn) | VAS during burn ↔ | |
| Area of secondary HA to pinprick and allodynia to stroking | Area of secondary HA to stroking and brushing ↔ | |||
| VAS to brushing in secondary HA area | VAS to brushing↓ | |||
| HA to static pinching to interdigital web VAS | VAS to static pinching ↓ | |||
| [21] (n= 24) | 3 × 400 mg p.o. and 3 × 800 mg p.o. at 2-h intervals | Freeze lesion | Mechanical HA after freeze lesion ↓ | |
| In area of primary HA: PDT to heat and mechanical stimuli | Thermal HA after freeze lesion ↔ | |||
| Topical capsaicin | Capsaicin induced allodynia ↔ | |||
| Allodynia to brushing | Increase in VAS (HA) from repetitive pinching was prevented | |||
| HA to repetitive pinching to interdigital web VAS | ||||
| [31] (n= 10) | 3 × 400 mg p.o. and 3 × topical application (5%) (for 3 days) | Post-exercise jaw-muscle soreness PDT and PTT to subsequent pressure algometry | PDT↓ (topical>systemic) | |
| PTT ↔ |
In the column ‘Model’ the method for pain assessment is normal font, and the method for pain induction is bold. PDT, pain detection threshold; PTT, pain tolerance threshold; AUC, area under curve; VAS, visual analogue scale; HA, hyperalgesia.
Acute models
Skin/nasal mucosa: Ibuprofen has been tested against various types of electrical pain and found to be effective. To be able to detect analgesia, electrical pain stimulation needs to be intense and evoke pain intensity well above the pain detection threshold [17, 18]. [19]. Here, as seen with other weak analgesics, only evoked potentials and not subjective pain ratings changed after drug administration [20]. Pain from repeated mechanical impact of the interdigital web was insensitive to ibuprofen, whereas pain was affected when the applied pressure was static [21, 22]. Pain from argon laser stimulation was also attenuated by ibuprofen. This could be because the repeated strong laser pulses caused sensitization of the skin [23].
Muscle: It is not known exactly from which tissues the pain from the cold pressor test arises, but the pain has a deep quality and hence this model is described in the section for models applied in the muscles. The cold pressor test has been tested with ibuprofen and found insensitive [24]. The test has a known sensitivity to many opioids, but is apparently not suitable for testing weak NSAIDs [25–30]. Eccentric jaw exercise caused muscle fatigue and low levels of post-exercise pain and soreness, which were attenuated by ibuprofen [31].
Models of hyperalgesia
Skin: Ibuprofen has been tested twice against hyperalgesia after freeze lesions. Topical application of ibuprofen showed an effect in the primary hyperalgesic area to pinprick (von Frey hair) demonstrating a local effect of the drug. Systemic administration decreased both primary and secondary hyperalgesia to pinprick, demonstrating both central and peripheral antihyperalgesic mechanisms of ibuprofen [32]. In the study by Kilo et al. the primary hyperalgesic area was tested with heat (thermode) and mechanical impact (metal cylinder) where only mechanical hyperalgesia was sensitive to ibuprofen analgesia [21, 32]. Capsaicin-induced allodynia to stroking was not affected by ibuprofen [21]. Repetitive pinching of the interdigital web has also been used in trials of ibuprofen and a good effect of the drug in preventing hyperalgesia was found [21, 33]. Ultraviolet B (UVB) radiation of the skin promotes hyperalgesia to heat and mechanical hyperalgesia, which was decreased by ibuprofen [33, 34]. This model slowly develops a mild skin inflammation producing hyperalgesic mechanisms of peripheral origin [33]. In the same category of pain models, the burn injury model has also been applied in the testing of ibuprofen, where pain to brushing but not to punctuate stimuli was reduced in the secondary hyperalgesic area [22].
Dosing regimes
The therapeutic dose for weak pain is 200–400 mg (three to four times daily); however, rheumatic pain needs doses ranging from 300 to 600 mg (three to four times daily). The experimental studies enrolled in this review applied supratherapeutic doses ranging from 400 to 800 mg. Two studies found equal effect of 400 and 800 mg in hyperalgesia, suggesting a plateau for the dose–response profile [21, 23]; however, another study applying the same doses found that evoked potentials responded in a dose-related manner [20]. Some studies have found a good sensitivity of evoked potentials compared with methods that apply subjective pain measures [8, 13, 35]. It could therefore be argued that the reason for the dose–response plateau seen by Kilo et al. and Nielsen et al. was due to a limited sensitivity of the methods [20, 21, 23].
Paracetamol (Table 3)
Table 3.
Schematic overview of studies involving paracetamol in human experimental pain models
| Reference | Dose | Model (pain assessment) | Findings | |
|---|---|---|---|---|
| Acute models | [39] (n= 18) | 1000 mg p.o. | Electrical skin and muscle stimulation, PTT | No parameters were affected |
| Deep pressure, PTT | ||||
| IM hypertonic saline AUCVAS | ||||
| [42] (n= 18) | 1000 mg p.o. | Cold pressor test | PTT↓ both before and after cold pressor test. Effect more pronounced after | |
| PTT to deep pressure before and after cold water immersion | ||||
| [40] (n= 24) | 1000 mg p.o. | Stimulation of nasal mucosa by CO2 (phasic pain) and dry air (tonic pain), VAS, evoked brain potentials (only phasic pain) | VAS after phasic pain ↔ | |
| VAS after tonic pain ↓ | ||||
| Amplitudes of evoked potentials ↓ | ||||
| [41] (n= 22) | 1000 mg i.v. (prodrug) | Deep pressure, PTT | PTT ↔ | |
| [37] (n= 10) | 500, 1000 (normal release) and 2000 (sustained release) mg p.o. | Laser skin stimulation, PDT | Normal release: PDT ↑ | |
| Sustained release: PDT ↔ | ||||
| [38] (n= 32) | 1000 mg p.o. | Intracutaneous electrical stimulation, VAS, evoked brain potentials | VAS ↓ | |
| Evoked potentials: power spectrum was altered and amplitudes ↓ | ||||
| [36] (n= 15) | 1000 mg p.o. normal release and 2000 mg p.o. slow release | Laser skin stimulation, PDT, evoked brain potentials | Pain threshold ↑ | |
| Evoked potentials ↔ | ||||
| Dosing regimes were alike | ||||
| Models inducing hyperalgesia | [32] (n= 24) | 1000 mg p.o. | Freeze lesion: | No parameters were affected |
| area of secondary HA to pinprick stimulation | ||||
| [43] (n= 17) | 75 mg p.o. | Continuous electrical skin stimulation Ongoing pain (numeric rating scale) | Area of HA and allodynia ↓ | |
| Ongoing pain ↔ | ||||
| Area of secondary HA to pinprick (von Frey) | ||||
| [44] (n= 14) | 1000 mg i.v. | Continuous intracutaneous electrical stimulation, ongoing pain (VAS), area of secondary HA to pinprick and allodynia to stroking | Area of HA and allodynia ↓ | |
| Ongoing pain ↔ |
In the column ‘Model’ the method for pain assessment is normal font, and the method for pain induction is bold. PDT, pain detection threshold; PTT, pain tolerance threshold; AUC, area under curve; VAS, visual analogue scale; HA, hyperalgesia.
Acute models
Skin/nasal mucosa: Paracetamol has been tested against pain from laser and electrical stimulation of the skin. The model involving laser was sensitive to paracetamol, but again, this was probably due to skin inflammation caused by repeated laser pulses [36, 37]. Models with electrical stimulation of the skin showed conflicting results regarding sensitivity to paracetamol. Pain from repeated electrical stimulation mimicking the previously mentioned central integration of the response was unaffected by paracetamol. In the study where the electrical pain was decreased by paracetamol, modulation was most distinct in the evoked potentials compared with the subjective pain rating [38, 39]. Pain in the nasal mucosa from gaseous CO2 stimulation and dry air has been tested. For the phasic pain induced by gaseous CO2 stimulation, only evoked potentials and not subjective pain ratings changed after drug administration. The subjective pain ratings were affected for the more tonic pain induced by dry air [40].
Muscle: Pain from pressure algometry used for testing analgesia from paracetamol was not sensitive to the analgesic in two studies [39, 41]. A third study found that paracetamol was effective against pressure pain, and this group found that immersing the hand into ice water gave a more pronounced analgesic effect for the pressure pain [42]. Furthermore, pain from intramuscular infusion of hypertonic saline has been tested, but was not decreased by the drug [39]. Likewise, a model involving both single and repeated intramuscular electrical stimulation was not sensitive to paracetamol [39].
Models of hyperalgesia
Skin: Pain and hyperalgesia from freeze was unaffected by paracetamol. Furthermore, paracetamol has been tested against hyperalgesia evoked by continuous electrical stimulation. Here the ongoing pain was unaffected, whereas the hyperalgesia was decreased [43]. The ongoing pain was assessed by subjective ratings and the limited sensitivity of this measuring method is in consensus with findings of Bromm et al.[38, 44].
Dosing regimes
The reviewed studies applied doses in the therapeutic range. This could partly explain the limited effects of this drug in experimental pain models.
Mechanistic aspects
NSAIDs and paracetamol work by different mechanisms and accordingly have different profiles in experimental pain models. NSAIDs work on several clinical pain conditions and accordingly they also attenuate several types of physiological activation of the pain system as seen in acute pain models. Paracetamol has limited effect in models with acute pain stimuli. Although acetylsalicylic acid is a weak analgesic working mainly in inflammatory pain, this drug is effective against several acute pain measures in the skin even in normal therapeutic doses. This suggests a central effect, since in the periphery cascades releasing prostaglandins will probably not be activated in short-lasting experimental pain [45]. Topical ibuprofen seemed to have some advantages over systemic ibuprofen for attenuation of exercise-induced jaw-muscle pain, indicating that a peripheral pain mechanism is important in this model [31].
Experimental models involving NSAIDs shows how a peripheral mediated analgesia can be detected by central measures, because the sum of incoming nociceptive activity to the central nervous system (CNS) is reduced [9]. Generally, it could be seen that evoked brain potentials from electrical skin stimulation were more sensitive than the subjective pain rating [13, 20, 38].
Models evoking tonic muscle pain show conflicting results. This is surprising, since such models mimic clinical inflammatory muscle pain, which is treated successfully with acetylsalicylic acid [16, 31, 39].
For the cold pressor test different results were seen for NSAIDs and paracetamol. Since immersing the hand into ice water is known to activate the descending noxious inhibition, the findings in this study indicate that paracetamol, but not NSAIDs, work through increasing this inhibition [24, 42].
Pain and hyperalgesia from freeze lesions was attenuated by NSAIDs, but paracetamol had no effect in this model. Compared with NSAIDs, paracetamol has a different mechanism of action with less affinity to the peripheral cyclooxygenases and a more pronounced central effect, and this could explain the different findings for, for example, the cold pressor test and freeze lesion [32, 42, 46, 47]. Furthermore, this is supported by findings in the continuous electrical hyperalgesia model, which probably activates central mechanisms that were attenuated by paracetamol [44]. However, NSAIDs also demonstrate effect towards central hyperalgesic mechanisms [15, 22].
N-methyl-D-aspartate antagonists in experimental pain
Ketamine
There are no clinical useful N-methyl-D-aspartate (NMDA) antagonists available for the treatment of pain, and ketamine has pronounced side-effects preventing wide clinical use as an analgesic. However, ketamine is used to some degree in difficult clinical cases and has been intensively investigated as a ‘model drug’ because of the substantial scientific interest in the NMDA receptors (Table 4).
Table 4.
Schematic overview of studies involving ketamine in human experimental pain models
| Reference | Dose | Model (pain assessments) | Findings | |
|---|---|---|---|---|
| Acute models | [48] (n= 9) | 50 mg topically | Heat skin stimulation VAS to set temperatures | VAS ↔ |
| [55] (n= 11) | Targeted i.v. infusion of 60 and 120 ng ml−1 | Heat skin stimulation (5 °C s−1) and oesophageal mechanical stimulation. pain and unpleasantness (VAS) | Oesophageal VAS ↓ Cutaneous VAS ↔ Unpleasantness ↓ for both oesophageal and cutaneous stimulation | |
| [54] (n= 12) | 0.375 mg kg−1 i.v. | Heat skin stimulation PDT and second pain to repeated stimulation (number of stimuli to PTT) (0.5 °C s−1) | PDT ↔ Number of stimuli to PTT ↑ | |
| [49, 59] (n= 12/11) | Targeted i.v. infusion of 150 ng ml−1 | Heat and cold skin stimulation detection threshold and PDT (1–1.5 °C s−1) Pinprick detection threshold | No parameters were affected | |
| [61] (n= 19) | 0.15 and 0.30 mg kg−1 h−1 for 135 min i.v. | Heat skin stimulation PDT (1 °C s−1) | PDT ↔ | |
| [30] (n= 15) | 10 µg kg−1 min−1 i.v. for 60 min | Skin and intramuscular single and repeated electrical stimulation, PDT, tonic pain to intramuscular electrical stimulation (pain to 1.5 × PDT for 10 s) IM injection of hypertonic saline, AUCVAS | Muscle and skin: PDT (single and repeated electrical stimulation) ↑ Tonic pain ↓ AUCVAS↓ | |
| [52] (n= 12) | Targeted i.v. infusion of 350 ng ml−1 racemic and 189 ng ml−1 (S+) | Laser skin stimulation (30 °C s−1) PDT to single, temporal and spatial summated stimulation, VAS to 2 s stimulation Electrical skin stimulation PDT to single and temporal summated stimulation, VAS to 1.2 × PDT Pressure algometry PDT, PTT | Laser PDT (both single, spatial and temporal summated stimulation)↑ VAS ↓ Electrical PDT (summated)↑, VAS ↓ Pressure PDT, PTT ↑ Pain of long duration was attenuated more than short-lasting pain The racemic ketamine had longer duration of action | |
| [51] (n= 12) | I.v. bolus of 20 µg kg−1 min−1 followed by an infusion | Argon laser pain PDT, PTT Single and repeated electrical skin stimulation nociceptive reflex and VAS, summation threshold, stimulus response curve Pressure algometry PDT, PTT | Laser pain ↔ Reflex threshold and VAS to a single electrical stimulus ↔ Reflex threshold summation threshold for repeated electrical stimulus ↑ VAS ↓ Pressure PDT, PTT ↑ | |
| [66] (n= 9) | 0.1 mg kg−1 i.v. | Ischaemic pain VAS | VAS ↓ | |
| Models inducing hyperalgesia | [48] (n= 9) | 50 mg topically | Intradermal capsaicin spontaneous pain, HA to heat and pinprick, allodynia to stroking (VAS) | Spontaneous pain after capsaicin ↔ Pinprick HA ↓, heat HA and allodynia ↔ |
| [49] (n= 12) | Targeted i.v. infusion of 150 ng ml−1 (after intradermal capsaicin) | Intradermal capsaicin spontaneous pain (VAS), area of secondary HA to pinprick, heat and allodynia to stroking | No parameters were affected | |
| [59] (n= 11) | Targeted i.v. infusion of 150 ng/ml (before and during capsaicin injection) | Intradermal capsaicin spontaneous pain (VAS) Area of secondary HA to pinprick, heat and allodynia to stroking | Spontaneous pain ↓ Area of HA to pinprick ↓ Area of HA to heat and allodynia to stroking ↔ | |
| [56, 57] (n= 12) | 2.5 mg ml−1 subcutaneous and 0.1 mg kg−1 i.v. followed by 7 µg kg−1 min−1 | Intradermal capsaicin spontaneous pain (VAS), area of secondary HA to pinprick and allodynia to stroking | Spontaneous VAS ↔ for both administrations Areas of HA and allodynia were affected after subcutaneous administration. Systemic administration decreased area of HA and allodynia | |
| [58] (n= 12) | 0.07 and 0.29 mg kg−1 i.v. | Intradermal capsaicin, spontaneous pain (VAS), area of secondary HA to pinprick and allodynia to stroking. HA to pinprick (VAS) | All parameters were unaffected | |
| [60] (n= 17) | I.v. bolus of 20 µg kg−1 min−1 followed by an infusion of 5 µg kg−1 min−1 | Topical capsaicin nociceptive reflexes to electrical stimulation In primary HA area: PDT to pinprick, heat, laser and single electrical stimulation In secondary HA area: PDT to single and repeated electrical stimulation in the area of secondary HA to pinprick | Pain under induction of nociceptive reflexes ↓ Nociceptive reflex size ↓ In primary HA area: heat and laser PDT ↔ Single electrical stimulation PDT ↑ Secondary HA area: summation PDT ↑, PDT to heat and single electrical stimulation and area of HA to pinprick size ↔ | |
| [62] (n= 11) | 9 µg kg−1 min−1 i.v. for 45 min | Burn injury PDT to pinprick in injured skin, area of secondary HA to pinprick Repeated pinprick AUCVAS | PDT to pinprick ↑ Area of secondary HA to pinprick ↓ AUCVAS from repetitive pinprick stimulation ↔ | |
| [50] (n= 12) | 0.15 mg kg−1 i.v. | Burn injury heat and cold (1 °C s−1) detection threshold and PDT in primary and secondary HA area Detection threshold to pinprick, area of secondary HA to pinprick,, wind-up like pain to repeated pinprick stimulation | All thermal thresholds ↔ The lowering of pinprick detection threshold was reversed Area of secondary HA to pinprick ↓ Wind-up like pain and primary HA to pinprick ↓ | |
| [61] (n= 19) | 0.15 and 0.30 mg kg−1 h−1 for 135 min i.v. | Burn injury pain during injury induction (VAS) Primary HA to heat (PDT) (1 °C s−1) Area of secondary HA to pinprick and allodynia to stroking. Brief thermal priming area of HA to pinprick | Dose-dependent reduction of pain during injury induction High dose: heat PDT ↑, areas of HA and allodynia ↓ Low dose: area of allodynia↓, heat PDT and pinprick HA ↔ Area of HA to after brief thermal priming ↓ for both doses | |
| [53] (n= 12) | 0.40 mg kg−1 i.v. | Continuous electrical skin stimulation Evoked pain (threshold to VAS5/10) Area of secondary HA to pinprick and allodynia to stroking | Evoked pain ↓ Area of HA to pinprick and allodynia ↓ | |
| [63] (n= 18) | 2 mM i.m. | I.m. injection of glutamate. HA tested by pressure algometry | Mechanical HA was decreased by ketamine | |
| [64] (n= 12/14) | Loading dose: 0.075 mg kg−1 i.v., followed by infusion of 0.005 mg kg−1 min−112/14 subjects had ketamine before/after induction of HA | Infusion of hydrochloric acid/electrical oesophageal stimulation PDT | Reduction in PDT (HA) was prevented (ketamine before induction of HA) and reversed (ketamine after induction of HA) |
In the column ‘Model’ the method for pain assessment is normal font, and the method for pain induction is bold. PDT, pain detection threshold; PTT, pain tolerance threshold; AUC, area under curve; VAS, visual analogue scale; HA, hyperalgesia.
Acute models
Skin: Ketamine has been tested against sensations and pain from heat, cold and electrical stimulation. In several studies ketamine was ineffective against acute experimental pain in the skin [48–50]. However, the drug showed effect against electrical pain (particularly strong pain intensities) and heat pain in some studies [30, 51–53]. Pricking ‘first pain’ from laser stimulation was unaffected by ketamine [51], whereas another study found effect on the affective components of heat pain and ‘second pain’ after thermal stimulation [54, 55]. Pain from repeated and continuous electrical stimulation was sensitive to ketamine [52, 53].
Muscle: Muscular pain from electrical stimulation, pressure pain and hypertonic saline infusion was decreased after ketamine treatment [30, 51, 52].
Viscera: Pain and unpleasantness from visceral distension was also decreased by ketamine [55].
Models of hyperalgesia
Skin: Hyperalgesia to stroking and pinprick were affected by systemic, but not locally applied ketamine [56–59]. One study evaluated the hyperalgesia with laser heat and electrical stimulation. Here only the electrical stimulation in the secondary hyperalgesic area was sensitive to the drug [60].
Hyperalgesia evoked by burn injury is decreased by ketamine, and substantial effect could be seen on hyperalgesia to pinprick and allodynia to stroking [50, 61, 62].
Allodynia and hyperalgesia from continuous electrical stimulation were all affected by ketamine [53], which should be expected since the inflicted pain is strong and long lasting and probably activates the NMDA receptors.
Muscle: Mechanical hyperalgesia after glutamate injection in the masseter muscle was attenuated by intramuscular ketamine, showing an effect of the drug on the peripheral nerves where NMDA receptors have also been demonstrated [63].
Viscera: Hyperalgesia to electrical pain has been induced in the oesophagus by infusion of hydrochloric acid. This study showed that ketamine was able to both prevent the development of hyperalgesia and reverse hyperalgesia already developed [64].
Dosing regimes
Ketamine is very potent, and the pronounced side-effects produced by this drug make dosing complex. Here a good example of the importance of the right timing of the dose was found. Willert et al. found that ketamine administered prior to induction of oesophageal hyperalgesia prevented the lowering of the pain detection threshold but, furthermore, reversal of induced hyperalgesia was seen if ketamine was administered after induction of hyperalgesia [64]. Hence the ability of ketamine to reverse hyperalgesia seems to be dependent on the model and tissue where the hyperalgesia is induced. Warncke et al. found that several of the decreased hyperalgesic responses to brushing and pinprick returned to the original hyperalgesic state 15 min after ketamine administration [50]. This could very well be caused by the short half-life of ketamine, meaning that the plasma level was markedly reduced after 15 min. It illustrated the importance of testing at time points where the drug is present in the body. Wallace et al. infused mean amounts of 0.33, 0.52 and 0.82 mg kg−1[59]. These doses were high compared with those applied by Sethna et al., which could explain why Wallace et al. found much more pronounced effects [58, 59]. The doses were administered at different rates giving different plasma concentrations, which was probably important for the effect.
Mechanistic aspects
The NMDA receptor is mainly activated under strong or repeated stimulation [51, 65] and accordingly temporal and spatial summation (central integration of the afferent barrage via the NMDA receptor) is likely to be affected by ketamine [52, 53]. This may explain why ketamine was not effective in models using single stimulations with brief, superficial pain.
Deep pain from muscle and viscera was affected more than superficial pain [55, 66]. This is in accordance with findings in other human studies, where deep pain activated central mechanisms (involving the NMDA receptor) more quickly than superficial pain [67].
Generally, models involving hyperalgesia induce long-lasting and strong pain and therefore could demonstrate analgesia from ketamine. Wallace et al. tried to administer ketamine both prior to and after intradermal injection of capsaicin. The group found that to provide analgesia the drug needed to be administered prior to the induction of hyperalgesia [49, 59]. This probably reflects that development of hyperalgesia was prevented rather than a reversal of the process. Hyperalgesia from application of capsaicin to the skin was sensitive to ketamine when evaluated by stroking and pinprick [58, 59]. This effect seemed to be central, since subcutaneous administration failed to attenuate the hyperalgesia and spontaneous pain [56, 57].
Anticonvulsive agents in experimental pain
Gabapentin
Gabapentin is widely used in neuropathic pain and has been the subject of great interest in experimental pain testing (Table 5).
Table 5.
Schematic overview of studies involving gabapentin in human experimental pain models
| Reference | Dose | Model | Main findings | |
|---|---|---|---|---|
| Acute models | [69] (n= 12) | 1800 mg p.o. | Repeated pinprick stimulation of the skin VAS, fMRI | VAS, fMRI was modulated |
| [68] (n= 25) | 1200 mg p.o. | Heat skin stimulation (1 °C s−1) PDT, pain intensity (VAS) after long-lasting heat stimulation | Heat PDT ↔ VAS after long-lasting heat stimulation ↔ | |
| [25] (n= 12) | 600 mg p.o. | Cold pressor test AUCVAS | AUCVAS↔ | |
| [71] (n= 16) | 1200 as single dose or 2600 as multiple doses mg p.o. | Intramuscular infusion of hypertonic saline peak pain (VAS), AUCVASPDT to repetitive pinprick stimulation | AUCVAS, peak VAS ↔ PDT ↔ | |
| [70] (n= 20) | 1200 mg p.o. | Single and repeated cutaneous and intramuscular electrical stimulation PDT, stimulus response curve Intramuscular injection of hypertonic saline AUCVAS, area of referred pain | Single and repeated electrical stimulation: –cutaneous PDT ↑ –muscle PDT ↔ Stimulus response curve ↔ Injection of hypertonic saline: AUCVAS and area of referred pain ↓ | |
| Models inducing hyperalgesia | [73] (n= 41) | 2400 mg p.o. daily (15 days of dosing) | Intradermal injection of capsaicin spontaneous pain rated (VAS) area of HA to pinprick, allodynia to brushing, intensity of pain to pinprick and brushing | Area of allodynia to brushing ↓ Spontaneous pain, area of HA to pinprick, brush and pinprick pain ↔ |
| [68] (n= 25) | 1200 mg p.o. | Heat-capsaicin sensitization In primary HA area: PDT to heat Area of secondary HA to pinprick and allodynia to brushing | Heat PDT ↑ Area of HA to pinprick and allodynia ↓ | |
| [69] (n= 12) | 1800 mg p.o. | Heat-capsaicin sensitization, spontaneous pain (VAS), fMRI, HA to repeated pinprick stimulation | Spontaneous pain ↔ fMRI: small decrease of pain/hyperalgesia induced deactivation of areas in the insula and somatosensory cortex | |
| [71] (n= 16) | 1200 as single dose or 2600 as multiple doses mg p.o. | Continuous intracutaneous electrical stimulation threshold of moderate pain, ongoing pain (VAS), area of secondary HA to pinprick, PDT to repetitive pinprick stimulation in HA area | Threshold to moderate pain↑, but only for 2600 mg Ongoing pain and PDT to repetitive pinprick ↔ (both doses) Area of HA to pinprick ↓ | |
| [74] (n= 16) | 600 mg p.o. | Burn injury (UVB) PDT and PTT to heat in the primary HA area and in control area, area of secondary HA to pinprick | Primary HA area: heat PDT ↔, PTT ↑ Control area: heat PDT and PTT ↔ Area of secondary HA to pinprick ↓ |
In the column ‘Model’ the method for pain assessment is normal font, and the method for pain induction is bold. PDT, pain detection threshold; PTT, pain tolerance threshold; AUC, area under curve; VAS, visual analogue scale; HA, hyperalgesia; fMRI, functional magnetic resonance imaging.
Acute models
Skin: Heat pain and pain from stimulation with von Frey filaments have been tested with gabapentin. The subjective pain ratings after such stimulations were unaffected by gabapentin, but in functional magnetic resonance imaging (fMRI) studies activations in the bilateral insula were modulated [68, 69].
Muscle: The cold pressor test, which is sensitive to opioids, was insensitive to gabapentin [25]. Arendt-Nielsen et al. found that pain from infusion of hypertonic saline was attenuated after gabapentin [70]. However, these findings were in contrast to another study using the same stimulation and assessment methods, but applying higher and multiple doses [71]. These conflicting results are hard to explain, since the study by Segerdahl et al. should have seen a stronger analgesic effect because of their dosing regimen (see section on Dose). However, the different results could be attributed to differences between laboratories such as instruction of subjects and assessments of the pain [72] (Figure 1).
Figure 1.
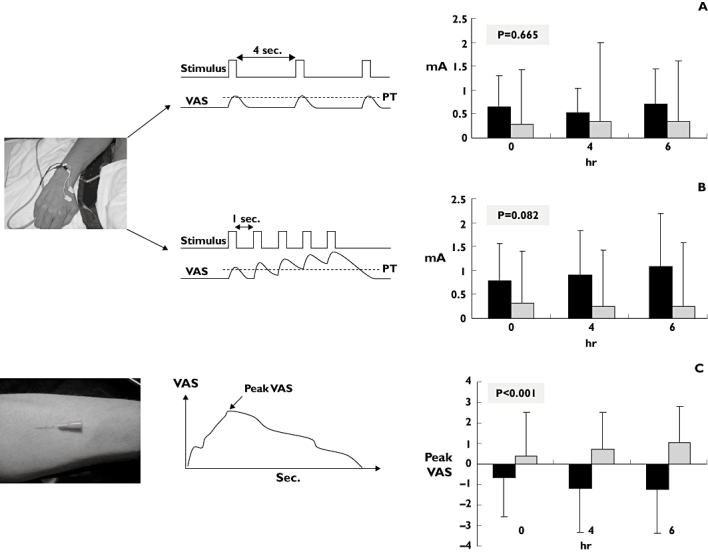
Illustration of how different types of pain induction have different sensitivities for detecting analgesia. The single electrical stimulation gave the least separation of the treatment effects of gabapentin; 1200 mg (black) and placebo (grey). Repeated electrical stimulation activating central pain (temporal summation) mechanisms gave a better separation (but still not significant). Of the three, the most tonic and deep pain type after intramuscular injection of hypertonic saline gave significant separation of placebo and gabapentin [75]
Models of hyperalgesia
Skin: Several groups have tested gabapentin against hyperalgesia/allodynia from cutaneous capsaicin stimulation. The results are variable, but all studies showed analgesic effect on at least one hyperalgesia parameter, where allodynia to brushing seemed to be more robust than secondary hyperalgesia to pinprick [68, 69, 73]. One study did not show effect on subjective pain ratings, but only on brainstem activation in an fMRI study. This study found that the modulation of the cerebral response was clearer for hyperalgesic pain than for acute pain [69]. Inflammation from UVB radiation was unaffected by gabapentin [74].
Dosing regimes
A straightforward dose–response relationship is not apparent from the trials of gabapentin. In the hypertonic saline model inducing muscle pain, an effect was seen in the study applying the lower dose. Segerdahl et al. used a single dose of 1200 or 2600 mg distributed over 24 h, whereas Arendt-Nielsen et al. used a single dose of 1200 mg [71, 75]. The study by Gottrup applied a daily dose of 2400 mg p.o. for 15 days, and this study revealed only effect on allodynia to brushing [73]. The study by Dirks et al., however, applied a rather low dose, and found an overall good effect on several pain parameters [68]. In the clinic this drug needs a slow titration to effect and can take weeks for the analgesia to appear. This complex pharmacodynamic/pharmacokinetic relationship may explain the nonlinear relation between dose and effect found in experimental pain models.
Lamotrigine
Lamotrigine is a sodium channel antagonist that has been characterized in various experimental pain models (Table 6).
Table 6.
Schematic overview of studies involving lamotrigine in human experimental pain models
| Reference | Dose | Model | Main findings | |
|---|---|---|---|---|
| Acute models | [77] (n= 18) | 400 mg p.o. | Heat skin stimulation (1 min 45 °C) VAS | No parameters were affected |
| [76] (n= 12) | 300 mg p.o. | Stimulation of nasal mucosa by CO2 VAS, evoked brain potentials | VAS ↔ Evoked potentials: latency of the P100↑, amplitudes ↔ | |
| [78] (n= 14) | 300 mg p.o. | Heat and cold skin stimulation 1–1.5 °C s−1) warm and cool sensation, PDT Mechanical skin stimulation threshold to touch | No parameters were affected | |
| [81] (n= 12) | 300 mg p.o. | Cold pressor test maximum pain (VAS) | Maximum pain ↓ | |
| Models inducing hyperalgesia | [77] (n= 18) | 400 mg p.o. | Heat-capsaicin sensitization area of secondary HA to pinprick and allodynia to brushing | No parameters were affected |
| [78] (n= 14) | 300 mg p.o. | Intradermal capsaicin: area of secondary HA to pinprick, stroking and heat | No parameters were affected |
In the column ‘Model’ the method for pain assessment is normal font, and the method for pain induction is bold. PDT, pain detection threshold; PTT, pain tolerance threshold; AUC, area under curve; VAS, visual analogue scale; HA, hyperalgesia.
Acute models
Skin/nasal mucosa: Lamotrigine has been tested against heat pain in the skin and in pain from CO2 stimulation of the nasal mucosa. The drug generally did not show effects in any of these models. Only the latency to P100 (a component of the evoked brain potential) was increased and this was seen as a coincidence of multiple testing of several parameters. [76–78].
Muscle: The cold pressor test, which is sensitive to opioids, was also sensitive to the analgesic effects of lamotrigine.
Models of hyperalgesia
Skin: Lamotrigine has been tested against capsaicin and capsaicin in combination with heat. In these models no effect was found on either spontaneous pain or on the subsequent hyperalgesia [77, 78].
Dosing regimes
In several of the reviewed studies, doses that produced profound side-effects were applied, and hence it does not seem that application of insufficient doses is the reason for the lack of effect in the various models [76, 77].
Mechanistic aspects
Both drugs in this class have limited effect in physiological pain mechanisms. Lamotrigine stabilizes neuronal membranes and reduces pathological spinal neurotransmitter release. The effect of gabapentin seem to be the result of a complex synergy between increased GABA synthesis, non-NMDA receptor antagonism and binding to the α2δ subunit of voltage-dependent calcium channels [79]. For both drugs the effect seems to depend on the reduction of pathological neurotransmitter release and hence the lack of effect in short-lasting acute pain models [80]. Interestingly, lamotrigine decreases pain from the cold pressor test. This could be due to the tonic (and relative long-lasting) nature of the evoked pain in this model or because lamotrigine affects the descending noxious inhibitory control [81].
Both drugs have been tested extensively in the hyperalgesia caused by capsaicin, where only gabapentin showed effect. Lamotrigine has a clinical effect comparable to gabapentin, but apparently the mechanisms of lamotrigine cannot be shown in experimental hyperalgesia, which is the case for gabapentin. It should be stressed that even in experimental models inducing hyperalgesia, central plasticity is not induced in the same manner and to the same extent as in chronic pain patients. For example, upregulation of calcium channels is unlikely to occur. These differences may partly explain the better effect of anticonvulsive agents in clinical conditions.
Antidepressants in experimental pain
The tricyclic antidepressants are primarily used in treatment of neuropathic pain, and a few tricyclic antidepressants have been tested in experimental pain models.
Imipramine (Table 7)
Table 7.
Schematic overview of studies involving imipramine in human experimental pain models
| Reference | Dose | Model (pain assessments) | Findings | |
|---|---|---|---|---|
| Acute models | [83] (n= 18) | 100 mg p.o. | Pressure algometry (phalanx) PTT, single and repeated electrical skin stimulation PDT, PTT Cold pressor test VAS, peak pain AUCVAS | PTT to pressure and electrical stimulation ↑ PDT to electrical stimulation ↔ Pain intensity during the cold pressor test ↔ |
| [85] (n= 12) | 100 mg p.o. | Heat skin stimulation (1 °C s−1) PDT, PTT Pressure algometry (phalanx) PTT Single and repeated electrical skin stimulation nociceptive reflex, PTT, summation threshold Cold pressor test VAS, peak pain AUCVAS, discomfort rating | PDT to heat and pressure↔, PTT to heat and pressure ↑ Nociceptive reflex threshold ↑, reflex amplitudes ↔, PTT to single and repeated electrical stimulation ↑ Cold pressor test: peak pain↓, other parameters unaffected | |
| [86] (n= 10) | 100 mg p.o. | Argon laser pain Pinprick pain and warmth threshold, evoked potentials | No parameters were affected | |
| [82] (n= 20) | 100 mg p.o. | Intracutaneous electrical stimulation VAS, evoked brain potentials, power spectrum | VAS ↓ Evoked brain potentials: power spectrum was altered and amplitudes ↓ | |
| [84] (n= 16) | 100 mg p.o. | Stimulation of nasal mucosa by CO2 VAS, evoked brain potentials, power spectrum | VAS ↓ Evoked potentials and power spectrum ↔ | |
| [89] (n= 15) | 75 mg p.o. | Oesophageal balloon distension first sensation, PDT | First sensation ↔ PDT ↑ |
In the column ‘model’ the method for pain assessment is normal font, and the method for pain induction is bold. PDT, pain detection threshold; PTT, pain tolerance threshold; AUC, area under curve; VAS, visual analogue scale; HA, hyperalgesia.
Acute models
Skin/nasal mucosa: Imipramine has been tested in models involving heat, electrical and laser stimulation of the skin as well as nasal gaseous CO2 stimulation [82–86]. Electrical pain was sensitive to the drug, but pain intensity had to be above the pain threshold for detection of analgesia [83]. Sensations from laser stimulation (reflecting a fast heat stimulus) were unaffected by imipramine, whereas the pain to heat from a thermode was decreased [85, 86]. The rate of the heating in the latter study was slow, securing more selective C-fibre activation [85, 87]. Sensation to intracutaneous electrical stimulation was decreased by imipramine. The effect was detectable on both subjective pain ratings and in the evoked potential [82]. The nociceptive withdrawal reflex is initiated by a nociceptive input (typically electrical stimulation) followed by secondary processing in the spinal cord, which initiates the generation of the withdrawal reflex [88]. The effect of the nociceptive withdrawal reflex to sural nerve stimulations was investigated by Poulsen et al.[85]. This group found a profound effect of imipramine on the reflex threshold and subjective pain ratings to single stimulations, whereas the effect on repeated stimulations (reflecting the central, NMDA-mediated integration of the response) was detected only by the subjective pain rating [85].
Muscle: Pain evoked by the cold pressor test was unaffected by imipramine [83]. Deep pressure pain was decreased by imipramine, but the stimulus intensity needed to exceed the pain threshold for a significant effect [83, 85].
Viscera: Non-nociceptive sensations and pain to distension of the oesophagus were investigated by Peghini et al., who found that only the painful sensations were affected [89].
Dosing regimes
Most studies applied high doses (100 mg) compared with clinically used doses (25–50 mg or less in chronic pain), and this could be part of the reason for the overall good effect of imipramine in the majority of studies. However, in the clinical situation there is often a delay of 1–2 weeks before the drug works.
Mechanistic aspects
The tricyclic antidepressants mainly inhibit the serotonergic and norepinephrinergic reuptake, but several mechanisms probably account for the analgesic properties of this drug [90]. The fact that imipramine works through many different analgesic mechanisms may also have contributed to the effects found in several experimental models. However, several studies show a pain-specific action of imipramine with a decrease of sensations at the pain detection threshold or of higher intensity [83, 85, 89].
Other types of analgesics
Delta-9-tetrahydrocannabinol
Acute models
Skin: Delta-9-tetrahydrocannabinol has been tested in models involving sensation to warmth and cold and pain evoked by mechanical [pinprick (von Frey) and brush], cold, heat and electrical stimulation [91–94]. The majority of studies found no effect on the pain parameters except for brush and pinprick evoked pain [94]. However, in an older study applying electrical skin pain and repeated pressure algometry, the pain detection thresholds, but not the pain tolerance thresholds to these pain stimulations, were increased after administration of delta-9-tetrahydrocannabinol.
Muscle: The cold pressor test has been used with different routes of administration of the drug, where no effect was found after either p.o., intravenous or pulmonal administration [91, 92] (Table 8).
Table 8.
Schematic overview of studies involving delta-9-tetrahydrocannabinol in human experimental pain models
| Reference | Dose | Model (pain assessments) | Findings | |
|---|---|---|---|---|
| Acute models | [94] (n= 18) | 2–8% smoked | Warm and cold skin stimulation sensation and pain (1–1.5 °C s−1) sensation threshold, PDT Brush and pinprick pain PDT | Warm and cold sensation and PDT ↔ PDT to brush and pinprick ↑ |
| [93] (n= 13) | 5 mg p.o. | Heat skin stimulation (preset temperature for 2 s) pain intensity and unpleasantness (VAS) | All parameters ↔ | |
| [92] (n= 8) | 0.053 mg kg−1 either i.v. or pulmonal | Cold pressor test AUCVAS, peak pain, mean pain VAS) | All parameters ↔ | |
| [91] (n= 12) | 20 mg p.o. | Pressure algometry (finger pulp) PTT Heat skin stimulation (2 °C s−1) PDT, PTT Cold pressor test AUCVAS, peak pain, mean pain (VAS) Single and repeated electrical skin stimulation PDT | All parameters ↔ | |
| [96] (n= 10) | 0.22 and 0.44 mg kg−1 i.v. | Repeated pressure algometry (glabella) PDT and PTT Transcutaneous electrical stimulation PDT and PTT | For both modalities: PDT ↑ PTT ↔ | |
| Models inducing hyperalgesia | [94] (n= 18) | 2–8% smoked | Intradermal capsaicin-evoked pain (VAS, McGill Pain Questionnaire), area of secondary HA to heat and pinprick and allodynia to stroking) | 2–4%: dose-dependent reduction of VAS 8%: VAS ↑ All doses: secondary HA and allodynia ↔ |
In the column ‘model’ the method for pain assessment is normal font, and the method for pain induction is bold. PDT, pain detection threshold; PTT, pain tolerance threshold; AUC, area under curve; VAS, visual analogue scale; HA, hyperalgesia.
Models of hyperalgesia
Skin: After intradermal injection of capsaicin there was no effect of smoking cannabis on secondary hyperalgesia to heat or pinprick or allodynia from stroking. However, different doses reduced and increased the spontaneous pain after capsaicin [94].
Dosing regimes
Since this drug has not been approved for pain treatment, no therapeutic range for clinical use exists. However, a dose range of 5–10 mg p.o. has been found effective in sclerosis [95]. On the other hand, even doses of 20 mg. p.o. did not produce significant analgesia in acute experimental pain [91]. The only model where delta-9-tetrahydrocannabinol produced significant analgesia (spontaneous pain from intradermal capsaicin) administered the drug via smoking of cannabis. This exposed the subjects to a mixture of cannabinoids where several possess activity in the CNS [94]. This makes comparison of the dose of this study with other studies using the clean compound difficult. Furthermore, one study found an inverse dose–response relation for delta-9-tetrahydrocannabinol [94].
Mechanistic aspects
The analgesic effect of delta-9-tetrahydrocannabinol is hard to show in acute experimental pain models. However, the drug has shown a complex pattern of analgesia with a stronger effect on pain intensities below the pain detection threshold than on those above [96]. This pattern is opposite to that of classic analgesics such as opioids working mainly on suprathreshold intensities, and could reflect that delta-9-tetrahydrocannabinol works on the sensory-discriminative rather than affective-motivational aspects of pain, which is in consensus of the findings of Wallace et al.[94, 96]. In hyperalgesic models pain from capsaicin, reflecting strong C-fibre activation, was decreased by delta-9-tetrahydrocannabinol
Discussion
Non-opioid analgesics in experimental pain
This type of analgesics exhibits different potencies and accordingly they show a more variable efficacy in experimental pain models compared with opioids. An overview of the findings in different experimental models is provided in Tables 9 and 10.
Table 9.
Schematic overview of the effect of non-opioid analgesics in acute human experimental pain models
| Model | Drugs where analgesia has been detected | Drugs where analgesia has not been detected | References |
|---|---|---|---|
| Models applied in the skin/mucosa | |||
| Heat stimulation (laser) | Acetylsalicylic acid, ibuprofen, (paracetamol), | Imipramine, ketamine, | [9, 23, 37, 51, 52, 86] |
| Heat stimulation (thermode) | (ketamine), imipramine | Gabapentin, lamotrigine, delta-9-tetrahydrocannabinol | [48, 49, 54, 55, 59, 61, 68, 77, 78, 85, 93, 94] |
| Cold | Ketamine, lamotrigine, delta-9-tetrahydrocannabinol | [49, 59, 78, 94] | |
| Single electrical skin stimulation | Acetylsalicylic acid, ibuprofen, (ketamine), gabapentin, imipramine, (delta-9-tetrahydrocannabinol) | (Paracetamol) | [8, 17, 18, 30, 38, 39, 51, 52, 70, 83, 85, 91, 96] |
| Repeated electrical skin stimulation | Ketamine, gabapentin, imipramine | Paracetamol, delta-9-tetrahydrocannabinol | [30, 39, 51, 52, 70, 83, 85, 91] |
| Electrical stimulation of dental mucosa | Acetylsalicylic acid | [12] | |
| Stimulation of nasal mucosa with CO2 | Acetylsalicylic acid, ibuprofen, paracetamol, imipramine | (Lamotrigine) | [13, 20, 40, 76, 84] |
| Pressure [interdigital web (IW) or phalanx (P)] | Ibuprofen (IW), imipramine (P) | [21, 22, 83] | |
| Intradermal infusion of phosphate buffered solution | Acetylsalicylic acid, ibuprofen | [10] | |
| Pinprick | (Gabapentin) (repeated stimulation), delta-9-tetrahydrocannabinol | Ibuprofen, ketamine | [32, 49, 59, 69, 94] |
| Models applied in the muscle | |||
| Deep pressure or fingerpulp (F) | Ketamine, delta-9-tetrahydrocannabinol (repeated) | (Paracetamol), delta-9-tetrahydrocannabinol (F) | [39, 41, 51, 52, 91, 96] |
| Cold pressor test | Paracetamol, lamotrigine, (imipramine) | Ibuprofen, gabapentin, delta-9-tetrahydrocannabinol | [24, 25, 42, 81, 83, 85, 91, 92] |
| Injection of hypertonic saline | Ketamine | Paracetamol, (gabapentin) | [30, 39, 70, 71] |
| Single electrical stimulation | Ketamine | Paracetamol, gabapentin | [30, 39, 70] |
| Repeated electrical stimulation | Ketamine | Paracetamol, gabapentin | [30, 39, 70] |
| Ischaemic pain | Ketamine | Acetylsalicylic acid | [14, 66] |
| Models applied in the viscera | |||
| Distension of oesophagus | Imipramine, ketamine | [55, 89] | |
The table concludes and summarizes findings found above. Drugs in parentheses have different efficacy in different trials; the drug is then put in the category where evidence is strongest.
Table 10.
Schematic overview of the effect of non-opioid analgesics in human experimental pain models inducing hyperalgesia
| Model | Drugs where analgesia has been detected | Drugs where analgesia has not been detected | References |
|---|---|---|---|
| Models applied in the skin/mucosa | |||
| Capsaicin | Acetylsalicylic acid, (ketamine), gabapentin, delta-9-tetrahydrocannabinol | Ibuprofen, lamotrigine | [15, 21, 48, 49, 58–60, 73, 78, 94] |
| Heat/capsaicin | Gabapentin | Lamotrigine | [68, 69, 77] |
| Continuous electrical stimulation | Paracetamol, ketamine, gabapentin, | [43, 44, 53, 71] | |
| Burn injury | Ibuprofen, ketamine, | [22, 50, 61, 62] | |
| UVB radiation | Ibuprofen, gabapentin | [33, 34, 74] | |
| Freeze lesion | Ibuprofen | Paracetamol | [32] |
| Long-lasting or repetitive pinching | Acetylsalicylic acid, ibuprofen | [11, 21, 22, 33] | |
| Repeated pinprick | Ketamine | [50, 62] | |
| Models applied in the muscle | |||
| Delayed onset muscle soreness | Ibuprofen | Acetylsalicylic acid | [16, 31] |
| Injection of glutamate | Ketamine | [63] | |
| Models applied in the viscera | |||
| Hydrochloric acid applied in oesophagus | Ketamine | [64] | |
The table concludes and summarizes findings found above. Drugs in parentheses have different efficacy in different trials; the drug is then put in the category where evidence is strongest.
Because of the relatively moderate analgesic potency of NSAIDs and paracetamol, the effects of these drugs are best demonstrated in models that have a large dynamic range and hence can detect small treatment effects. Particularly for paracetamol, it is problematic to prove efficacy in acute experimental pain [36, 39, 41]. Good models for detecting analgesia of NSAIDs include heat pain from laser stimulation, pain from nasal stimulation of the nasal mucosa or long-lasting or repetitive pinching of the interdigital web.
Ketamine that is a strong agonist for particularly the NMDA receptor works well in models that activate the NMDA receptor, including models with repeated stimulation or strong and long-lasting pain [30, 51, 52, 66]. However, this compound also decreases more short-lasting pain, despite the questionable activation of the NMDA receptor in such models [30, 52]. An explanation for these findings could be that sedation influences the subject's pain scoring. Pain from deep structures seems to be more affected by this compound, illustrating a tissue different profile for this compound [55].
Anticonvulsive agents are generally not very efficacious in experimental pain, although gabapentin has shown effect mainly in models evoking hyperalgesia [68, 69, 73]. In the clinic these compounds needs to dosed for >1 week before the effect appears, and this complex pharmacokinetic/dynamic relation could partly explain the limited success seen in studies involving anticonvulsive agents and experimental pain.
Imipramine affected several types of experimental acute pain. However, the most sensitive models include pain intensities above the pain detection threshold or pain assessment by the mean of evoked brain potentials [82–85].
Cannabinoids (delta-9-tetrahydrocannabinol) are not approved for pain treatment, and paradoxical findings regarding analgesia from these compounds exist, e.g. nonpainful sensations were affected but not the painful sensations [96]. Such findings could reflect that pain processing is affected differently compared with the traditional analgesics.
Designing experimental studies involving non-opioid analgesics
A good trial of an unknown drug in an experimental pain model is best designed with careful consideration of the pharmacological mechanisms and kinetics of the drug [49, 59]. To obtain a good trial design, at least three factors needs to be considered: (i) a model (including an appropriate induction and assessment method) that activates mechanisms and pain pathways being sensitive to the analgesic in question; (ii) correct dose, which ensures sufficient efficacy combined with limited side-effects; and (iii) correct dosing regime (single dose/multiple dose) and time points of testing for analgesia (Figure 2).
Figure 2.
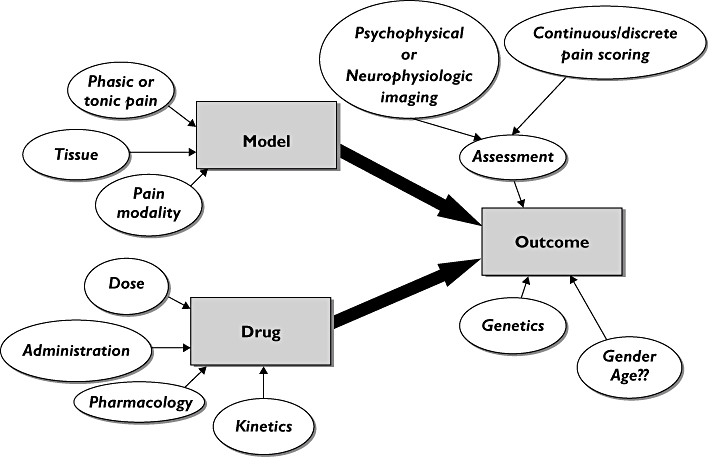
Schematic overview over how several parameters can affect the outcome of trials involving drug testing in human experimental pain models. The information in this review gives the possibility of going backwards in the flowchart and deciding on the appropriate model, assessment method and population to make a realistic prognosis of the outcome for a drug with a known pharmacological profile
Choosing the right model
Since non-opioid analgesics are less potent than opioids, detection of analgesia in experimental pain can be problematic and models recruited should have high sensitivity. High sensitivity comes from combining a model that recruits pain mechanisms that are modulated by the compound in question with a robust, sensitive method of pain assessment. However, for some analgesics this is not obvious and, for example, it seems less clear-cut which mechanisms are attenuated by, for example, NSAIDs and paracetamol [10, 13, 20, 38]. The same is true for the anticonvulsive agents.
Some assessment methods summarize the pain over time, whereas others register the peak pain. Normally, summarized pain measures are more robust, giving a high signal-to-noise ratio, which determines the sensitivity of the pain model [97]. It is seen in several studies that evoked potentials and brain imaging can provide more sensitive measures of analgesia when compared with subjective pain rating on various scales [8, 13, 38, 69]. However, it should be kept in mind that amplitudes of evoked potentials reflect only a few components (e.g. intensity) of the pain experience (Figure 3).
Figure 3.
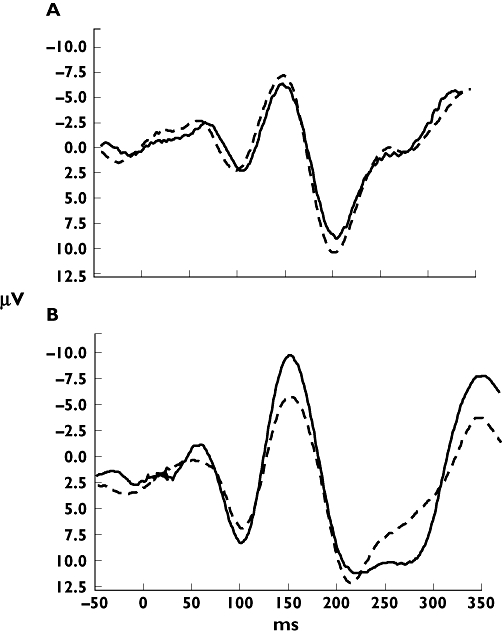
Example of an evoked potential after electrical stimulation of the skin. (A) The electroencephalogram after placebo at baseline (—) and after 90 min (---). (B) The electroencephalogram after an analgesic at baseline (—) and after 90 min (---). It can be seen here that the method is reproducible and how the amplitudes but not the latencies of the peaks change after administration of an analgesic
Furthermore, the reproducibility of the model is an important factor in testing analgesics, where it is necessary to repeat the pain stimulation several times during active and placebo treatments. If the reproducibility is low, then the change in the pain measure needs to be large for the model to detect it [97].
When the right model is chosen one should bear in mind that the stimulation intensity in the experimental model should be well controlled, so that the impact on the pain system is stable [1, 4]. This also ensures repeatability, which is crucial for good sensitivity towards analgesic modulation of the pain in the model [6]. In the case of cross-over designed studies it is important that the investigator is the same in all pain assessments, since gender and appearance of the investigator can influence the pain rating of the volunteers [98].
If the molecular targets of an analgesic are known, then selection of the appropriate model can be done by looking into previous trials of drugs working on similar targets. However, as non-opioid analgesics are less efficacious than opioids, this does not guarantee selection of a sensitive test. To overcome this, as many test modalities as possible (e.g. mechanical, thermal and electrical) can be included to ensure the best possible investigation of the drug. However, with several stimulus modalities it is important to select appropriate primary outcomes and statistical methods to avoid the problem with multiple testing [99]. Differential knowledge of a drug's pharmacological properties can be achieved from inclusion of models that activate both peripheral and central pain mechanisms [51]. It is also an advantage to include models that evoke sensitization of the pain system, since many non-opioid analgesics seem to show more robust analgesia in the sensitized pain system [32, 43, 68, 69].
Choosing the right dosing regime and time points for testing the analgesia
The kinetic profile is needed to determine when it is optimal to perform the pain tests, bearing in mind that bad timing of the pain testing can jeopardize an otherwise well-designed trial.
The dosing regime is important, particularly for drugs that work on pain mechanisms prevalent for chronic pain conditions, but also for centrally working drugs that cross the blood–brain barrier slowly. It could be speculated that multiple dosing is necessary for drugs such as lamotrigine or tricyclic antidepressants, because these drug works in the clinic, but need time to develop the analgesia [73, 76–78].
Choosing the right dosing regime
The dosing of the analgesic is an important factor for the findings of the model [10, 59, 100, 101]. It has been suggested that supratherapeutic doses are needed for detectable analgesic effects, especially for non-opioid analgesics [10, 20]. This could be explained by the more short-lasting limited and/or differential activation of the pain system in experimental pain [102–104]. It can be speculated that limited activation the pain system produces a smaller pain response compared with the clinical situation and that a relatively large decrease (which can be induced by a high dose of the analgesic) in this pain response is necessary to be statistical significant.
It can be difficult to decide the dose of a new compound, and safety often decides the upper limit of a dose. The dose should be selected as high as possible, well within a safe dose range.
Furthermore, the dose applied should reach the site of action. This can be problematic when topical formulations of polar drugs are applied, for example. This is seen with studies involving acetylsalicylic acid, which would not diffuse across skin in clinically significant amounts unless it hydrolyses first to salicylic acid. Topical administration of ibuprofen does not present these problems, since this drug is much more diffusible and does not hydrolyse.
The role of experimental pain in drug testing
There is need for translational studies between animal experiments and expensive and complex Phase III studies in patients. Typically, when studying the effect of a new analgesic compound, Phase I safety studies in healthy volunteers lead more or less directly to studies in patients. The experimental pain models in healthy volunteers can provide additional information about drugs overcoming species differences and avoiding the bias seen in clinical trials involving analgesics. As experimental pain models recruit only part of the various mechanisms working in clinical pain, it is possible to investigate on a mechanistic basis how analgesics work. Hence, some of the pharmacological mechanisms found in animal testing, such as involvement of the NMDA receptors in temporal summation, are also valid in human models [51, 52]. This has the potential to define the right target pain mechanisms and hence patient groups in Phase II and III trials, which in the end could reduce costs and lead to better and more mechanism-based pain treatment. Considering the many experimental data summarized in this review, there are still major problems in exact determination of the activated pathways and pain mechanisms [105]. Nevertheless, if several modalities and tissues are stimulated, experimental human models open the possibility of obtaining reproducible results in test–retest experiments and hence are useful for drug screening [106]. This justifies the use of human experimental pain models for trials involving compounds that exhibit large interspecies differences (e.g. have effect in rats, but not in humans). Such compounds can then be excluded from further clinical studies. Furthermore, the current knowledge of molecular pharmacological mechanisms for clinically available analgesics is limited and there is still a need for basic investigations of several analgesics in well-designed human experimental pain models. However, this field of research has so far produced many contradictory findings even in studies using the same pain stimulus. Differences are often caused by different pain assessment methods (where the intensity and modality are often poorly controlled) and different populations of volunteers and/or dosing regimes, and the use of more homogenous trial designs in this respect would make study comparisons more useful.
Competing interests
None declared.
REFERENCES
- 1.Drewes AM, Gregersen H, Arendt-Nielsen L. Experimental pain in gastroenterology: a reappraisal of human studies. Scand J Gastroenterol. 2003;38:1115–30. doi: 10.1080/00365520310004399. [DOI] [PubMed] [Google Scholar]
- 2.Arendt-Nielsen L, Curatolo M, Drewes A. Human experimental pain models in drug development: translational pain research. Curr Opin Investig Drugs. 2007;8:41–53. [PubMed] [Google Scholar]
- 3.Graven-Nielsen T, Mense S. The peripheral apparatus of muscle pain: evidence from animal and human studies. Clin J Pain. 2001;17:2–10. doi: 10.1097/00002508-200103000-00002. [DOI] [PubMed] [Google Scholar]
- 4.Staahl C, Drewes AM. Experimental human pain models: a review of standardised methods for preclinical testing of analgesics. Basic Clin Pharmacol Toxicol. 2004;95:97–111. doi: 10.1111/j.1742-7843.2004.950301.x. [DOI] [PubMed] [Google Scholar]
- 5.Staahl C, Olesen AE, Andresen T, Arendt-Nielsen L, Drewes AM. Assessing analgesic actions of opioids by experimental pain models in healthy volunteers – an updated review. Br J Clin Pharmacol. 2009;68:149–68. doi: 10.1111/j.1365-2125.2009.03456.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Staahl C, Reddy H, Andersen SD, Arendt-Nielsen L, Drewes AM. Multi-modal and tissue-differentiated experimental pain assessment: reproducibility of a new concept for assessment of analgesics. Basic Clin Pharmacol Toxicol. 2006;98:201–11. doi: 10.1111/j.1742-7843.2006.pto_211.x. [DOI] [PubMed] [Google Scholar]
- 7.Luginbuhl M, Schnider TW, Petersen-Felix S, Arendt-Nielsen L, Zbinden AM. Comparison of five experimental pain tests to measure analgesic effects of alfentanil. Anesthesiology. 2001;95:22–9. doi: 10.1097/00000542-200107000-00009. [DOI] [PubMed] [Google Scholar]
- 8.Bromm B, Rundshagen I, Scharein E. Central analgesic effects of acetylsalicylic acid in healthy men. Arzneimittelforschung. 1991;41:1123–9. [PubMed] [Google Scholar]
- 9.Schaffler K, Reeh PW, Zentzis K, Hamperl W. Analgesic effect of acetylsalicylic acid (ASA) versus a lithium–ASA combination: an evoked potential study employing radiant heat stimulation with a CO2 laser. Pharmacopsychiatry. 1987;20:217–21. doi: 10.1055/s-2007-1017107. [DOI] [PubMed] [Google Scholar]
- 10.Steen KH, Reeh PW, Kreysel HW. Dose-dependent competitive block by topical acetylsalicylic and salicylic acid of low pH-induced cutaneous pain. Pain. 1996;64:71–82. doi: 10.1016/0304-3959(95)00087-9. [DOI] [PubMed] [Google Scholar]
- 11.Forster C, Anton F, Reeh PW, Weber E, Handwerker HO. Measurement of the analgesic effects of aspirin with a new experimental algesimetric procedure. Pain. 1988;32:215–22. doi: 10.1016/0304-3959(88)90070-X. [DOI] [PubMed] [Google Scholar]
- 12.Cruccu G, Bini G, Accornero N, Berardelli A, Manfredi M. Analgesic activity of carprofen on human experimental dental pain. Arzneimittelforschung. 1982;32:1146–9. [PubMed] [Google Scholar]
- 13.Kobal G, Hummel C, Nuernberg B, Brune K. Effects of pentazocine and acetylsalicylic acid on pain-rating, pain-related evoked potentials and vigilance in relationship to pharmacokinetic parameters. Agents Actions. 1990;29:342–59. doi: 10.1007/BF01966467. [DOI] [PubMed] [Google Scholar]
- 14.Posner J. A modified submaximal effort tourniquet test for evaluation of analgesics in healthy volunteers. Pain. 1984;19:143–51. doi: 10.1016/0304-3959(84)90834-0. [DOI] [PubMed] [Google Scholar]
- 15.Schmelz M, Kress M. Topical acetylsalicylate attenuates capsaicin induced pain, flare and allodynia but not thermal hyperalgesia. Neurosci Lett. 1996;214:72–4. doi: 10.1016/0304-3940(96)12868-8. [DOI] [PubMed] [Google Scholar]
- 16.Barlas P, Craig JA, Robinson J, Walsh DM, Baxter GD, Allen JM. Managing delayed-onset muscle soreness: lack of effect of selected oral systemic analgesics. Arch Phys Med Rehabil. 2000;81:966–72. doi: 10.1053/apmr.2000.6277. [DOI] [PubMed] [Google Scholar]
- 17.Sandrini G, Ruiz L, Capararo M, Garofoli F, Beretta A, Nappi G. Central analgesic activity of ibuprofen. A neurophysiological study in humans. Int J Clin Pharmacol Res. 1992;12:197–204. [PubMed] [Google Scholar]
- 18.Walker JS, Carmody JJ. Experimental pain in healthy human subjects: gender differences in nociception and in response to ibuprofen. Anesth Analg. 1998;86:1257–62. doi: 10.1097/00000539-199806000-00023. [DOI] [PubMed] [Google Scholar]
- 19.Oertel BG, Preibisch C, Wallenhorst T, Hummel T, Geisslinger G, Lanfermann H, Lotsch J. Differential opioid action on sensory and affective cerebral pain processing. Clin Pharmacol Ther. 2008;83:577–88. doi: 10.1038/sj.clpt.6100441. [DOI] [PubMed] [Google Scholar]
- 20.Kobal G, Hummel C, Gruber M, Geisslinger G, Hummel T. Dose-related effects of ibuprofen on pain-related potentials. Br J Clin Pharmacol. 1994;37:445–52. doi: 10.1111/j.1365-2125.1994.tb05712.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kilo S, Forster C, Geisslinger G, Brune K, Handwerker HO. Inflammatory models of cutaneous hyperalgesia are sensitive to effects of ibuprofen in man. Pain. 1995;62:187–93. doi: 10.1016/0304-3959(94)00265-G. [DOI] [PubMed] [Google Scholar]
- 22.Petersen KL, Brennum J, Dahl JB. Experimental evaluation of the analgesic effect of ibuprofen on primary and secondary hyperalgesia. Pain. 1997;70:167–74. doi: 10.1016/s0304-3959(96)03316-7. [DOI] [PubMed] [Google Scholar]
- 23.Nielsen JC, Bjerring P, Arendt-Nielsen L, Petterson KJ. A double-blind, placebo controlled, cross-over comparison of the analgesic effect of ibuprofen 400 mg and 800 mg on laser-induced pain. Br J Clin Pharmacol. 1990;30:711–5. doi: 10.1111/j.1365-2125.1990.tb03840.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jones SF, McQuay HJ, Moore RA, Hand CW. Morphine and ibuprofen compared using the cold pressor test. Pain. 1988;34:117–22. doi: 10.1016/0304-3959(88)90156-X. [DOI] [PubMed] [Google Scholar]
- 25.Eckhardt K, Ammon S, Hofmann U, Riebe A, Gugeler N, Mikus G. Gabapentin enhances the analgesic effect of morphine in healthy volunteers. Anesth Analg. 2000;91:185–91. doi: 10.1097/00000539-200007000-00035. [DOI] [PubMed] [Google Scholar]
- 26.Enggaard TP, Poulsen L, Arendt-Nielsen L, Brosen K, Ossig J, Sindrup SH. The analgesic effect of tramadol after intravenous injection in healthy volunteers in relation to CYP2D6. Anesth Analg. 2006;102:146–50. doi: 10.1213/01.ane.0000189613.61910.32. [DOI] [PubMed] [Google Scholar]
- 27.Grach M, Massalha W, Pud D, Adler R, Eisenberg E. Can coadministration of oxycodone and morphine produce analgesic synergy in humans? An experimental cold pain study. Br J Clin Pharmacol. 2004;58:235–42. doi: 10.1111/j.1365-2125.2004.02141.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Poulsen L, Arendt-Nielsen L, Brosen K, Sindrup SH. The hypoalgesic effect of tramadol in relation to CYP2D6. Clin Pharmacol Ther. 1996;60:636–44. doi: 10.1016/S0009-9236(96)90211-8. [DOI] [PubMed] [Google Scholar]
- 29.Pud D, Yarnitsky D, Sprecher E, Rogowski Z, Adler R, Eisenberg E. Can personality traits and gender predict the response to morphine? An experimental cold pain study. Eur J Pain. 2006;10:103–12. doi: 10.1016/j.ejpain.2005.01.010. [DOI] [PubMed] [Google Scholar]
- 30.Schulte H, Graven-Nielsen T, Sollevi A, Jansson Y, Arendt-Nielsen L, Segerdahl M. Pharmacological modulation of experimental phasic and tonic muscle pain by morphine, alfentanil and ketamine in healthy volunteers. Acta Anaesthesiol Scand. 2003;47:1020–30. doi: 10.1034/j.1399-6576.2003.00204.x. [DOI] [PubMed] [Google Scholar]
- 31.Svensson P, Houe L, Arendt-Nielsen L. Effect of systemic versus topical nonsteroidal anti-inflammatory drugs on postexercise jaw-muscle soreness: a placebo-controlled study. J Orofac Pain. 1997;11:353–62. [PubMed] [Google Scholar]
- 32.Chassaing C, Schmidt J, Eschalier A, Cardot JM, Dubray C. Hyperalgesia induced by cutaneous freeze injury for testing analgesics in healthy volunteers. Br J Clin Pharmacol. 2006;61:389–97. doi: 10.1111/j.1365-2125.2006.02582.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bickel A, Dorfs S, Schmelz M, Forster C, Uhl W, Handwerker HO. Effects of antihyperalgesic drugs on experimentally induced hyperalgesia in man. Pain. 1998;76:317–25. doi: 10.1016/S0304-3959(98)00062-1. [DOI] [PubMed] [Google Scholar]
- 34.Sycha T, Gustorff B, Lehr S, Tanew A, Eichler HG, Schmetterer L. A simple pain model for the evaluation of analgesic effects of NSAIDs in healthy subjects. Br J Clin Pharmacol. 2003;56:165–72. doi: 10.1046/j.0306-5251.2003.01869.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bromm B, Treede RD. Laser-evoked cerebral potentials in the assessment of cutaneous pain sensitivity in normal subjects and patients. Rev Neurol (Paris) 1991;147:625–43. [PubMed] [Google Scholar]
- 36.Nielsen JC, Bjerring P, Arendt-Nielsen L. A comparison of the hypoalgesic effect of paracetamol in slow-release and plain tablets on laser-induced pain. Br J Clin Pharmacol. 1991;31:267–70. doi: 10.1111/j.1365-2125.1991.tb05528.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nielsen JC, Bjerring P, Arendt-Nielsen L, Petterson KJ. Analgesic efficacy of immediate and sustained release paracetamol and plasma concentration of paracetamol. Double blind, placebo-controlled evaluation using painful laser stimulation. Eur J Clin Pharmacol. 1992;42:261–4. doi: 10.1007/BF00266345. [DOI] [PubMed] [Google Scholar]
- 38.Bromm B, Forth W, Richter E, Scharein E. Effects of acetaminophen and antipyrine on non-inflammatory pain and EEG activity. Pain. 1992;50:213–21. doi: 10.1016/0304-3959(92)90165-8. [DOI] [PubMed] [Google Scholar]
- 39.Olesen AE, Staahl C, Ali Z, Drewes AM, Arendt-Nielsen L. Effects of paracetamol combined with dextromethorphan in human experimental muscle and skin pain. Basic Clin Pharmacol Toxicol. 2007;101:172–6. doi: 10.1111/j.1742-7843.2007.00095.x. [DOI] [PubMed] [Google Scholar]
- 40.Renner B, Clarke G, Grattan T, Beisel A, Mueller C, Werner U, Kobal G, Brune K. Caffeine accelerates absorption and enhances the analgesic effect of acetaminophen. J Clin Pharmacol. 2007;47:715–26. doi: 10.1177/0091270007299762. [DOI] [PubMed] [Google Scholar]
- 41.Romundstad L, Stubhaug A, Niemi G, Rosseland LA, Breivik H. Adding propacetamol to ketorolac increases the tolerance to painful pressure. Eur J Pain. 2006;10:177–83. doi: 10.1016/j.ejpain.2005.03.003. [DOI] [PubMed] [Google Scholar]
- 42.Pickering G, Esteve V, Loriot MA, Eschalier A, Dubray C. Acetaminophen reinforces descending inhibitory pain pathways. Clin Pharmacol Ther. 2008;84:47–51. doi: 10.1038/sj.clpt.6100403. [DOI] [PubMed] [Google Scholar]
- 43.Filitz J, Ihmsen H, Gunther W, Troster A, Schwilden H, Schuttler J, Koppert W. Supra-additive effects of tramadol and acetaminophen in a human pain model. Pain. 2008;136:262–70. doi: 10.1016/j.pain.2007.06.036. [DOI] [PubMed] [Google Scholar]
- 44.Koppert W, Wehrfritz A, Korber N, Sittl R, Albrecht S, Schuttler J, Schmelz M. The cyclooxygenase isozyme inhibitors parecoxib and paracetamol reduce central hyperalgesia in humans. Pain. 2004;108:148–53. doi: 10.1016/j.pain.2003.12.017. [DOI] [PubMed] [Google Scholar]
- 45.Arendt-Nielsen L, Drewes AM, Svendsen L, Brennum J. Quantitative assessment of joint pain following treatment of rheumatoid arthritis with ibuprofen cream. Scand J Rheumatol. 1994;23:334–7. doi: 10.3109/03009749409099283. [DOI] [PubMed] [Google Scholar]
- 46.Kis B, Snipes JA, Busija DW. Acetaminophen and the cyclooxygenase-3 puzzle: sorting out facts, fictions, and uncertainties. J Pharmacol Exp Ther. 2005;315:1–7. doi: 10.1124/jpet.105.085431. [DOI] [PubMed] [Google Scholar]
- 47.Steinmeyer J. Pharmacological basis for the therapy of pain and inflammation with nonsteroidal anti-inflammatory drugs. Arthritis Res. 2000;2:379–85. doi: 10.1186/ar116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Poyhia R, Vainio A. Topically administered ketamine reduces capsaicin-evoked mechanical hyperalgesia. Clin J Pain. 2006;22:32–6. doi: 10.1097/01.ajp.0000149800.39240.95. [DOI] [PubMed] [Google Scholar]
- 49.Wallace MS, Braun J, Schulteis G. Postdelivery of alfentanil and ketamine has no effect on intradermal capsaicin-induced pain and hyperalgesia. Clin J Pain. 2002;18:373–9. doi: 10.1097/00002508-200211000-00005. [DOI] [PubMed] [Google Scholar]
- 50.Warncke T, Stubhaug A, Jorum E. Ketamine, an NMDA receptor antagonist, suppresses spatial and temporal properties of burn-induced secondary hyperalgesia in man: a double-blind, cross-over comparison with morphine and placebo. Pain. 1997;72:99–106. doi: 10.1016/s0304-3959(97)00006-7. [DOI] [PubMed] [Google Scholar]
- 51.Arendt-Nielsen L, Petersen-Felix S, Fischer M, Bak P, Bjerring P, Zbinden AM. The effect of N-methyl-D-aspartate antagonist (ketamine) on single and repeated nociceptive stimuli: a placebo-controlled experimental human study. Anesth Analg. 1995;81:63–8. doi: 10.1097/00000539-199507000-00013. [DOI] [PubMed] [Google Scholar]
- 52.Arendt-Nielsen L, Nielsen J, Petersen-Felix S, Schnider TW, Zbinden AM. Effect of racemic mixture and the (S+)-isomer of ketamine on temporal and spatial summation of pain. Br J Anaesth. 1996;77:625–31. doi: 10.1093/bja/77.5.625. [DOI] [PubMed] [Google Scholar]
- 53.Koppert W, Dern SK, Sittl R, Albrecht S, Schuttler J, Schmelz M. A new model of electrically evoked pain and hyperalgesia in human skin: the effects of intravenous alfentanil, S(+)-ketamine, and lidocaine. Anesthesiology. 2001;95:395–402. doi: 10.1097/00000542-200108000-00022. [DOI] [PubMed] [Google Scholar]
- 54.Hughes AM, Rhodes J, Fisher G, Sellers M, Growcott JW. Assessment of the effect of dextromethorphan and ketamine on the acute nociceptive threshold and wind-up of the second pain response in healthy male volunteers. Br J Clin Pharmacol. 2002;53:604–12. doi: 10.1046/j.1365-2125.2002.01602.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Strigo IA, Duncan GH, Bushnell MC, Boivin M, Wainer I, Rodriguez Rosas ME, Persson J. The effects of racemic ketamine on painful stimulation of skin and viscera in human subjects. Pain. 2005;113:255–64. doi: 10.1016/j.pain.2004.10.023. [DOI] [PubMed] [Google Scholar]
- 56.Gottrup H, Bach FW, Arendt-Nielsen L, Jensen TS. Peripheral lidocaine but not ketamine inhibits capsaicin-induced hyperalgesia in humans. Br J Anaesth. 2000;85:520–8. doi: 10.1093/bja/85.4.520. [DOI] [PubMed] [Google Scholar]
- 57.Gottrup H, Hansen PO, Arendt-Nielsen L, Jensen TS. Differential effects of systemically administered ketamine and lidocaine on dynamic and static hyperalgesia induced by intradermal capsaicin in humans. Br J Anaesth. 2000;84:155–62. doi: 10.1093/oxfordjournals.bja.a013396. [DOI] [PubMed] [Google Scholar]
- 58.Sethna NF, Liu M, Gracely R, Bennett GJ, Max MB. Analgesic and cognitive effects of intravenous ketamine–alfentanil combinations versus either drug alone after intradermal capsaicin in normal subjects. Anesth Analg. 1998;86:1250–6. doi: 10.1097/00000539-199806000-00022. [DOI] [PubMed] [Google Scholar]
- 59.Wallace MS, Ridgeway B, III, Leung A, Schulteis G, Yaksh TL. Concentration–effect relationships for intravenous alfentanil and ketamine infusions in human volunteers: effects on acute thresholds and capsaicin-evoked hyperpathia. J Clin Pharmacol. 2002;42:70–80. doi: 10.1177/0091270002042001008. [DOI] [PubMed] [Google Scholar]
- 60.Andersen OK, Felsby S, Nicolaisen L, Bjerring P, Jensen TS, Arendt-Nielsen L. The effect of ketamine on stimulation of primary and secondary hyperalgesic areas induced by capsaicin – a double-blind, placebo-controlled, human experimental study. Pain. 1996;66:51–62. doi: 10.1016/0304-3959(96)02995-8. [DOI] [PubMed] [Google Scholar]
- 61.Ilkjaer S, Petersen KL, Brennum J, Wernberg M, Dahl JB. Effect of systemic N-methyl-D-aspartate receptor antagonist (ketamine) on primary and secondary hyperalgesia in humans. Br J Anaesth. 1996;76:829–34. doi: 10.1093/bja/76.6.829. [DOI] [PubMed] [Google Scholar]
- 62.Schulte H, Sollevi A, Segerdahl M. The synergistic effect of combined treatment with systemic ketamine and morphine on experimentally induced windup-like pain in humans. Anesth Analg. 2004;98:1574–80. doi: 10.1213/01.ANE.0000113237.89875.5D. table. [DOI] [PubMed] [Google Scholar]
- 63.Cairns BE, Svensson P, Wang K, Castrillon E, Hupfeld S, Sessle BJ, Arendt-Nielsen L. Ketamine attenuates glutamate-induced mechanical sensitization of the masseter muscle in human males. Exp Brain Res. 2006;169:467–72. doi: 10.1007/s00221-005-0158-z. [DOI] [PubMed] [Google Scholar]
- 64.Willert RP, Woolf CJ, Hobson AR, Delaney C, Thompson DG, Aziz Q. The development and maintenance of human visceral pain hypersensitivity is dependent on the N-methyl-D-aspartate receptor. Gastroenterology. 2004;126:683–92. doi: 10.1053/j.gastro.2003.11.047. [DOI] [PubMed] [Google Scholar]
- 65.Dickenson AH. Spinal cord pharmacology of pain. Br J Anaesth. 1995;75:193–200. doi: 10.1093/bja/75.2.193. [DOI] [PubMed] [Google Scholar]
- 66.Segerdahl M, Ekblom A, Sollevi A. The influence of adenosine, ketamine, and morphine on experimentally induced ischemic pain in healthy volunteers. Anesth Analg. 1994;79:787–91. doi: 10.1213/00000539-199410000-00029. [DOI] [PubMed] [Google Scholar]
- 67.Nie H, Arendt-Nielsen L, Andersen H, Graven-Nielsen T. Temporal summation of pain evoked by mechanical stimulation in deep and superficial tissue. J Pain. 2005;6:348–55. doi: 10.1016/j.jpain.2005.01.352. [DOI] [PubMed] [Google Scholar]
- 68.Dirks J, Petersen KL, Rowbotham MC, Dahl JB. Gabapentin suppresses cutaneous hyperalgesia following heat-capsaicin sensitization. Anesthesiology. 2002;97:102–7. doi: 10.1097/00000542-200207000-00015. [DOI] [PubMed] [Google Scholar]
- 69.Iannetti GD, Zambreanu L, Wise RG, Buchanan TJ, Huggins JP, Smart TS, Vennart W, Tracey I. Pharmacological modulation of pain-related brain activity during normal and central sensitization states in humans. Proc Natl Acad Sci USA. 2005;102:18195–200. doi: 10.1073/pnas.0506624102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Arendt-Nielsen L, Frokjaer JB, Staahl C, Graven-Nielsen T, Huggins JP, Smart TS, Drewes AM. Effects of gabapentin on experimental somatic pain and temporal summation. Reg Anesth Pain Med. 2007;32:382–8. doi: 10.1016/j.rapm.2007.05.002. [DOI] [PubMed] [Google Scholar]
- 71.Segerdahl M. Multiple dose gabapentin attenuates cutaneous pain and central sensitisation but not muscle pain in healthy volunteers. Pain. 2006;125:158–64. doi: 10.1016/j.pain.2006.05.008. [DOI] [PubMed] [Google Scholar]
- 72.Levine FM, De Simone LL. The effects of experimenter gender on pain report in male and female subjects. Pain. 1991;44:69–72. doi: 10.1016/0304-3959(91)90149-R. [DOI] [PubMed] [Google Scholar]
- 73.Gottrup H, Juhl G, Kristensen AD, Lai R, Chizh BA, Brown J, Bach FW, Jensen TS. Chronic oral gabapentin reduces elements of central sensitization in human experimental hyperalgesia. Anesthesiology. 2004;101:1400–8. doi: 10.1097/00000542-200412000-00021. [DOI] [PubMed] [Google Scholar]
- 74.Gustorff B, Hoechtl K, Sycha T, Felouzis E, Lehr S, Kress HG. The effects of remifentanil and gabapentin on hyperalgesia in a new extended inflammatory skin pain model in healthy volunteers. Anesth Analg. 2004;98:401–7. doi: 10.1213/01.ANE.0000095150.76735.5D. table. [DOI] [PubMed] [Google Scholar]
- 75.Arendt-Nielsen L, Froekjaer J, Staahl C, Graven-Nielsen T, Huggins JP, Smart TS, Drewes AM. The effects of gabapentin on experimental somatic pain and temporal summation. Reg Anesth Pain Med. 2007;32:382–8. doi: 10.1016/j.rapm.2007.05.002. [DOI] [PubMed] [Google Scholar]
- 76.Klamt JG, Posner J. Effects of lamotrigine on pain-induced chemo-somatosensory evoked potentials. Anaesthesia. 1999;54:774–7. doi: 10.1046/j.1365-2044.1999.00959.x. [DOI] [PubMed] [Google Scholar]
- 77.Petersen KL, Maloney A, Hoke F, Dahl JB, Rowbotham MC. A randomized study of the effect of oral lamotrigine and hydromorphone on pain and hyperalgesia following heat/capsaicin sensitization. J Pain. 2003;4:400–6. doi: 10.1016/s1526-5900(03)00718-1. [DOI] [PubMed] [Google Scholar]
- 78.Wallace MS, Quessy S, Schulteis G. Lack of effect of two oral sodium channel antagonists, lamotrigine and 4030W92, on intradermal capsaicin-induced hyperalgesia model. Pharmacol Biochem Behav. 2004;78:349–55. doi: 10.1016/j.pbb.2004.04.007. [DOI] [PubMed] [Google Scholar]
- 79.Bennett MI, Simpson KH. Gabapentin in the treatment of neuropathic pain. Palliat Med. 2004;18:5–11. doi: 10.1191/0269216304pm845ra. [DOI] [PubMed] [Google Scholar]
- 80.Laughlin TM, Tram KV, Wilcox GL, Birnbaum AK. Comparison of antiepileptic drugs tiagabine, lamotrigine, and gabapentin in mouse models of acute, prolonged, and chronic nociception. J Pharmacol Exp Ther. 2002;302:1168–75. doi: 10.1124/jpet.302.3.1168. [DOI] [PubMed] [Google Scholar]
- 81.Webb J, Kamali F. Analgesic effects of lamotrigine and phenytoin on cold-induced pain: a crossover placebo-controlled study in healthy volunteers. Pain. 1998;76:357–63. doi: 10.1016/S0304-3959(98)00068-2. [DOI] [PubMed] [Google Scholar]
- 82.Bromm B, Meier W, Scharein E. Imipramine reduces experimental pain. Pain. 1986;25:245–57. doi: 10.1016/0304-3959(86)90100-4. [DOI] [PubMed] [Google Scholar]
- 83.Enggaard TP, Poulsen L, Arendt-Nielsen L, Hansen SH, Bjornsdottir I, Gram LF, Sindrup SH. The analgesic effect of codeine as compared to imipramine in different human experimental pain models. Pain. 2001;92:277–82. doi: 10.1016/s0304-3959(01)00267-6. [DOI] [PubMed] [Google Scholar]
- 84.Hummel T, Hummel C, Friedel I, Pauli E, Kobal G. A comparison of the antinociceptive effects of imipramine, tramadol and anpirtoline. Br J Clin Pharmacol. 1994;37:325–33. doi: 10.1111/j.1365-2125.1994.tb04285.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Poulsen L, Arendt-Nielsen L, Brosen K, Nielsen KK, Gram LF, Sindrup SH. The hypoalgesic effect of imipramine in different human experimental pain models. Pain. 1995;60:287–93. doi: 10.1016/0304-3959(94)00142-2. [DOI] [PubMed] [Google Scholar]
- 86.Sindrup SH, Nielsen JC, Bjerring P, Arendt-Nielsen L. Imipramine does not affect argon-laser-induced pin-prick pain thresholds and laser-evoked cerebral potentials. Eur J Pain. 1998;2:127–32. doi: 10.1016/s1090-3801(98)90005-2. [DOI] [PubMed] [Google Scholar]
- 87.Le Bars D, Gozariu M, Cadden SW. Animal models of nociception. Pharmacol Rev. 2001;53:597–652. [PubMed] [Google Scholar]
- 88.Sandrini G, Serrao M, Rossi P, Romaniello A, Cruccu G, Willer JC. The lower limb flexion reflex in humans. Prog Neurobiol. 2005;77:353–95. doi: 10.1016/j.pneurobio.2005.11.003. [DOI] [PubMed] [Google Scholar]
- 89.Peghini PL, Katz PO, Castell DO. Imipramine decreases oesophageal pain perception in human male volunteers. Gut. 1998;42:807–13. doi: 10.1136/gut.42.6.807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Sindrup SH, Jensen TS. Efficacy of pharmacological treatments of neuropathic pain: an update and effect related to mechanism of drug action. Pain. 1999;83:389–400. doi: 10.1016/S0304-3959(99)00154-2. [DOI] [PubMed] [Google Scholar]
- 91.Naef M, Curatolo M, Petersen-Felix S, Arendt-Nielsen L, Zbinden A, Brenneisen R. The analgesic effect of oral delta-9-tetrahydrocannabinol (THC), morphine, and a THC–morphine combination in healthy subjects under experimental pain conditions. Pain. 2003;105:79–88. doi: 10.1016/s0304-3959(03)00163-5. [DOI] [PubMed] [Google Scholar]
- 92.Naef M, Russmann S, Petersen-Felix S, Brenneisen R. Development and pharmacokinetic characterization of pulmonal and intravenous delta-9-tetrahydrocannabinol (THC) in humans. J Pharm Sci. 2004;93:1176–84. doi: 10.1002/jps.20037. [DOI] [PubMed] [Google Scholar]
- 93.Roberts JD, Gennings C, Shih M. Synergistic affective analgesic interaction between delta-9-tetrahydrocannabinol and morphine. Eur J Pharmacol. 2006;530:54–8. doi: 10.1016/j.ejphar.2005.11.036. [DOI] [PubMed] [Google Scholar]
- 94.Wallace M, Schulteis G, Atkinson JH, Wolfson T, Lazzaretto D, Bentley H, Gouaux B, Abramson I. Dose-dependent effects of smoked cannabis on capsaicin-induced pain and hyperalgesia in healthy volunteers. Anesthesiology. 2007;107:785–96. doi: 10.1097/01.anes.0000286986.92475.b7. [DOI] [PubMed] [Google Scholar]
- 95.Svendsen KB, Jensen TS, Bach FW. Does the cannabinoid dronabinol reduce central pain in multiple sclerosis? Randomised double blind placebo controlled crossover trial. BMJ. 2004;329:253. doi: 10.1136/bmj.38149.566979.AE. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Raft D, Gregg J, Ghia J, Harris L. Effects of intravenous tetrahydrocannabinol on experimental and surgical pain. Psychological correlates of the analgesic response. Clin Pharmacol Ther. 1977;21:26–33. doi: 10.1002/cpt197721126. [DOI] [PubMed] [Google Scholar]
- 97.Angst MS, Clark JD. Comment on Koltzenburg et al.: differential sensitivity of three experimental pain models in detecting the analgesic effects of transdermal fentanyl and buprenorphine. Pain. 2007;128:292–4. doi: 10.1016/j.pain.2006.12.024. Pain 2006; 126: 165–74. [DOI] [PubMed] [Google Scholar]
- 98.Gijsbers K, Nicholson F. Experimental pain thresholds influenced by sex of experimenter. Percept Mot Skills. 2005;101:803–7. doi: 10.2466/pms.101.3.803-807. [DOI] [PubMed] [Google Scholar]
- 99.Farcomeni A. A review of modern multiple hypothesis testing, with particular attention to the false discovery proportion. Stat Methods Med Res. 2008;17:347–88. doi: 10.1177/0962280206079046. [DOI] [PubMed] [Google Scholar]
- 100.Schulte H, Sollevi A, Segerdahl M. Dose-dependent effects of morphine on experimentally induced cutaneous pain in healthy volunteers. Pain. 2005;116:366–74. doi: 10.1016/j.pain.2005.05.005. [DOI] [PubMed] [Google Scholar]
- 101.Thurauf N, Fleischer WK, Liefhold J, Schmid O, Kobal G. Dose dependent time course of the analgesic effect of a sustained-release preparation of tramadol on experimental phasic and tonic pain. Br J Clin Pharmacol. 1996;41:115–23. doi: 10.1111/j.1365-2125.1996.tb00168.x. [DOI] [PubMed] [Google Scholar]
- 102.Apkarian AV, Bushnell MC, Treede RD, Zubieta JK. Human brain mechanisms of pain perception and regulation in health and disease. Eur J Pain. 2005;9:463–84. doi: 10.1016/j.ejpain.2004.11.001. [DOI] [PubMed] [Google Scholar]
- 103.May A. Neuroimaging: visualising the brain in pain. Neurol Sci. 2007;28(Suppl. 2):S101–7. doi: 10.1007/s10072-007-0760-x. [DOI] [PubMed] [Google Scholar]
- 104.Petersen-Felix S, Arendt-Nielsen L. From pain research to pain treatment: the role of human experimental pain models. Best Pract Res Clin Anaesthesiol. 2002;16:667–80. doi: 10.1053/bean.2002.0258. [DOI] [PubMed] [Google Scholar]
- 105.Woolf CJ, Max MB. Mechanism-based pain diagnosis: issues for analgesic drug development. Anesthesiology. 2001;95:241–9. doi: 10.1097/00000542-200107000-00034. [DOI] [PubMed] [Google Scholar]
- 106.Handwerker HO, Kobal G. Psychophysiology of experimentally induced pain. Physiol Rev. 1993;73:639–71. doi: 10.1152/physrev.1993.73.3.639. [DOI] [PubMed] [Google Scholar]