

Abstract
AIMS
ABCB1, some ABCCs and SLCOs have been reported to affect the intracellular accumulation of various protease inhibitors in vitro and ex vivo. Darunavir is the most recently licensed protease inhibitor and we sought to investigate the ability of transport inhibitors to influence its intracellular accumulation in lymphocytes from healthy volunteers.
METHODS
The intracellular accumulation of radiolabelled darunavir was assessed using CEM cells and ABCB1-overexpressing CEMVBL cells. Apical and basolateral transport of radiolabelled darunavir through MDCKII monolayers was also studied. Finally the ability of known inhibitors to influence intracellular accumulation of darunavir in peripheral blood mononuclear cells (PBMC) was investigated.
RESULTS
CEMVBL cells (1.4 ± 0.6, P < 0.001, 95% CI for the difference = 0.46, 0.80, n= 7) had significantly lower accumulation of darunavir compared with CEM cells (5.6 ± 0.7, n= 7) and this was reversed by addition of tariquidar (30 nm, 4.6 ± 0.8, P < 0.001, 95% CI =−0.64, −0.41, n= 4). In MDCKII-ABCBI cells, transport from the basal to the apical compartment was observed and this was also reversible with the addition of tariquidar. In PBMCs, dipyridamole (6.9 ± 1.3, P < 0.01, 95% CI for the difference =−1.16, −0.30, (n= 8) significantly increased whilst montelukast (5.7 ± 1.0, P < 0.01, 95% CI for the difference = 0.16, 0.79, n= 8) significantly decreased the intracellular accumulation of darunavir when compared with control (6.2 ± 1.1, n= 8).
CONCLUSIONS
Darunavir is a substrate for efflux and influx transporters in PBMC and intracellular concentrations can be manipulated using known inhibitors.
Keywords: darunavir, efflux transport, PBMCs
WHAT IS ALREADY KNOWN ABOUT THIS SUBJECT
Antiretroviral protease inhibitors such as lopinavir and saquinavir have been shown to be substrates of ABCB1. Co-administration with the potent ABCB1 and CYP3A4 inhibitor, ritonavir, has shown improved pharmacokinetics and subsequent therapeutic effects of protease inhibitors. Darunavir is a recently licensed protease inhibitor with potent antiretroviral effects but has yet to be characterized as a potential substrate for drug transporters.
WHAT THIS STUDY ADDS
Darunavir was shown to be a substrate of ABCB1 using different in vitro models. Inhibition of ABCB1 alone does not increase cellular accumulation of darunavir in PBMCs. However, inhibition of ABCB1 and ABCCs significantly increased cellular accumulation of darunavir whereas inhibition of influx transporters significantly reduced the cellular accumulation of darunavir in PBMCs.
Introduction
Darunavir (DRV, TMC114) is a protease inhibitor (PI) with potent antiretroviral activity against PI resistant HIV. Intracellular drug concentrations of other PIs can be influenced by drug transport [1, 2] with ABCB1 being the most extensively researched transporter protein. ABCB1 has broad substrate specificity and wide tissue distribution [3], [4] with expression in the intestine and liver affecting bioavailability of substrates by efflux into the gut lumen and bile respectively [4]. At the cellular level, ABCB1 expression has been demonstrated in lymphocyte subsets including natural killer cells, CD8 and CD4+ cells [5]. Here, drug efflux may serve to decrease drug concentrations at sites vital for the control of HIV replication. PIs are also substrates for ABCC1 and ABCC2 and the ABCC family of proteins vary in tissue distribution but often share substrate specificity and transport a wide range of biological molecules and xenobiotics [6].
More recently, the importance of influx transporters has also been demonstrated with many xenobiotics including statins [7–9], antibiotics [10] and anti-cancer drugs [11] shown to be substrates for influx transporters. We previously examined the mRNA expression of SLCO1A2, SLCO1B1, SLCO1B3, SLCO2B1, SLCO3A1 and SLCO4A1 in peripheral blood mononuclear, CEM and CEMVBL cells and showed that of these, SLCO3A1 was the only isoform expressed in all cell types [2]. We have also shown darunavir to be a substrate for SLCO1A2 and SLCO1B1 [12]. Functional polymorphisms found in some influx transporters have been shown to alter the pharmacokinetics of substrates [13, 14]. The net effect of influx and efflux transporters in a cell is likely to determine overall cellular drug accumulation.
Given the clear importance of drug transport in determining bioavailability, drug distribution and ultimately intracellular concentration of antiretroviral drugs, the aim of this study was to determine whether intracellular concentrations of darunavir at its site of action are modifiable by known inhibitors of efflux [tariquidar (inhibits ABCB1 and ABCG2); MK571 (inhibits ABCC1 and ABCC2), dipyridamole (inhibits ABCB1 and ABCC1), frusemide (inhibits ABCC1 and ABCC2), GF120918 (inhibits ABCG2 and ABCB1), probenecid (inhibits ABCC2 and organic anion transporters), and verapamil (non-specific inhibitor of drug efflux proteins)] and influx [estrone-3-sulphate and montelukast (substrates/competitive inhibibitors of multiple SLCO transporters)].Interactions of darunavir as a substrate and/or inhibitor of ABCB1 were also assessed.
Methods
Materials
[C14]-darunavir (specific activity, 7.6 Bq mmol−1) was provided by Tibotec BVBA (Mechelen, Belgium). Foetal calf serum (FCS) was purchased from Biosera. Tariquidar was a gift from Xenova Group Plc. (Berkshire, UK), GF120918 was obtained from GlaxoSmithKline (Greenford, UK), MK571 and montelukast were gifts from Merck Frosst (Quebec, Canada). Ultima Gold scintillation fluid was purchased from Perkin Elmer (Boston, USA). Nucleocounter cassettes and Nucleocounter reagents were purchased from Sartorius Ltd. (Surrey, UK).
Peripheral blood mononuclear cells (PBMC) were isolated from blood buffy coats obtained from the regional blood transfusion centre (Liverpool, UK). CEM (parental) and CEMVBL100 (P-gp over-expressing) cells were gifts from Dr R. Davey, University of Queensland, Australia. MDCKII-ABCB1 was a gift from Professor P. Borst (The Netherlands Cancer Institute, Amsterdam).
Cell culture
CEM cells are human T-lymphoblastoid cells from which CEMVBL100 cells are derived. CEMVBL100 cells were selected for P-gp over-expression by stepwise selection with vinblastine to a final concentration of 100 ng ml−1[15, 16]. CEM and CEMVBL100 were cultured in RPMI 1640 medium with 10% FCS and incubated at 37.5°C in the presence of 5% CO2. CEMVBL cells were treated routinely with 100 ng ml−1 of vinblastine to maintain their phenotype. Cells were passaged 1 : 6 every 3–4 days and passaged at least twice in the absence of selecting compound prior to use in experiments.
The MDCKII cell lines are canine kidney derived cells, from which the MDCKII-ABCB1 cell was generated by transfection with the plasmid containing the ABCB1 gene [17]. The MDCKII cells were cultured with DMEM supplemented with 10% FCS and incubated at 37.5°C, 5% CO2). MDCKII-ABCB1 was routinely treated with the antibiotic G418 to select for cells containing the ABCB1 plasmid.
Determining the toxicity of darunavir
The cellular toxicity of darunavir was assessed using the MTT (thiazolyl blue tetrazolium bromide) cytotoxicity method [18]. CEM and CEMVBL cells were counted using the NucleoCounter (Chemometec) and cells were resuspended in RPMI 1640 media containing 10% FCS, 1 × 106 cells/ml. Darunavir was serially diluted in RPMI 1640 media containing 10% FCS to give a range of concentrations (100–0.2 µm). Drug dilutions (50 µl) were added to sterile 96-well plates, which were followed by the addition of CEM or CEMVBL cells (50 µl). The plates were then incubated (3 days, 37.5°C, 5% CO2). The assays were terminated by adding MTT (20 µl, 5 mg ml−1 dissolved in HBSS) to each well. The plates were returned to the incubator (37.5°C, 5% CO2, 2 h). Lysis solution (50% v : v dimethylformide in water, 20% w : v lauryl sulphate, 100 µl) was added to each well and plates were incubated overnight (37.5°C, 5% CO2). Plates were analyzed using the Genios XFLUOR4 fluorescence plate reader (560 nmλ). There was no evidence of reduced cell viability in the CEM or the CEMVBL cells when cultured in concentrations of up to 50 µm of darunavir (data not shown).
Characterizing cellular accumulation of darunavir in CEM cell lines
The impact of ABCB1 on darunavir transport was assessed in CEM and CEMVBL cell lines and the ABCB1 inhibitor, tariquidar was used to confirm the involvement of ABCB1. CEM and CEMVBL100 cells were counted using a NucleoCounter. The cells were centrifuged (2000 rev min−1, 5 min) and resuspended with RPMI 1640 medium (5 × 106 cells ml−1). Aliquots of cell suspension (500 µl) were incubated (37.5°C, 5% CO2, 20 min) with RPMI 1640 (500 µl) containing [14C]-darunavir (1 µm) with or without tariquidar (1 µm). After incubation, the samples were centrifuged (9000 rev min−1, 1 min, 4°C) and the supernatant (100 µl) was transferred from each sample to scintillation vials. The remaining supernatant was discarded and the cell pellet was washed with 1 ml of HBSS (9000 rev min−1, 1 min, 4°C). Cell pellets were solubilized with tap water and transferred to scintillation vials. Scintillation fluid (4 ml) was added to each vial and the samples were counted. Following a correction for volume, a comparable value called the cellular accumulation ratio (CAR) was calculated using the following formula:
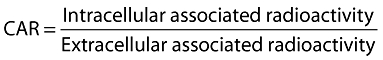 |
Inhibition of darunavir transport in vitro
To observe the inhibitory effects of darunavir on transport by P-gp, the toxicity of vinblastine in the presence of darunavir was assessed. CEM and CEMVBL cells were counted using the NucleoCounter and cells were resuspended in RPMI 1640 media containing 10% FCS (1 × 106 cells ml−1). Vinblastine was serially diluted in RPMI 1640 media containing 10% FCS to give a range of concentrations (2000–10 ng ml−1). In one set of dilutions, tariquidar (final concentration, 1 µm) was added. In another set of dilutions, darunavir (final concentration, 25 µm) was added. Drug (50 µl) was added to sterile 96-well plates, which was followed by the addition of CEM or CEMVBL cells (50 µl). The plates were incubated (3 days, 37.5°C, 5% CO2) and assays were terminated by adding MTT (20 µl, 5 mg ml−1 dissolved in HBSS) to each well. The plates were returned to the incubator (37.5°C, 5% CO2, 2 h). Lysis solution (100 µl) was added to each well and plates were incubated overnight (37.5°C, 5% CO2). The plates were analyzed using the Genios XFLUOR4 fluorescence plate reader (560 nm λ).
Characterizing cellular accumulation of darunavir in MDCKII cell lines
Firstly, transwell plate inserts (3 µm pore size, 24 mm diameter, Costar) were incubated with DMEM supplemented with 10% FCS (30 min, 37°C, 5% CO2). The media was aspirated and MDCKII-ABCB1 cells (1.5 × 106) suspended in DMEM containing 10% FCS were seeded onto inserts in transwell plates (2 ml) and media (2 ml) was added to the bottom chamber. The cells were then cultured with fresh DMEM supplemented with 10% FCS in the apical (AP) and basolateral (BL) chambers each day (3 days, 37°C, 5% CO2). Confluence was measured by transepithelial resistance (above 150 Ω was accepted). The media was aspirated and DMEM containing [14C]-darunavir (0.125 µCi, 2 ml) alone or [14C]-darunavir with tariquidar ([14C]-darunavir 0.125 µCi, tariquidar 1 µm, 2 ml) was added to the AP chamber to measure AP transport and to the BL chamber to measure BL transport. Cells were incubated (37°C, 5% CO2) for pre-determined time intervals (30 min, 1 h, 2 h, 3 h and 4 h), when samples from the AP and BL chambers were taken (100 µl and 4 ml scintillation fluid) and measured by scintillation spectrometry.
Characterizing cellular accumulation of darunavir in PBMCs
Cellular accumulation of darunavir was assessed ex vivo to observe the effects of inhibition of drug transporters on intracellular accumulation in primary PBMC. PBMC were isolated from blood buffy coats by gradient density centrifugation. Blood buffy coat (30 ml) was slowly added to lymphoprep (15 ml) and was centrifuged (2000 rev min−1, 30 min). PBMC were extracted from the solution and washed with HBSS (50 ml, 2000 rev min−1, 5 min). The cells were resuspended and cultured overnight in RPMI 1640 media containing 15% FCS in the presence of phytohaemagglutinin (10 µg ml−1).
Intracellular accumulation was conducted as described above for cell lines using 1 × 107 cells ml−1. Tissue solubilizing solution (50 µl, Optisolve : glacial acetic acid : hydrogen peroxide in a 5 : 5 : 2.5 ratio) was used in combination with water (100 µl) to solubilize the cell pellets. CAR values were calculated as described using the average volume of a PBMC as 0.4 pl [1, 19]. To ascertain the effects of transport proteins present in PBMC on darunavir CAR, a range of inhibitors (tariquidar 1 µm, MK571 50 µm, GF120918 5 µm, dipyridamole 50 µm, furosemide 50 µm, estrone-3-sulphate 100 µm, montelukast 50 µm, verapamil 50 µm, probenecid 50 µm) which were used in previously published work [1]) were co-incubated with the cells and radiolabelled darunavir.
Statistical analysis
All data are given as mean ± SD. For each comparison a P value less than 0.05 was taken as significant and the 95% confidence interval for the difference between means is also given (95% CI). The Shapiro-Wilk test was used to assess normality of the data. Darunavir accumulation in CEM and CEMVBL, in the presence or absence of tariquidar data were log transformed and tested analyzed by paired t-test. The paired t-test was also used to assess differences in darunavir PBMC accumulation for each inhibitor vs controls. The darunavir inhibition of transport data was analyzed using the GraphPad Prism 3.0 software. Log transformation and non-linear regression analysis was used to obtain EC50 concentrations.
Results
Cellular transport of darunavir in CEM cells
The CAR of darunavir was significantly higher in CEM cells (5.6 ± 0.7) than in CEMVBL cells (1.4 ± 0.6, P < 0.001, 95% CI 0.46, 0.80, n= 7). In CEMVBL cells, CAR was significantly increased in the presence of tariquidar in a concentration dependant manner (Figure 1a), with significant increases of CAR at 20 nm tariquidar (2.9 ± 0.5, P < 0.01, 95% CI −0.47, −0.38, n= 4), 30 nm tariquidar (4.6 ± 0.8, P < 0.001, 95% CI −0.64, −0.41, n= 4) and higher concentrations of tariquidar (100 nm, 300 nm, 1000 nm) showing trends towards further increase (4.8, 5.2, 5.2, n= 3).
Figure 1.
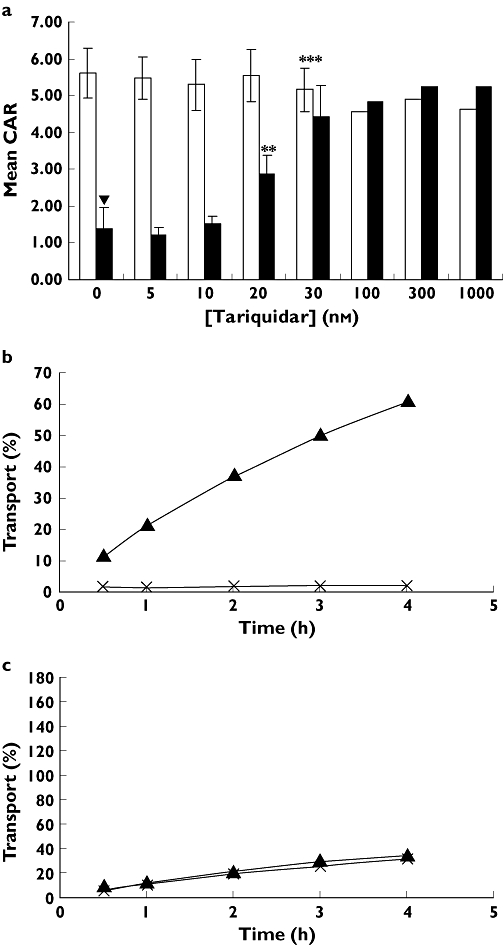
a) The effect of tariquidar on [C14]-darunavir CAR in CEM (open bars) and CEMVBL (closed bars) cell lines. ▾P < 0.001 (n = 7), compared with CEM control. *P < 0.05 (n= 4), **P < 0.01 (n= 4), ***P < 0.001 (n= 7) compared with CEMVBL control. b) The transport of [C14]–darunavir through MDCKII-ABCB1 monolayer (n= 3) AP to BL ( ) and BL to AP (
) and BL to AP ( ). c) MDCKII-ABCB1 monolayer in the presence of tariquidar (n= 3), AP to BL (
). c) MDCKII-ABCB1 monolayer in the presence of tariquidar (n= 3), AP to BL ( ) and BL to AP (
) and BL to AP ( )
)
Cellular transport of darunavir in MDCKII-ABCB1 cells
Transport of [14C]-darunavir from the BL chamber to the AP chamber was observed in the MDCKII-ABCB1 transwell experiments (Figure 1b). The transcelluar transport was reversed with the addition of tariquidar and minimal transport was observed from the AP to the BL chamber (Figure 1c).
Inhibition of ABCB1 transport by darunavir
A lower vinblastine EC50 was observed in CEM than in CEMVBL cells (0.02 ± 0.01 ng ml−1, compared with 35.3 ± 9.8 ng ml−1, P < 0.001, 95% CI, −2.09, −1.06). In the presence of tariquidar, the vinblastine EC50 in CEMVBL cells was reduced (0.45 ± 0.08 ng ml−1, P < 0.001, 95% CI 1.68, 2.09). No effect on vinblastine toxicity was observed with tariquidar (EC50 0.017 ± 0.002 ng ml−1) or darunavir (EC50 0.004 ± 0.004 ng ml−1) in CEM cells. However, vinblastine EC50 in CEMVBL cells was significantly reduced by darunavir (3.50 ± 1.22 ng ml−1, P < 0.001, 95% CI 0.90, 1.11).
Accumulation of darunavir in PBMC in the presence of known inhibitors
In PBMC, the only inhibitors having a significant effect on darunavir CAR were dipyridamole (6.9 ± 1.3, P < 0.01, 95% CI −1.16, −0.30, n= 8) and montelukast (5.7 ± 1.0, P < 0.01, 95% CI 0.16, 0.79, n= 8) compared with baseline CAR (6.2 ± 1.1, n= 8, Figure 2). Dipyridamole increased CAR in seven out of eight samples by a mean CAR change of 11.8 ± 9.4%, whilst montelukast decreased CAR in all eight samples by a mean of −7.5% ± 5.44. The mean CAR of darunavir was not significantly altered by tariquidar (6.6 ± 1.7, n= 8), verapamil (6.5 ± 0.8, n= 8), probenecid (6.3 ± 1.2, n= 8), MK571 (6.4 ± 1.4, n= 8), GF120918 (6.6 ± 1.0, n= 4), furosemide (6.1 ± 1.0, n= 8) or estrone-3-sulphate (6.0 ± 1.0, n= 8).
Figure 2.
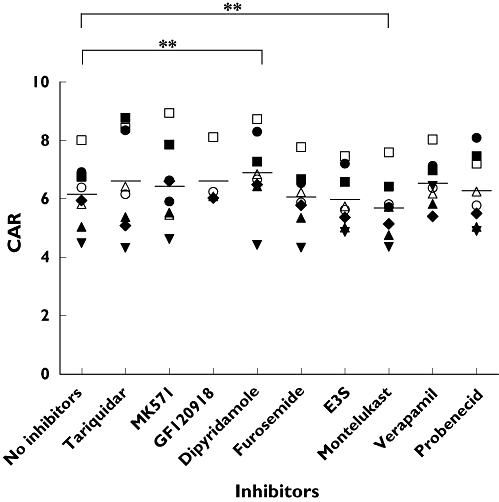
The effect of various inhibitors on the CAR of darunavir in PBMC, **P < 0.01
Discussion
Drug transporter proteins may modulate intracellular drug concentrations, which for an antiretroviral may ultimately affect a drug's ability to suppress HIV replication. Darunavir is a relatively new protease inhibitor with limited characterization of its interaction with transport proteins. Initially, the toxicity of darunavir was assessed in CEM and CEMVBL cells but the highest concentration used (50 µm, dictated by available stocks and solubility) was not toxic in these cells, which is consistent with the lack of toxicity observed by De Meyer and colleagues. in a different cell line [20].
A significantly lower cellular accumulation of [C14]-darunavir was observed in CEMVBL cells compared with parental CEM cells indicating that darunavir is an ABCB1 substrate. This was further supported by the reversal in CEMVBL with increasing concentrations of tariquidar. An increase in transport of darunavir between MDCKII-ABCB1 basolateral compared with apical chambers in the transwell system was also observed, and this increase was reversed with the addition of 1 µm tariquidar. Furthermore, no transport was observed from the apical to the basolateral chamber, which is consistent with other studies which found ABCB1 to be expressed on the basolateral membrane [21].
CEMVBL cells were more resistant to vinblastine toxicity than CEM cells due to over-expression of P-gp. These cells, with vinblastine were therefore used to assess whether darunavir was an ABCB1 inhibitor. Tariquidar was used as a positive control. CEMVBL cell viability was reduced when darunavir was included in vinblastine cultures confirming the previous observation that darunavir is an inhibitor of ABCB1 [22]. CEM cells were cultured with vinblastine and tariquidar or darunavir to act as a negative control and confirm that changes in cell viability were attributable to ABCB1. Neither tariquidar nor darunavir altered cell viability of CEM cells cultured with vinblastine.
In PBMC, the only changes in CAR were in the presence of dipyridamole and montelukast. Montelukast significantly lowered CAR of darunavir in all of the PBMC samples (although to a very modest extent), which is consistent with our previous observation that darunavir is a substrate for influx transporters [12]. This also suggests that influx does have an impact on darunavir cellular accumulation ex vivo. The P-gp inhibitors tariquidar and verapamil did not alter darunavir CAR in PBMC samples. This may be due to lower levels of P-gp expression in PBMC compared with CEMVBL (where the protein is greatly overexpressed) and suggests that P-gp may not be a major contributor of change in intracellular accumulation in PBMC. MK571, the MRP inhibitor also did not alter cellular accumulation of darunavir in PBMC. Dipyridamole is a known inhibitor of multiple ABC transporters and increased CAR in seven out of eight PBMC samples, suggesting that the inhibition of multiple transport systems may be required to increase drug accumulation in these cells.
These data should be interpreted with awareness that lipophilic compounds like darunavir have a tendency to accumulate in the cell membrane. Therefore, it is difficult to determine accurately actual intracellular drug concentration that is able to reach its pharmacological target or that is subject to transport. Therefore, membrane-association may mask larger effects within the cell and it is possible that a proportion of the cell-associated compound is not subject to transport. A small difference in CAR may actually reflect a large difference in intracellular darunavir.
In conclusion, although darunavir is a substrate and inhibitor of ABCB1 in some in vitro models, the effect of ABCB1 on darunavir transport in PBMCs is unlikely to be important. It is possible that ABCB1 may not be the main transporter altering cellular concentrations of darunavir in these cells. However since dipyridamole only increased the cellular accumulation of darunavir by 11.8% it is unlikely that this will prove to have clinical utility as an intracellular booster. Inhibition of influx transporters also impacted to a small degree on darunavir accumulation in PBMC. Although it is clear that intracellular concentrations of darunavir are dependent on complex interactions with multiple transport proteins the ability to manipulate pharmacologically remains a challenge.
Competing interests
None declared.
This work was supported by the MRC, the NIHR Biomedical Research Centre for Microbial Diseases and Tibotec. WSK is a Tibotec funded PhD student.
REFERENCES
- 1.Janneh O, Owen A, Chandler B, Hartkoorn RC, Hart CA, Bray PG, Ward SA, Back DJ, Khoo SH. Modulation of the intracellular accumulation of saquinavir in peripheral blood mononuclear cells by inhibitors of mrp1, mrp2, p-gp and BCRP. AIDS. 2005;19:2097–102. doi: 10.1097/01.aids.0000194793.36175.40. [DOI] [PubMed] [Google Scholar]
- 2.Janneh O, Hartkoorn RC, Jones E, Owen A, Ward SA, Davey R, Back DJ, Khoo SH. Cultured CD4T cells and primary human lymphocytes express hOATPs: intracellular accumulation of saquinavir and lopinavir. Br J Pharmacol. 2008;155:875–83. doi: 10.1038/bjp.2008.320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hoggard PG, Owen A. The mechanisms that control intracellular penetration of the HIV protease inhibitors. J Antimicrob Chemother. 2003;51:493–6. doi: 10.1093/jac/dkg137. [DOI] [PubMed] [Google Scholar]
- 4.Schellens JH, Malingre MM, Kruijtzer CM, Bardelmeijer HA, van Tellingen O, Schinkel AH, Beijnen JH. Modulation of oral bioavailability of anticancer drugs: from mouse to man. Eur J Pharm Sci. 2000;12:103–10. doi: 10.1016/s0928-0987(00)00153-6. [DOI] [PubMed] [Google Scholar]
- 5.Ford J, Meaden ER, Hoggard PG, Dalton M, Newton P, Williams I, Khoo SH, Back DJ. Effect of protease inhibitor-containing regimens on lymphocyte multidrug resistance transporter expression. J Antimicrob Chemother. 2003;52:354–8. doi: 10.1093/jac/dkg381. [DOI] [PubMed] [Google Scholar]
- 6.Leslie EM, Deeley RG, Cole SP. Toxicological relevance of the multidrug resistance protein 1, MRP1 (ABCC1) and related transporters. Toxicology. 2001;167:3–23. doi: 10.1016/s0300-483x(01)00454-1. [DOI] [PubMed] [Google Scholar]
- 7.Hirano M, Maeda K, Shitara Y, Sugiyama Y. Contribution of OATP2 (OATP1B1) and OATP8 (OATP1B3) to the hepatic uptake of pitavastatin in humans. J Pharmacol Exp Ther. 2004;311:139–46. doi: 10.1124/jpet.104.068056. [DOI] [PubMed] [Google Scholar]
- 8.Kopplow K, Letschert K, Konig J, Walter B, Keppler D. Human hepatobiliary transport of organic anions analyzed by quadruple-transfected cells. Mol Pharmacol. 2005;68:1031–8. doi: 10.1124/mol.105.014605. [DOI] [PubMed] [Google Scholar]
- 9.Schneck DW, Birmingham BK, Zalikowski JA, Mitchell PD, Wang Y, Martin PD, Lasseter KC, Brown CD, Windass AS, Raza A. The effect of gemfibrozil on the pharmacokinetics of rosuvastatin. Clin Pharmacol Ther. 2004;75:455–63. doi: 10.1016/j.clpt.2003.12.014. [DOI] [PubMed] [Google Scholar]
- 10.Tamai I, Nezu J, Uchino H, Sai Y, Oku A, Shimane M, Tsuji A. Molecular identification and characterization of novel members of the human organic anion transporter (OATP) family. Biochem Biophys Res Commun. 2000;273:251–60. doi: 10.1006/bbrc.2000.2922. [DOI] [PubMed] [Google Scholar]
- 11.Abe T, Kakyo M, Tokui T, Nakagomi R, Nishio T, Nakai D, Nomura H, Unno M, Suzuki M, Naitoh T, Matsuno S, Yawo H. Identification of a novel gene family encoding human liver-specific organic anion transporter LST-1. J Biol Chem. 1999;274:17159–63. doi: 10.1074/jbc.274.24.17159. [DOI] [PubMed] [Google Scholar]
- 12.Kwan W, Hartkoorn R, Salcedo-Sora E, Bray P, Khoo S, Back D, Owen A. Determining the substrate specificities of SLCO1A2 and SLCO1B1 for antiretroviral drugs. In: 9th International Workshop on Clinical Pharmacology of HIV Therapy; 2008 April 7–9, 2008; New Orleans, USA; 2008.
- 13.Tirona RG, Leake BF, Merino G, Kim RB. Polymorphisms in OATP-C: identification of multiple allelic variants associated with altered transport activity among European- and African-Americans. J Biol Chem. 2001;276:35669–75. doi: 10.1074/jbc.M103792200. [DOI] [PubMed] [Google Scholar]
- 14.Lee W, Glaeser H, Smith LH, Roberts RL, Moeckel GW, Gervasini G, Leake BF, Kim RB. Polymorphisms in human organic anion-transporting polypeptide 1A2 (OATP1A2): implications for altered drug disposition and central nervous system drug entry. J Biol Chem. 2005;280:9610–7. doi: 10.1074/jbc.M411092200. [DOI] [PubMed] [Google Scholar]
- 15.Davey MW, Hargrave RM, Davey RA. Comparison of drug accumulation in P-glycoprotein-expressing and MRP-expressing human leukaemia cells. Leuk Res. 1996;20:657–64. doi: 10.1016/0145-2126(96)00023-9. [DOI] [PubMed] [Google Scholar]
- 16.Beck WT, Mueller TJ, Tanzer LR. Altered surface membrane glycoproteins in Vinca alkaloid-resistant human leukemic lymphoblasts. Cancer Res. 1979;39:2070–6. [PubMed] [Google Scholar]
- 17.Bakos E, Evers R, Szakacs G, Tusnady GE, Welker E, Szabo K, de Haas M, van Deemter L, Borst P, Varadi A, Sarkadi B. Functional multidrug resistance protein (MRP1) lacking the N-terminal transmembrane domain. J Biol Chem. 1998;273:32167–75. doi: 10.1074/jbc.273.48.32167. [DOI] [PubMed] [Google Scholar]
- 18.Mosmann T. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J Immunol Methods. 1983;65:55–63. doi: 10.1016/0022-1759(83)90303-4. [DOI] [PubMed] [Google Scholar]
- 19.Jones K, Hoggard PG, Sales SD, Khoo S, Davey R, Back DJ. Differences in the intracellular accumulation of HIV protease inhibitors in vitro and the effect of active transport. Aids. 2001;15:675–81. doi: 10.1097/00002030-200104130-00002. [DOI] [PubMed] [Google Scholar]
- 20.De Meyer S, Azijn H, Surleraux D, Jochmans D, Tahri A, Pauwels R, Wigerinck P, de Bethune MP. TMC114, a novel human immunodeficiency virus type 1 protease inhibitor active against protease inhibitor-resistant viruses, including a broad range of clinical isolates. Antimicrob Agents Chemother. 2005;49:2314–21. doi: 10.1128/AAC.49.6.2314-2321.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Horio M, Chin KV, Currier SJ, Goldenberg S, Williams C, Pastan I, Gottesman MM, Handler J. Transepithelial transport of drugs by the multidrug transporter in cultured Madin-Darby canine kidney cell epithelia. J Biol Chem. 1989;264:14880–4. [PubMed] [Google Scholar]
- 22.Tong L, Phan TK, Robinson KL, Babusis D, Strab R, Bhoopathy S, Hidalgo IJ, Rhodes GR, Ray AS. Effects of human immunodeficiency virus protease inhibitors on the intestinal absorption of tenofovir disoproxil fumarate in vitro. Antimicrob Agents Chemother. 2007;51:3498–504. doi: 10.1128/AAC.00671-07. [DOI] [PMC free article] [PubMed] [Google Scholar]