

Abstract
AIMS
Sotrastaurin is an immunosuppressant that reduces T-lymphocyte activation via protein kinase C inhibition. The effect of CYP3A4 inhibition by ketoconazole on the pharmacokinetics of sotrastaurin, a CYP3A4 substrate, was investigated.
METHODS
This was a two-period, single-sequence crossover study in 18 healthy subjects. They received a single 50 mg oral dose of sotrastaurin in period 1 followed by a 14-day inter-treatment phase. In period 2 they received ketoconazole 200 mg twice daily for 6 days and a single 50 mg dose of sotrastaurin on the fourth day of ketoconazole administration.
RESULTS
Co-administration of single-dose sotrastaurin during steady-state ketoconazole increased sotrastaurin Cmax by 2.5-fold (90% confidence interval 2.2, 2.9) from 285 ± 128 to 678 ± 189 ng ml−1 and increased AUC by 4.6-fold (4.1, 5.2) from 1666 ± 808 to 7378 ± 3011 ng ml−1 h. Sotrastaurin half-life was nearly doubled from 5.9 ± 1.7 to 10.6 ± 2.5 h. The AUC of the active metabolite N-desmethyl-sotrastaurin was increased by 6.8-fold. Sotrastaurin did not alter ketoconazole steady-state predose plasma concentrations.
CONCLUSIONS
The strong CYP3A4 inhibitor ketoconazole increased sotrastaurin AUC by 4.6-fold. A compensatory reduction in the dose of sotrastaurin is warranted when strong CYP3A4 inhibitors are co-administered.
Keywords: drug interactions, immunomodulators, ketoconazole, protein kinase C, sotrastaurin
WHAT IS ALREADY KNOWN ABOUT THIS SUBJECT?
• Sotrastaurin is an investigational protein kinase C inhibitor. This represents a new mechanism of action for immunosuppression to prevent acute rejection after organ transplantation. In vitro experiments indicate that sotrastaurin is a substrate of CYP3A4.
WHAT THIS STUDY ADDS
• This study provides clinical pharmacokinetic information to quantify the effect of the strong CYP3A4 inhibitor ketoconazole on sotrastaurin. The susceptibility of sotrastaurin to CYP3A4-related drug interactions is placed in the context of similar information about calcineurin inhibitors (tacrolimus, ciclosporin) and T-cell proliferation inhibitors (everolimus, sirolimus).
Introduction
Sotrastaurin (AEB071) is a novel small molecular weight immunosuppressant that blocks T-lymphocyte activation by selectively inhibiting protein kinase C. It is being developed as an immunosuppressant to prevent rejection after solid organ transplantation and for the treatment of various autoimmune diseases. In a 1 month proof-of-concept study in patients with severe psoriasis, for example, a significant reduction from baseline in the severity of psoriasis by up to 70% relative to placebo was observed [1].
Sotrastaurin is orally administered with a tmax ranging from 0.5 to 3 h post-dose and dose-proportional increases in Cmax and AUC after single doses up to 750 mg. It is >98% bound to plasma proteins, primarily α1-acid-glycoprotein. Renal excretion of sotrastaurin is negligible but it is extensively metabolized with metabolites excreted in the bile. In vitro experiments with human liver microsomes and recombinant CYP450 isozymes demonstrated that sotrastaurin is metabolized primarily by CYP3A4. The elimination half-life averages 6 h. There is one active metabolite, N-desmethyl-sotrastaurin, with a potency in mixed lymphocyte reaction tests similar to that of the parent compound. However, blood concentrations of this metabolite are relatively low compared with the parent drug (<5%).
Given these biotransformation data, it is reasonable to assume that the clearance of sotrastaurin may be decreased by inhibitors of CYP3A4. According to current practice in drug development, a clinical drug interaction study using ketoconazole as a strong CYP3A4 inhibitor is a logical first step to test an extreme scenario and thereafter to test other CYP3A4 inhibitors, if warranted by the ketoconazole results [2, 3]. The study described herein conforms to this strategy and yields insight into the sensitivity of sotrastaurin to inhibition via the CYP3A4 pathway.
Methods
Study design
This was an open-label, two period, single-sequence crossover study planned for 18 healthy subjects. In period 1 pre-study clinical assessments were performed on day −1. On day 1 subjects received 50 mg sotrastaurin with clinical and pharmacokinetic assessments to day 3. After a 14 day inter-treatment phase, baseline clinical assessments for period 2 were performed on day 14. Ketoconazole was administered orally 200 mg every 12 h from day 15 to 20. On day 18, a single dose of 50 mg sotrastaurin was co-administered. Clinical and pharmacokinetic assessments were performed from day 15 to day 21. The protocol was reviewed and approved by the Independent Investigational Review Board, Inc, Plantation, FL, USA. Subjects gave written informed consent to participate.
Study population and disposition
We enrolled 23 subjects of whom 18 completed the study. Of the five subjects who did not complete the study one withdrew consent for personal reasons, two violated the protocol having positive urine cotinine at the period 2 baseline, one was withdrawn for unco-operative behaviour and one was withdrawn for an adverse event as described below. In the full study population, there were 21 men and two women aged 31.1 ± 6.7 years (range 21–43), weighing 75.0 ± 9.8 kg (range 60–91.5), with a body mass index of 25.0 ± 2.4 kg m−2 (range 19.8–28.7). There were 21 Whites and two Blacks.
Domiciling and study drug administration
Subjects stayed at the study site from the evening of day −2 to day 3 in period 1 and from the evening of day 13 to day 21 of period 2. Sotrastaurin was administered as a single oral dose of a simple powder mixture in a capsule of 50 mg strength with 240 ml water after a 10 h fast on days 1 and 18. Ketoconazole tablets (Teva Pharmaceuticals) were administered orally 200 mg every 12 h from day 15 to day 20.
Clinical assessments
Standard biochemistry, haematology and urinalysis laboratory parameters were assessed at baseline before each period and at the end of the study. Vital signs (blood pressure and pulse) were recorded frequently for 48 h after each sotrastaurin dose and at the end of the study. Electrocardiograms were recorded at pre-dose and 2 and 6 h post-dose on the 2 days of sotrastaurin administration and at the end of the study.
Pharmacokinetic assessments and bioanalytics
Blood samples for the determination of sotrastaurin and N-desmethyl-sotrastaurin were collected pre-dose and then 0.25, 0.5, 1, 1.5, 2, 2.5, 3, 4, 6, 9, 12, 16, 24, 36, and 48 h post-dose. For ketoconazole, venous blood samples were collected before the morning dose on days 17−21.
Sotrastaurin and N-desmethyl-sotrastaurin were extracted from whole blood with tert-butyl methyl ether under alkaline conditions. Reconstituted samples were analyzed by a validated liquid chromatography method with tandem mass spectrometry using turbo ion spray in positive mode. As applied in this study for both analytes, there were seven calibration concentrations (range 3−2400 ng ml−1) and three quality control concentrations (9, 200, 2000 ng ml−1). For sotrastaurin, quality control precision ranged from 5.5%−8.7% and bias from −2.5%−4.7%. For N-desmethyl-sotrastaurin, quality control precision ranged from 4.9%−9.9% and bias from 2.0%−10.1%. The lower quantification limit was 3 ng ml−1 for both analytes. Ketoconazole plasma concentrations were determined by reverse phase high performance liquid chromatography with ultraviolet detection (wavelength 254 nm). There were seven calibration concentrations (range, 0.01−5 µg ml−1) and three quality control concentrations (0.02, 2.5, 4 µg ml−1). Quality control precision ranged from 3.2%−10.5% and bias from −3.2%−10.5%. The lower quantification limit was 0.01 µg ml−1.
Standard noncompartmental pharmacokinetic parameters were derived in WinNonlin (Pharsight Corp, Mountain View, CA, USA). For sotrastaurin, they included the time post-dose when the maximum concentration occurred (tmax), the maximum concentration (Cmax), the area under the concentration−time curve by trapezoidal summation (AUC), and the terminal half-life by unweighted log-linear regression (t1/2). Given the lower blood concentrations of N-desmethyl-sotrastaurin, the terminal half-life could not be estimated and the AUC parameter was calculated to the last time point with a quantifiable concentration designated as AUC(0,t). The metabolic ratio was calculated as the metabolite : parent ratio of AUC(0,t) expressed as molar equivalents.
Statistical evaluation
Pharmacokinetic parameters were log-transformed and compared between treatments using a linear mixed-effects model with treatment as a fixed effect and subject as a random effect in SAS (SAS Institute, Cary, NC, USA). Test : reference ratios of the parameter geometric means and their 90% confidence intervals were generated.
Results
Pharmacokinetics of sotrastaurin
Pharmacokinetic parameters are summarized in Table 1 and mean concentration−time profiles are shown in Figure 1. Co-administration of ketoconazole with sotrastaurin increased sotrastaurin Cmax in all subjects ranging from 1.5−4.2-fold with an overall average increase of 2.5-fold (90% confidence interval 2.2, 2.9). Intersubject variability of Cmax was numerically lower during co-administration (28%) compared with administration alone (45%). AUC increased in all subjects ranging from 3.1−7.7-fold with an overall increase of 4.6-fold (90% confidence interval 4.1, 5.2). Intersubject variability of AUC was largely unaffected by the drug interaction: 48% (alone) vs 41% (combination). The half-life was nearly doubled in the presence of ketoconazole by an average of 88%.
Table 1.
Sotrastaurin pharmacokinetics
| Parameter | Sotrastaurin alone | Sotrastaurin with ketoconazole |
|---|---|---|
| Sotrastaurin | ||
| tmax (h) | 2 (0.5–3) | 2.5 (1.5–4) |
| Cmax (ng ml−1) | 285 ± 128 | 678 ± 189 |
| AUC(0,t) (ng ml−1 h) | 1619 ± 807 | 7282 ± 2972 |
| AUC (ng ml−1 h) | 1666 ± 808 | 7378 ± 3011 |
| t1/2 (h) | 5.9 ± 1.7 | 10.6 ± 2.5 |
| N-desmethyl-sotrastaurin | ||
| tmax (h) | 2.5 (1–3.1) | 4 (2–6) |
| Cmax (ng ml−1) | 7.5 ± 3.6 | 9.6 ± 2.9 |
| AUC(0,t) (ng ml−1 h) | 20 ± 13 | 98 ± 49 |
| Metabolic ratio | 0.012 ± 0.007 | 0.014 ± 0.008 |
Values are mean ± SD except for tmax which is median (range). tmax is time to reach maximum concentration; Cmax is maximum concentration; AUC is area under the concentration–time curve; t1/2 is half-life.
Figure 1.
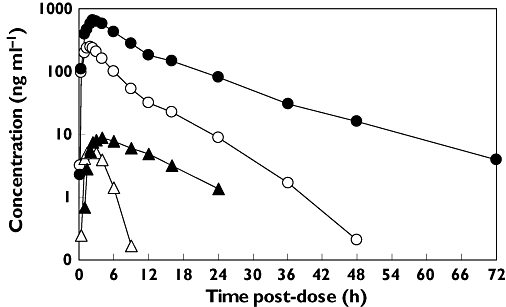
Mean concentration−time profiles of sotrastaurin alone (○) and with ketoconazole (•) and N-desmethyl-sotrastaurin alone (▵) and with ketoconazole (▴)
Peak N-desmethyl-sotrastaurin concentrations were delayed a median 1.5 h in the presence of ketoconazole, from 2.5 to 4 h post-dose. Of the 17 evaluable subjects, eight had no change in Cmax and the remaining nine subjects had increases ranging from 1.3−2.4-fold. The overall increase in Cmax across all subjects was 1.4-fold. AUC(0,t) increased in all subjects ranging from 2.4−24.4-fold with an average increase of 6.8-fold. Intersubject variability in AUC(0,t) was numerically lower in the presence of ketoconazole (50%) vs in its absence (65%). The metabolic ratio, calculated as the metabolite : parent AUC ratio corrected for molecular weight, was slightly higher by 35% in the presence of ketoconazole.
Ketoconazole pharmacokinetics
Ketoconazole blood samples were obtained before the morning dose on days 17 and 18 in the absence of sotrastaurin and days 19−21 in the presence of sotrastaurin. They are plotted in Figure 2. Mean ketoconazole pre-dose concentrations were stable over the observation period regardless of whether sotrastaurin was present systemically or not. They averaged 0.87 ± 0.73 µg ml−1 on day 17 and 1.11 ± 1.08 µg ml−1 on day 21. Regression analysis of ketoconazole troughs vs time across days 17−21 yielded a slope of −0.0008 ng ml−1 day−1 (90% CI −0.04, 0.04), not different from a horizontal line. Hence, ketoconazole exposure was at steady-state before the administration of sotrastaurin and concentrations did not change during co-administration.
Figure 2.
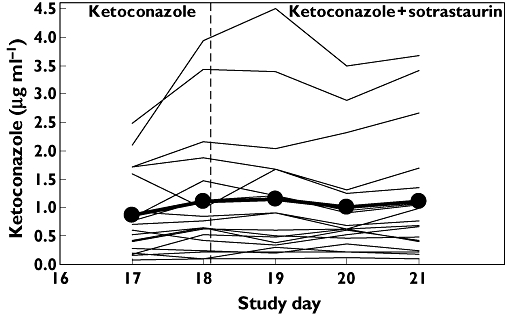
Mean ketoconazole pre-dose concentration trajectory (•) and individual trajectories (lines). Sotrastaurin was administered just after the ketoconazole trough on day 18
Exposure-response associations
A plot of the ketoconazole pre-dose concentration on day 18 vs the fold-change in sotrastaurin AUC (not shown) did not reveal any meaningful pattern indicating that ketoconazole exposure was not predictive of the magnitude of the interaction on sotrastaurin. Figure 3 shows that subjects with a low baseline sotrastaurin AUC tended to have a greater increase in sotrastaurin exposure in the presence of ketoconazole than subjects with a high baseline AUC.
Figure 3.
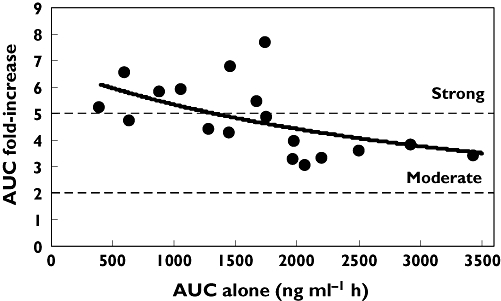
Scatter plot of baseline sotrastaurin AUC vs the fold-change in AUC in the presence of ketoconazole. Shown is a trendline based on a standard Imax inhibitory model (r2= 0.30). Demarcated is the fold-change for a moderate interaction (≥2-fold) and a strong interaction (>5-fold)
Clinical observations
A total of 12 adverse events were reported by nine subjects. Adverse events suspected to be study drug related consisted mainly of mild headache and nausea and did not show any association with a particular treatment (sotrastaurin, ketoconazole, or the combination). Furthermore one subject had an elevated alanine aminotransferase concentration of 74 U l−1 (normal range 3–60 U l−1) at the end of the study which was suspected to be study drug related. He was requested to return for a re-assessment but did not report back until 2 months after study completion at which time the value was 18 U l−1. There were no clinically relevant or drug-related changes over the course of the study in other laboratory parameters, vital signs or electrocardiograms.
Discussion
In vitro experiments indicated that sotrastaurin is a substrate of CYP3A4. Because this isozyme plays a role in the biotransformation of many medications, it is of relevance to determine how sensitive sotrastaurin is to inhibition by co-medications that are CYP3A4 inhibitors. The conventional approach is to use ketoconazole, a strong CYP3A4 inhibitor, as a probe to quantify the maximum expected change in the substrate systemic exposure during co-administration. Our clinical study demonstrated that ketoconazole caused a 2.5-fold increase in sotrastaurin Cmax and a 4.6-fold increase in sotrastaurin AUC. Even with elevated sotrastaurin blood concentrations, this drug combination was well tolerated and safe under the conditions studied. It was reassuring to note that the intersubject pharmacokinetic variability of sotrastaurin exposure was not increased as a result of the interaction. The magnitude of the interaction on sotrastaurin was not related to the ketoconazole plasma concentration which was consistent with ketoconazole exposure being above the inhibitory constant for CYP3A4 inhibition from the dose regimen used in this study. By contrast, we noted that subjects whose baseline sotrastaurin AUC was at the lower end of the population range tended to have a stronger interaction when expressed as the fold-increase in sotrastaurin AUC. Our interpretation is that subjects with a low baseline AUC have a high extraction ratio (which is probably due to high CYP3A4 expression) and therefore a low bioavailability. Consequently, they would have a greater potential for an increase in bioavailability after administration of a CYP3A4 inhibitor and this may explain the larger interaction magnitude. Sotrastaurin is also a substrate of MDR1 in vitro and ketoconazole is an inhibitor of this transporter. Hence, inhibition via this pathway may also have contributed to the interaction on sotrastaurin, but a precise quantitation of MDR1 inhibition cannot be made based on the data from this study.
Although the blood concentrations of N-desmethyl-sotrastaurin were low, they provided some additional insights into this drug interaction. The delayed appearance of the metabolite in blood may reflect the acute influence of CYP3A4 inhibition on the formation of this metabolite. Subsequently, metabolite blood concentrations in the presence of continued CYP3A4 inhibition by ketoconazole exceeded those when sotrastaurin was given without an inhibitor. The exact reasons for this are not discernible from the data but may indicate that biliary clearance of the metabolite is reduced due to transporter inhibition by ketoconazole and/or that subsequent CYP3A4-related metabolite biotransformation steps are inhibited by ketoconazole. Regardless, the metabolic ratio increased only slightly in the presence of ketoconazole suggesting that the overall proportion of systemic exposure to parent and to the N-desmethyl metabolite remains largely unchanged during the interaction.
According to the current classification system for the magnitude of drug interactions [2, 3], the influence of strong CYP3A4 inhibition by ketoconazole on sotrastaurin AUC is categorized as moderate (≥2–<5-fold). With respect to other immunosuppressants, this is in the range of the influence of ketoconazole on ciclosporin AUC which increases 5-fold [4] and tacrolimus which increases 2-fold [5], but it is less than the marked influence of ketoconazole on sirolimus AUC of an 11-fold increase [6] and everolimus of a 15-fold increase [7]. Based on the above data, it is recommended to avoid the use of strong CYP3A4 inhibitors in patients taking sirolimus or everolimus; whereas, dose reductions are generally feasible to compensate for the drug interactions with ciclosporin [8] or tacrolimus [9]. The present study indicates that for sotrastaurin an average 4−5-fold dose reduction appears warranted when sotrastaurin is co-administered with a strong CYP3A4 inhibitor. It is not yet known whether sotrastaurin dosing will be guided by therapeutic drug monitoring. If so, this could aid in individually adjusting the dose of sotrastaurin upon adding or removing a strong CYP3A4 inhibitor from a patient's drug regimen. In addition, clinical monitoring could focus on nausea and vomiting, the dose-limiting adverse events for sotrastaurin in clinical trials. Alternatively, the use of a co-medication which is not a CYP3A4 inhibitor, if available, could be considered depending on the therapeutic options.
Competing interests
J.M.K., H.-L.A.H., A.S. and N.S. are employees of Novartis with rights to stock options. P.A.C. declared no conflicts of interest.
We thank Francoise Pommier for performing the bioanalysis of the blood samples.
REFERENCES
- 1.Skvara H, Dawid M, Kleyn CE, Wolff B, Meingassner JG, Knight H, Dumortier T, Kopp T, Fallahi N, Stary G, Burkhart C, Grenet O, Wagner J, Hijazi Y, Morris RE, McGeown C, Rordorf C, Griffiths CEM, Stingl G, Jung T. Potential therapeutic option for psoriasis with aeb071, a novel protein kinase c inhibitor. J Clin Invest. 2008;118:3151–9. doi: 10.1172/JCI35636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bjornsson TD, Callaghan JT, Einholf HJ. The conduct of in vitro and in vivo drug-drug interaction studies: a PhRMA perspective. J Clin Pharmacol. 2003;43:443–69. [PubMed] [Google Scholar]
- 3.United States Food and Drug Administration. Draft guidance for industry: drug interaction studies – study design, data analysis, and implications for dosing and labeling. 2006. Available at http://www.fda.gov/drugs/guidancecomplianceregulatoryinformation/guidances (last accessed 17 June 2009.
- 4.Choc MG, Mueller EA, Robinson WT, Kuhmle A, Smith HT, Charnick SB. Relative bioavailability of Neoral ver. us Sandimmun in the presence of a P450IIIA and P-glycoprotein inhibitor. Transplant Proc. 1998;30:1664–5. doi: 10.1016/s0041-1345(98)00383-2. [DOI] [PubMed] [Google Scholar]
- 5.Floren LC, Bekersky I, Benet LZ, Mekki Q, Dressler D, Lee JW, Roberts JP, Herbert MF. Tacrolimus oral bioavailability doubles with co-administration of ketoconazole. Clin Pharmacol Ther. 1997;62:41–9. doi: 10.1016/S0009-9236(97)90150-8. [DOI] [PubMed] [Google Scholar]
- 6.Floren LC, Christians U, Zimmerman JJ, Neefe L, Schorer R, Rushworth D, Harper D, Renz J, Benet LZ. Sirolimus oral bioavailability increases ten-fold with concomitant ketoconazole (abstract) Clin Pharmacol Ther. 1999;65:159. [Google Scholar]
- 7.Kovarik JM, Beyer D, Bizot MN, Jiang Q, Shenouda M, Schmouder RL. Blood concentrations of everolimus are markedly increased by ketoconazole. J Clin Pharmacol. 2005;45:514–8. doi: 10.1177/0091270005275368. [DOI] [PubMed] [Google Scholar]
- 8.el-Agroudy AE, Sobh AE, Hamdy MA, Ghoneim MA. A prospective, randomized study of coadministration of ketoconazole and cyclosporine A in kidney transplant recipients: ten-year follow-up. Transplantation. 2004;77:1371–6. doi: 10.1097/01.tp.0000121133.84763.26. [DOI] [PubMed] [Google Scholar]
- 9.Tuteja S, Alloway RR, Johnson JA, Gaber O. The effect of gut metabolism on tacrolimus bioavailability in renal transplant recipients. Transplantation. 2001;71:1303–7. doi: 10.1097/00007890-200105150-00021. [DOI] [PubMed] [Google Scholar]