

Abstract
AIMS
To assess the association between polymorphisms of the ABCB1 gene and the pharmacokinetics of verapamil among healthy Chinese Han ethnic subjects.
METHODS
Based on polymorphisms of the ABCB1 gene at positions 2677 and 3435, 24 healthy male participants were divided into three groups: 2677GG/3435CC (n = 6), 2677GT/3435CT (n = 12) and 2677TT/3435TT (n = 6). Each subject had received a single oral dose of verapamil (80 mg) under fasting conditions. Multiple blood samples were collected over 24 h, and plasma concentrations of verapamil were determined by HPLC. Pharmacokinetic characteristics were compared between the different genotypic groups.
RESULTS
The pharmacokinetics parameters of verapamil differed significantly among the three genotypic groups. AUC(last) was significantly lower among individuals with the 2677TT/3435TT (159.5 ± 79.0 ng ml−1 h) and 2677GT/3435CT (189.3 ± 73.1 ng ml−1 h) genotypes than those with the 2677GG/3435CC genotype (303.1 ± 83.7 ng ml−1 h) (P= 0.004 and P= 0.008, respectively). However, the CL/F value was higher among subjects with the 2677TT/3435TT (523.0 ± 173.7 l h−1) genotype than those with the 2677GT/3435CT (452.2 ± 188.6 l h−1) or 2677GG/3435CC (265.4 ± 72.8 l h−1) genotypes. A significant difference was also found between the latter two groups (P= 0.034). In addition, the Cmax tended to be higher among subjects with the 2677GG/3435CC genotype than those with the 2677GT/3435CT or 2677TT/3435TT genotypes (42.2 ± 3.9 vs 32.2 ± 16.2 vs 38.1 ± 13.7 ng ml−1).
CONCLUSIONS
Our study showed for the first time that verapamil pharmacokinetics may be influenced by particular genetic polymorphisms of the ABCB1 gene among healthy Chinese Han ethnic subjects. An individualized dosage regimen design incorporating such information may improve the efficacy of the drug whilst reducing adverse reactions.
Keywords: ABCB1, pharmacokinetics, SNP, verapamil
WHAT IS ALREADY KNOWN ABOUT THIS SUBJECT
The pharmacokinetics of verapamil have been assessed in a number of studies.
As a substrate of P-glycoprotein (P-gp), verapamil shows a large inter-individual variability in terms of plasma concentrations.
It has been confirmed that polymorphisms of the ABCB1 gene at positions 2677 and 3435 are related to the expression and function of P-gp.
WHAT THIS STUDY ADDS
This study has confirmed that polymorphisms of ABCB1 gene may influence the pharmacokinetics of verapamil among healthy Chinese Han ethnic subjects.
Introduction
Verapamil is one of the most widely used calcium ion influx inhibitors with anti-arrhythmic, anti-anginal, and antihypertensive properties, and is approved for the treatment of angina (including vasospastic and unstable types), atrial arrhythmias, and hypertension. Clinical use of the drug, however, has been complicated by a narrow therapeutic range and highly variable pharmacokinetics in individual patients [1, 2]. Various factors including age, gender and drug interactions may influence the efficacy of verapamil [3–6], among which genetic factors are thought to contribute substantially to inter-individual variations in the disposition of the drug. Identification of genetic parameters predictive of optimal dosage is therefore of great clinical interest.
Verapamil is a substrate for P-glycoprotein (P-gp, a multi-drug resistance transporter), product of the ABCB1 (ATP-binding cassette 1) gene. P-gp is widely expressed in tumour cells, but is also found on the apical surface of intestinal epithelial cells, the biliary canalicular membrane of hepatocytes, the luminal surface of the capillary cells forming the blood brain barrier, the brush border membranes of proximal tubules in the kidneys, and the adrenal cortex and placenta. As a transporter, it plays a significant role in drug disposition, i.e. absorption, distribution, and excretion, and may also be involved in secretion of steroids [7–9]. Although comparison of the uptake of two P-gp substrates, verapamil and vinblastine, showed that the intestinal absorption profile of verapamil was not affected by P-gp [10], several groups have concluded that the effect of P-gp on intestinal absorption of the drug may not be predicted based on in vitro data [11, 12].
The ABCB1 gene has been found to be highly polymorphic between individuals and different ethnic groups. Three most frequently occurring single nucleotide polymorphisms (SNPs) of the gene included 1236T in exon 12, 2677T/A in exon 21 and 3435T in exon 26 [13]. In recent years, clinical studies have been conducted worldwide to investigate the association between polymorphisms of the ABCB1 gene, expression and function of P-gp and pharmacokinetics of its substrates. Discrepancies, however, seem to exist between the results [14–23]. As genotypes comprising particular SNPs may be responsible for the alteration in the functions of P-gp [16, 24, 25], two SNPs of the ABCB1 gene, i.e. 2677T/A and 3435T, are currently considered as the main genetic factors implicated. In this study, we attempted to assess the influence of such SNPs on the pharmacokinetics of verapamil among Chinese Han ethnic subjects.
As verapamil is also known to be a substrate of CYP3A4, oral clearance of the drug, therefore, may be influenced by not only its absorption and/or transportation via P-gp, but also metabolic pathways. Considering the relatively low prevalence of the mutant alleles in the Chinese populations and uncertain in vivo function(s) [26, 27], we did not assess the effect of CYP3A4 polymorphisms in present study. On the other hand, CYP3A5 genotypes have recently been associated with oral clearance and verapamil response in healthy subjects as well as patients [28, 29]. The CYP3A5*3 allele is characterized by an A→G SNP in intron 3 of the gene, which has created a cryptic consensus splice site and exon 3B. The mRNA of this splice variant contains a premature stop codon that can result in almost complete absence of CYP3A5 protein [30]. As a common mutant, the allelic frequency of CYP3A5*3 reached 93% in Caucasians, 72% in Asians, and 32% in African Americans. To exclude the effect of the CYP3A5*3 allele on our result, we also detected the CYP3A5*3 allele in our samples.
Methods
Subjects
Among 202 subjects who had been previously genotyped for ABCB1 exons including polymorphisms 2677T/A and 3435T, 24 healthy males (Han ethnics) were enrolled. These included six individuals with 2677GG/3435CC (age 22.3 ± 0.8 years, BMI 21.54 ± 1.99 kg m−2), six with 2677TT/3435TT (age 22.2 ± 1.0 years, BMI 21.22 ± 1.47 kg m−2) and 12 with 2677GT/3435CT (age 21.9 ± 1.1 years, BMI 21.24 ± 1.71 kg m−2) genotypes, and no significant difference was detected between different genotypic groups. The study protocol was approved by the ethics committee of Shengjing Hospital affiliated to China Medical University and performed according to the rules of the Helsinki declaration. Participants were given both oral and written information about the study protocol before a written consent was obtained.
All subjects were non-smokers and in good health as determined by medical history, physical examination, ECG evaluation and routine laboratory tests (blood chemistry, haematology and urine analysis). They were asked not to take any prescription or non-prescription medication from 2 weeks before and throughout the study, and were instructed to abstain from grapefruit, grapefruit juice, herbal dietary supplements, and caffeine-containing beverages including coffee and green tea 3 days before and throughout the study. Only subjects fulfilling above criteria were included.
Genotyping of ABCB1 polymorphism
A blood sample (2 ml) was obtained from each subject, and DNA was extracted from peripheral whole blood of each subject using an EZ-10 Spin Column Genomic DNA Minipreps Kit (for blood) (Bio Basic Inc.). The genotypes of 2677T/A, 3435T and CYP3A5*3/*3 were determined, respectively, with allele-specific polymerase chain reaction (AS-PCR), polymerase chain reaction-restriction fragment length polymorphism analysis (RFLP-PCR), and direct sequencing [31].
To verify the results obtained from gel electrophoresis analysis of AS-PCR or RFLP-PCR, samples of each genotype (homozygous wild-type, heterozygous, and homozygous polymorphism, a total of 15 samples) were sequenced. The confirmed genotypes allowed us to use the common nomenclature 2677T/A, and 3435T for such sites (GenBank accession numbers NC000007, NM000927).
Pharmacokinetic study
After overnight fasting for 10 h, each subject received a single oral dose of 80 mg verapamil with 240 ml of water (verapamil (immediate-release tablet, 40 mg, Batch no. 061001, Central Pharmaceutical Co., Ltd of Tianjin, China)). Standardized meals were served 4 h and 10 h after dosing. Venous blood samples were collected at pre-dose (0 h) and 0.33, 0.67, 1, 1.5, 2, 3, 4, 6, 8, 12 and 24 h post-dosing. All blood samples were collected into lithium heparin-coated tubes and centrifuged immediately. Plasma samples were stored at −20°C until use.
Plasma verapamil concentrations were analyzed using validated high-performance liquid chromatography (HPLC) with fluorescence detection as described previously with a slight modification. A linearity calibration curve in the range of 1–100 ng ml−1 was established for verapamil (r2= 0.9973). Intra- and inter-day coefficients of variation (CV) were < 15% and < 10%, respectively.
The peak plasma concentration (Cmax) and the time to reach Cmax(tmax) were estimated directly from the observed data. The plasma concentration–time curve for time 0–24 h (AUC(last)) was calculated using the linear trapezoidal rule. The AUC from time 0 to infinity (AUC(0,∞) was calculated as AUC(0,∞) = AUC(last) +Ct/ke, where Ctwas the last-measured plasma concentration and ke was determined using linear regression analysis of the logarithm-linear part of the plasma concentration–time curve. The half-life (t1/2) of verapamil was calculated as t1/2= ln2/ke. The apparent oral clearance (CL/F) of verapamil was calculated as CL/F= dose/AUCinf.
Statistical analysis
All data were expressed as mean ± SD. Normally distributed data were analyzed with one-way anova (for multi-group comparison) or LSD or S-N-K-test (for two-group comparison). Non-normally distributed data were analyzed with Mann-Whitney U-test (for two-group comparison) or Kruskal-Wallis H-test (for multi-group comparison). Statistical analysis was carried out using the SPSS package (version 11.0; SPSS Inc., Chicago, IL, USA). A P value < 0.05 was considered to be statistically significant.
Results
All subjects had completed the study without clinically important adverse effects.
Genotype-phenotype correlations
Pharmacokinetic parameters of verapamil for different genotypic groups were summarized. As shown in Table 1, plasma concentrations in the 2677GT/3435CT and 2677TT/3435TT groups were lower than those of the 2677GG/3435CC group (Figure 1). A statistical difference (P < 0.01) was observed between all paired groups except for the 2677TT/3435TT and 2677GT/3435CT groups (P > 0.05). Average values of AUC(last) were significantly higher (P < 0.01) in the 2677GG/3435CC group (303.1 ± 83.7 ng ml−1 h) as compared with the 2677GT/3435CT (189.3 ± 73.1 ng ml−1 h) and 2677TT/3435TT groups (159.5 ± 79.0 ng ml−1 h). In addition, apparent oral clearance of verapamil was, respectively, 44% and 52% lower in the 2677GG/3435CC group than in the 2677GT/3435CT and 2677TT/3435TT groups (P < 0.01). The elimination half-life (t1/2) of verapamil also appeared to be greater in the 2677GG/3435CC group (7.33 h vs 6.23 h and 6.48 h). This difference, however, was not statistically significant (P > 0.05), possibly due to inter-individual variability as well as the small number of recruited subjects. No significant difference was found with other pharmacokinetic parameters among the three groups.
Table 1.
Pharmacokinetic profiles of verapamil in different genotypic groups following administration of a single dose of verapamil (80 mg)
| Genotype group | Multiple comparison | |||||
|---|---|---|---|---|---|---|
| Parameter | 2677GG/3435CC (Group I, n= 6) | 2677GT/3435CT (Group II, n= 12) | 2677TT/3435TT (Group III, n= 6) | II vs I | III vs I | III vs II |
| Cmax (ng ml−1) | 42.2 ± 3.9 | 32.2 ± 16.2 | 38.1 ± 13.7 | 0.156 | 0.607 | 0.396 |
| (95% CI) | (30.6, 53.8) | (24.0, 40.3) | (26.5, 49.6) | |||
| tmax | 3.00 ± 1.79 | 2.29 ± 0.92 | 1.75 ± 0.76 | 0.234 | 0.075 | 0.360 |
| (95% CI) | (2.00, 3.98) | (1.60, 2.99) | (0.77, 2.73) | |||
| Half-life (h) | 7.33 ± 2.18 | 6.23 ± 1.55 | 6.48 ± 1.35 | 0.204 | 0.391 | 0.769 |
| (95% CI) | (5.91, 8.76) | (5.23, 7.24) | (5.06, 7.91) | |||
| AUC(last) (ng ml−1 h) | 303.1 ± 83.7 | 189.3 ± 73.1 | 159.5 ± 79.0 | 0.008 | 0.004 | 0.449 |
| (95% CI) | (237.6, 368.6) | (142.9, 235.6) | (94.0, 225.0) | |||
| AUC(0, ∞) (ng ml−1 h) | 322.4 ± 93.7 | 203.0 ± 73.8 | 172.1 ± 74.1 | 0.007 | 0.003 | 0.443 |
| (95% CI) | (255.3, 389.5) | (155.5, 250.5) | (105.0, 239.0) | |||
| CL/F (l h−1) | 265.4 ± 72.8 | 452.2 ± 188.6 | 523.0 ± 173.7 | 0.034 | 0.013 | 0.399 |
| (95% CI) | (125.6, 405.1) | (353.4, 551.0) | (383.3, 662.7) | |||
Data are shown as mean ± SD. tmax is given as median (range). Cmax, Peak plasma concentration; tmax, time to Cmax; AUC(last), area under the concentration–time curve from 0 to 24 h; AUC(0,∞), area under the concentration–time curve from 0 to infinity; CL/F, oral clearance.
Figure 1.
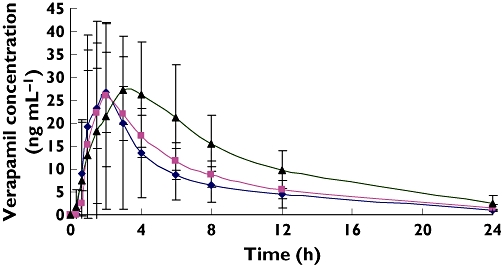
Plasma concentration–time curve following oral administration of 80 mg verapamil. Values are given as mean ± SD. 2677GG/3435CC (▴); 2677GT/3435CT( ); 2677TT/3435TT(♦)
); 2677TT/3435TT(♦)
As shown by the result, all subjects had a CYP3A5*3/*3 genotype. This frequency (24/24, 100%) was higher than that reported (72%) partly because our subjects were non-randomly recruited.
Discussion
Inter-individual variability in drug efficacy and toxicity, which may result in unpredictable drug responses, is common in clinical settings. This may be due to sequence variants in genes encoding drug metabolism enzymes, drug transporters and/or drug targets [32]. At least 105 variants have been identified with the ABCB1 gene, together with significant differences in their frequencies among different ethnic groups. Given the known inter-population differences in drug response for verapamil, it may be important to consider variability among ethnic groups by characterizing variability in genotypes, linkage disequilibrium and recombination within and between ethnic populations. 1236T has been regarded as one of the most frequent SNPs contributing to the ABCB1-related phenotypes [33]. During the past few years, researchers have studied the effect of ABCB1 haplotypes on the pharmacokinetics of digoxin [16, 34], cyclosporin [24, 35], tacrolimus [36] and amlodipine [37]. Notably, in such studies, all haplotype construction had been limited to two SNPs, i.e. 3435T and 2677T/A. Therefore, we decided to recruit subjects simultaneously featuring wild, heterozygous or mutant types for such polymorphisms in present study.
Pharmacokinetics parameters of verapamil (Cmax 36.14 ± 13.73 ng ml−1, AUC 210.3 ± 92.7 ng ml−1 h, tmax 2.3 ± 1.2 h, and t1/2 6.6 ± 1.7 h) from our results were consistent with those reported by McAllister et al. [38], but with lower Cmax and AUC when compared with those reported by Horne et al. and Hla et al. [39, 40]. This may be due in part to factors such as ethnicity, sample size and the large inter-individual variability of the drug.
As suggested from our results, individuals carrying two copies of mutant ABCB1 alleles (2677TT/3435TT) have an increased apparent oral clearance of verapamil when compared with heterozygotes (2677GT/3435CT) or carriers of wild-type alleles (2677GG/3435CC). Meanwhile, AUC values in such subjects were 47.3% and 37.5% lower than in those with 2677GT/3435CT or 2677GG/3435CC genotypes. This seems to suggest that 2677/3435 genotypes are likely to be a major factor responsible for the inter-individual differences in pharmacokinetics of verapamil.
In accordance with our results, higher amlodipine plasma concentrations, AUC and lower clearance have also been associated with the 2677GG/3435CC genotype in healthy Korean volunteers [38]. In another report, it was shown that patients with the 2677G/3435C genotype had significantly better response to chemotherapy when compared with those with other genotypes [41]. By contrast, in a recent study, Pan et al. showed that 2677TT/3435TT subjects had slightly, but not significantly higher Cmaxand AUC and lower tmax for verapamil than did 2677GG/3435CC subjects [42]. The absorption/first-pass effect of oral immediate-release tablets of verapamil is characterized by a saturable process that results in nonlinear pharmacokinetics and may show considerable inter- and intra-subject variability [43]. Therefore, the discrepancy between Pan et al. and our results may be attributed to saturable transporter kinetics with higher verapamil doses. Similar results have been shown with digoxin [15, 16, 44].
Clinically available verapamil is a racemic mixture of equal amounts of the R- and S-verapamil. In vitro and in vivo studies, however, have shown that the R-verapamil is 10-fold less potent than the S-verapamil as a calcium antagonist. Due to stereoselective first-pass metabolism, the plasma R-verapamil concentration was five times greater than that of the S-verapamil following oral administration. On the other hand, Hollt et al. found that both enantiomers had almost equivalent P-gp-inhibitory activities [10]. In this study, the calculated EC50 of the R-verapamil appeared to be less than that of S-verapamil, i.e. 2.6 µmvs 2.9 µm, but this was not statistically significant. Enantioselective interactions with P-gp were also observed in a study that had determined the IC50 values associated with the P-gp binding affinities of the R- and S-verapamil [45]. These seemed in keeping with earlier findings by Gruber et al. [46], Plumb et al. [47] and Keilhauer et al. [48]. Based on the above findings, we may expect that the effects of the R- and S-verapamil on P-gp are similar to verapamil enantiomers among different genotypic groups.
Additional studies involving a larger sample size and stratification based on haplotypes may still be necessary for the understanding of the influence of ABCB1 gene variants on the disposition, therapeutic response and toxicity of verapamil. To reduce the risk of spurious association between ABCB1 genotypes and in vivo phenotypes, demographic data of the selected subjects as well as sample size and environmental factors should also be considered carefully. Notably, factors other than the investigated genotypes may also contribute to the observed inter-individual variations. Although most nucleotide variants, including the studied ones, are mapped within the coding regions of ABCB1, variations in the promoter region of the gene have also been suggested to be important for inter-individual differences in terms of P-gp expression, luciferase activity, and placental and hepatic mRNA levels [49].
In conclusion, we have provided the first evidence that ABCB1 gene polymorphism may have a considerable impact on the pharmacokinetics of verapamil among healthy Chinese Han ethnic subjects. Genotyping the gene may provide useful information for individualizing the treatment by optimizing drug dosage.
Competing interests
None declared.
We thank all our subjects for their participation in this study. We also thank our technicians for their excellent assistance with chemical analysis.
REFERENCES
- 1.Halperin AK, Cubeddu LX. The role of calcium channel blockers in the treatment of hypertension. Am Heart J. 1986;111:363–82. doi: 10.1016/0002-8703(86)90154-7. [DOI] [PubMed] [Google Scholar]
- 2.McAllister RG, Jr, Hamann SR, Blouin RA. Pharmacokinetics of calcium-entry blockers. Am J Cardiol. 1985;55:30B–40B. doi: 10.1016/0002-9149(85)90611-3. [DOI] [PubMed] [Google Scholar]
- 3.Dadashzadeh S, Javadian B, Sadeghian S. The effect of gender on the pharmacokinetics of verapamil and norverapamil in humans. Biopharm Drug Dispos. 2006;27:329–34. doi: 10.1002/bdd.512. [DOI] [PubMed] [Google Scholar]
- 4.Choi DH, Shin WG, Choi JS. Drug interaction between oral atorvastatin and verapamil in healthy subjects: effects of atorvastatin on the pharmacokinetics of verapamil and norverapamil. Eur J Clin Pharmacol. 2008;64:445–9. doi: 10.1007/s00228-007-0447-5. [DOI] [PubMed] [Google Scholar]
- 5.Choi JS, Han HK. The effect of quercetin on the pharmacokinetics of verapamil and its major metabolite, norverapamil in rabbits. J Pharm Pharmacol. 2004;56:1537–42. doi: 10.1211/0022357044814. [DOI] [PubMed] [Google Scholar]
- 6.Ho PC, Ghose K, Saville D, Wanwimolruk S. Effect of grapefruit juice on pharmacokinetics and pharmacodynamics of verapamil enantiomers in healthy volunteers. Eur J Clin Pharmacol. 2000;56:693–8. doi: 10.1007/s002280000189. [DOI] [PubMed] [Google Scholar]
- 7.Kerb R, Hoffmeyer S, Brinkmann U. ABC drug transporters: hereditary polymorphisms and pharmacological impact in MDR1, MRP1 and MRP2. Pharmacogenomics. 2001;2:51–64. doi: 10.1517/14622416.2.1.51. [DOI] [PubMed] [Google Scholar]
- 8.Sakaeda T, Nakamura T, Okumura K. MDR1 genotype-related pharmacokinetics and pharmacodynamics. Biol Pharm Bull. 2002;25:1391–400. doi: 10.1248/bpb.25.1391. [DOI] [PubMed] [Google Scholar]
- 9.Leonard GD, Fojo T, Bates SE. The role of ABC transporters in clinical practice. Oncologist. 2003;8:411–24. doi: 10.1634/theoncologist.8-5-411. [DOI] [PubMed] [Google Scholar]
- 10.Hollt V, Kouba M, Dietel M, Vogt G. Stereoisomers of calcium antagonists which differ markedly in their potencies as calcium blockers are equally effective in modulating drug transport by p-glycoprotein. Biochemistry and. Pharmacology. 1992;40:2601–8. doi: 10.1016/0006-2952(92)90149-d. [DOI] [PubMed] [Google Scholar]
- 11.Chiou WL, Chung SM, Wu TC, Ma C. A comprehensive account on the role of efflux transporters in the gastrointestinal absorption of 13 commonly used substrate drugs in humans. Int J Clin Pharmacol Ther. 2001;39:93–101. doi: 10.5414/cpp39093. [DOI] [PubMed] [Google Scholar]
- 12.Yumoto R, Murakami T, Takano M. Differential effect of acute hepatic failure on in vivo and in vitro P-glycoprotein functions in the intestine. Pharm Res. 2003;20:765–71. doi: 10.1023/a:1023485519485. [DOI] [PubMed] [Google Scholar]
- 13.Bosch TM, Meijerman I, Beijnen JH, Schellens JH. Genetic polymorphisms of drug-metabolizing enzymes and drug transporters in the chemotherapeutic treatment of cancer. Clin Pharmacokinet. 2006;45:253–85. doi: 10.2165/00003088-200645030-00003. [DOI] [PubMed] [Google Scholar]
- 14.Becquemont L, Verstuyft C, Kerb R, Brinkmann U, Lebot M, Jaillon P, Funck-Brentano C. Effect of grapefruit juice on digoxin pharmacokinetics in humans. Clin Pharmacol Ther. 2001;70:311–6. [PubMed] [Google Scholar]
- 15.Gerloff T, Schaefer M, Johne A, Oselin K, Meisel C, Cascorbi I, Roots I. MDR1 genotypes do not influence the absorption of a single oral dose of 1 mg digoxin in healthy white males. Br J Clin Pharmacol. 2002;54:610–6. doi: 10.1046/j.1365-2125.2002.01691.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Johne A, Kopke K, Gerloff T, Mai I, Rietbrock S, Meisel C, Hoffmeyer S, Kerb R, Fromm MF, Brinkmann U, Eichelbaum M, Brockmoller J, Cascorbi I, Roots I. Modulation of steady-state kinetics of digoxin by haplotypes of the P-glycoprotein MDR1 gene. Clin Pharmacol Ther. 2002;72:584–94. doi: 10.1067/mcp.2002.129196. [DOI] [PubMed] [Google Scholar]
- 17.Kurata Y, Ieiri I, Kimura M, Morita T, Irie S, Urae A, Ohdo S, Ohtani H, Sawada Y, Hiquchi S, Otsubo K. Role of human MDR1 gene polymorphism in bioavailability and interaction of digoxin, a substrate of P-glycoprotein. Clin Pharmacol Ther. 2002;72:209–19. doi: 10.1067/mcp.2002.126177. [DOI] [PubMed] [Google Scholar]
- 18.Verstuyft C, Schwab M, Schaeffeler E, Kerb R, Brinkmann U, Jaillon P, Funck-Brentano C, Becquemont L. Digoxin pharmacokinetics and MDR1 genetic polymorphisms. Eur J Clin Pharmacol. 2003;58:809–12. doi: 10.1007/s00228-003-0567-5. [DOI] [PubMed] [Google Scholar]
- 19.Verstuyft C, Strabach S, El-Morabet H, Kerb R, Brinkmann U, Dubert L, Jaillon P, Funck-Brentano C, Truqnan G, Becquemont L. Dipyridamole enhances digoxin bioavailability via P-glycoprotein inhibition. Clin Pharmacol Ther. 2003;73:51–60. doi: 10.1067/mcp.2003.8. [DOI] [PubMed] [Google Scholar]
- 20.Keller F, Rietbrock N. Bioavailability of digoxin: some pitfalls and problems. Int J Clin Pharmacol Biopharm. 1977;15:549–56. [PubMed] [Google Scholar]
- 21.Sakaeda T, Nakamura T, Horinouchi M, Kakumoto M, Ohmoto N, Sakai T, Morita Y, Tamura T, Aoyama N, Hirai M, Kasuqa M, Okumura K. MDR1 genotype-related pharmacokinetics of digoxin after single oral administration in healthy Japanese subjects. Pharm Res. 2001;18:1400–4. doi: 10.1023/a:1012244520615. [DOI] [PubMed] [Google Scholar]
- 22.Kim RB, Leake BF, Choo EF, Dresser GK, Kubba SV, Schwarz UI, Taylor A, Xie HG, McKinsey J, Zhou S, Lan LB, Schuetz JD, Schuetz EG, Wilkinson GR. Identification of functionally variant MDR1 alleles among European Americans and African Americans. Clin Pharmacol Ther. 2001;70:189–99. doi: 10.1067/mcp.2001.117412. [DOI] [PubMed] [Google Scholar]
- 23.Horinouchi M, Sakaeda T, Nakamura T, Morita Y, Tamura T, Aoyama N, Kasuga M, Okumura K. Significant genetic linkage of MDR1 polymorphisms at positions 3435 and 2677: functional relevance to pharmacokinetics of digoxin. Pharm Res. 2002;19:1581–5. doi: 10.1023/a:1020433422259. [DOI] [PubMed] [Google Scholar]
- 24.Chowbay B, Cumaraswamy S, Cheung YB, Zhou Q, Lee EJ. Genetic polymorphisms in MDR1 and CYP3A4 genes in Asians and the influence of MDR1 haplotypes on cyclosporin disposition in heart transplant recipients. Pharmacogenetics. 2003;13:89–95. doi: 10.1097/00008571-200302000-00005. [DOI] [PubMed] [Google Scholar]
- 25.Tan EK, Chan DK, Ng PW, Woo J, Teo YY, Tang K, Wong LP, Chong SS, Tan C, Shen H, Zhao Y, Lee CG. Effect of MDR1 haplotype on risk of Parkinson disease. Arch Neurol. 2005;62:460–4. doi: 10.1001/archneur.62.3.460. [DOI] [PubMed] [Google Scholar]
- 26.Sata F, Sapone A, Elizondo G, Stocker P, Miller VP, Zheng W, Raunio H, Crespi CL, Gonzalez FJ. CYP3A4 allelic variants with amino acid substitutions in exons 7 and 12: evidence for an allelic variant with altered catalytic activity. Clin Pharmacol Ther. 2000;67:48–56. doi: 10.1067/mcp.2000.104391. [DOI] [PubMed] [Google Scholar]
- 27.Ball SE, Scatina J, Kao J, Ferron GM, Fruncillo R, Mayer P, Weinrvb I, Guida M, Hopkins PJ, Warner N, Hall J. Population distribution and effects on drug metabolism of a genetic variant in the 5′promoter region of CYP3A4. Clin Pharmacol Ther. 1999;66:288–94. doi: 10.1016/S0009-9236(99)70037-8. [DOI] [PubMed] [Google Scholar]
- 28.Jin Y, Wang YH, Miao J, Li L, Kovacs RJ, Marunde R, Hamman MA, Phillips S, Hilligoss J, Hall SD. Cytochrome P450 3A5 genotype is associated with verapamil response in healthy subjects. Clin Pharmacol Ther. 2007;82:579–85. doi: 10.1038/sj.clpt.6100208. [DOI] [PubMed] [Google Scholar]
- 29.Langaee TY, Gong Y, Yarandi HN, Katz DA, Cooper-DeHoff RM, Pepine CJ, Johnson JA. Association of CYP3A5 polymorphisms with hypertension and antihypertensive response to verapamil. Clin Pharmacol Ther. 2007;81:386–91. doi: 10.1038/sj.clpt.6100090. [DOI] [PubMed] [Google Scholar]
- 30.Kuehl P, Zhang J, Lin Y, Lamba J, Assem M, Schuetz J, Watkins PB, Daly A, Wrighton SA, Hall SD, Maurel P, Relling M, Brimer C, Yasuda K, Venkataramanan R, Strom S, Thummel K, Boquski MS, Schuetz E. Sequence diversity in CYP3A promoters and characterization of the genetic basis of polymorphic CYP3A5 expression. Nature Genet. 2001;27:383–91. doi: 10.1038/86882. [DOI] [PubMed] [Google Scholar]
- 31.Kurzawski M, Pawlik A, Gornik W, Drozdzik M. Frequency of common MDR1 gene variants in a Polish population. Pharmacol Rep. 2006;58:35–40. [PubMed] [Google Scholar]
- 32.Evans WE, Relling MV. Pharmacogenomics: translating functional genomics into rational therapeutics. Science. 1999;286:487–91. doi: 10.1126/science.286.5439.487. [DOI] [PubMed] [Google Scholar]
- 33.Anglicheau D, Thervet E, Etienne I, Hurault de Ligny B, Le Meur Y, Touchard G, Büchler M, Laurent-Puig P, Tregouet D, Beaune P, Daly A, Legendre C, Marquet P. CYP3A5 and MDR1 genetic polymorphisms and cyclosporine pharmacokinetics after renal transplantation. Clin Pharmacol Ther. 2004;75:422–33. doi: 10.1016/j.clpt.2004.01.009. [DOI] [PubMed] [Google Scholar]
- 34.Kurzawski M, Bartnicka L, Florczak M, Górnik W, Drozdzik M. Impact of ABCB1 (MDR1) gene polymorphism and P-glycoprotein inhibitors on digoxin serum concentration in congestive heart failure patients. Pharmacol Rep. 2007;59:107–11. [PubMed] [Google Scholar]
- 35.Mai I, Störmer E, Goldammer M, Johne A, Krüger H, Budde K, Roots I. MDR1 haplotypes do not affect the steady-state pharmacokinetics of cyclosporine in renal transplant patients. J Clin Pharmacol. 2003;43:1101–7. doi: 10.1177/0091270003257222. [DOI] [PubMed] [Google Scholar]
- 36.Mai I, Perloff ES, Bauer S, Goldammer M, Johne A, Filler G, Budde K, Roots I. MDR1 haplotypes derived from exons 21 and 26 do not affect the steady-state pharmacokinetics of tacrolimus in renal transplant patients. Br J Clin Pharmacol. 2004;58:548–53. doi: 10.1111/j.1365-2125.2004.02182.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kim KA, Park PW, Park JY. Effect of ABCB1 (MDR1) haplotypes derived from G2677T/C3435T on the pharmacokinetics of amlodipine in healthy subjects. Br J Clin Pharmacol. 2007;63:53–8. doi: 10.1111/j.1365-2125.2006.02733.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.McAllister RG, Jr, Kirsten EB. The pharmacology of verapamil. IV. Kinetic and dynamic effects after single intravenous and oral doses. Clin Pharmacol Ther. 1982;31:418–26. doi: 10.1038/clpt.1982.54. [DOI] [PubMed] [Google Scholar]
- 39.Horne C, Stenzhorn G, Blume H, Knauf H, Mutschler E. Bioavailability study of two different verapamil formulations. Arch Pharm (Weinheim) 1992;325:531–6. doi: 10.1002/ardp.19923250814. [DOI] [PubMed] [Google Scholar]
- 40.Hla KK, Latham AN, Henry JA. Influence of time of administration on verapamil pharmacokinetics. Clin Pharmacol Ther. 1992;51:366–70. doi: 10.1038/clpt.1992.35. [DOI] [PubMed] [Google Scholar]
- 41.Sohn JW, Lee SY, Lee SJ, Kim EJ, Cha SI, Kim CH, Lee JT, Jung TH, Park JY. MDR1 polymorphisms predict the response to etoposide-cisplatin combination chemotherapy in small cell lung cancer. Jpn J Clin Oncol. 2006;36:137–41. doi: 10.1093/jjco/hyi231. [DOI] [PubMed] [Google Scholar]
- 42.Pan W, Ryu JY, Shon JH, Song IS, Liu KH, Sunwoo YE, Kang W, Shin JG. Dietary salt dose not influence the disposition of verapamil enantiomers in relation to efflux transporter ABCB1 genetic polymorphism in healthy Korean subjects. Xenobiotica. 2008;38:422–34. doi: 10.1080/00498250701832446. [DOI] [PubMed] [Google Scholar]
- 43.Harder S, Thurmann P, Siewert M, Blume H, Huber TH, Rietbrock N. Pharmacodynamic profile of verapamil in relation to absolute bioavailability: investigations with a conventional and controlled release formulation. J Cardiovasc Pharmacol. 1991;17:207–12. doi: 10.1097/00005344-199102000-00005. [DOI] [PubMed] [Google Scholar]
- 44.Hoffmeyer S, Burk O, von Richter O, Arnold HP, Brockmoller J, Johne A, Cascorbi I, Gerloff T, Roots I, Eichelbaum M, Brinkmann U. Functional polymorphisms of the human multidrug-resistance gene:multiple sequence variations and correlation of one allele with P-glycoprotein expression and activity in vivo. Proc Natl Acad Sci U S A. 2000;97:3473–8. doi: 10.1073/pnas.050585397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Neuhoff S, Langguth P, Dressler C, Andersson TB, Regardh CG, Spahn-Langguth H. Affinities at the verapamil binding site of MDR1-encoded P-glycoprotein: Drugs and analogs, stereoisomers and metabolites. Int J Clin Pharmacol Ther. 2000;38:168–79. doi: 10.5414/cpp38168. [DOI] [PubMed] [Google Scholar]
- 46.Gruber A, Peterson C, Reizenstein P. D-verapamil and L-verapamil are equally effective in increasing vincristine accumulation in leukemic cells in vitro. Int J Cancer. 1988;41:224–6. doi: 10.1002/ijc.2910410211. [DOI] [PubMed] [Google Scholar]
- 47.Plumb JA, Milroy R, Kaye SB. The activity of verapamil as a resistance modifier in vitro in drug resistant human tumor cell lines is not stereospecific. Biochem Pharmacol. 1990;39:787–92. doi: 10.1016/0006-2952(90)90160-m. [DOI] [PubMed] [Google Scholar]
- 48.Keilhauer C, Emling F, Raschak M, Gries J, Schlick E. The use of R-verapamil (R-VPM) is superior to racemic VPM in breaking multidrug resistance (MDR) of malignant cells. Proc Am Assoc Cancer Res. 1989;30:503. Abstract. [Google Scholar]
- 49.Takane H, Kobayashi D, Hirota T, Kiqawa J, Terakawa N, Otsubo K, Ieiri I. Haplotype-oriented genetic analysis and functional assessment of promoter variants in the MDR1 (ABCB1) gene. J Pharmacol Exp Ther. 2004;311:1179–87. doi: 10.1124/jpet.104.069724. [DOI] [PubMed] [Google Scholar]