

Abstract
AIMS
To characterize: i) the kinetics of aldosterone (ALDO) 18β-glucuronidation using human liver and human kidney microsomes and identify the human UGT enzyme(s) responsible for ALDO 18β-glucuronidation and ii) the inhibition of ALDO 18β-glucuronidation by non-selective NSAIDs.
METHODS
Using HPLC and LC-MS methods, ALDO 18β-glucuronidation was characterized using human liver (n= 6), human kidney microsomes (n= 5) and recombinant human UGT 1A1, 1A3, 1A4, 1A5, 1A6, 1A7, 1A8, 1A9, 1A10, 2B4, 2B7, 2B10, 2B15, 2B17 and 2B28 as the enzyme sources. Inhibition of ALDO 18β-glucuronidation was investigated using alclofenac, cicloprofen, diclofenac, diflunisal, fenoprofen, R- and S-ibuprofen, indomethacin, ketoprofen, ketorolac, meclofenamic acid, mefenamic acid, S-naproxen, pirprofen and tiaprofenic acid. A rank order of inhibition (IC50) was established and the mechanism of inhibition investigated using diclofenac, S-ibuprofen, indomethacin, mefenamic acid and S-naproxen.
RESULTS
ALDO 18β-glucuronidation by hepatic and renal microsomes exhibited Michaelis-Menten kinetics. Mean (±SD) Km, Vmax and CLint values for HLM and HKCM were 509 ± 137 and 367 ± 170 µm, 1075 ± 429 and 1110 ± 522 pmol min−1 mg−1, and 2.36 ± 1.12 and 3.91 ± 2.35 µl min−1 mg−1, respectively. Of the UGT proteins, only UGT1A10 and UGT2B7 converted ALDO to its 18β-glucuronide. All NSAIDs investigated inhibited ALDO 18β-G formation by HLM, HKCM and UGT2B7. The rank order of inhibition (IC50) of renal and hepatic ALDO 18β-glucuronidation followed the general trend: fenamates > diclofenac > arylpropionates.
CONCLUSION
A NSAID-ALDO interaction in vivo may result in elevated intra-renal concentrations of ALDO that may contribute to the adverse renal effects of NSAIDs and their effects on antihypertensive drug response.
Keywords: aldosterone, glucuronidation, non-steroidal anti-inflammatory drugs, renal drug metabolism
WHAT IS ALREADY KNOWN ABOUT THIS SUBJECT
Carboxylic acid NSAIDs are extensively glucuronidated as either the parent drug or hydroxylated metabolites and UGT2B7 is ranked highest in terms of NSAID-glucuronidation activity.
NSAIDs cause adverse renal effects including sodium and water retention and hyperkalaemia.
In human kidney the mineralocorticoid aldosterone is glucuronidated directly to form aldosterone 18β-glucuronide.
WHAT THIS STUDY ADDS
Human liver and kidney microsomes and UGT1A10 and UGT2B7 catalyze aldosterone18β-glucuronidation.
Non-selective NSAIDs inhibit renal and hepatic aldosterone18β-glucuronidation and in vivo this may lead to elevated intra-renal concentrations of this hormone.
Common involvement of UGT2B7 in NSAID and aldosterone glucuronidation predicates an intra-renal NSAID-aldosterone interaction that may explain in part the clinical observations of variable effects of NSAIDs on electrolytes, fluid retention and blood pressure.
Introduction
Non-steroidal anti-inflammatory drugs (NSAIDs) comprise a structurally diverse group of compounds that includes salicylates, arylpropionates, oxicams, fenamates, indoles, phenylacetic acids, coxibs and pyrazoles. With the exception of the oxicams, coxibs and pyrazoles, the majority of NSAIDs contain a carboxylic acid functional group. Carboxylate NSAIDs are excreted to a variable extent in vivo as glucuronides, either as the conjugate of the parent carboxylic acid or a hydroxylated metabolite, with only a small fraction of the dose eliminated as unchanged drug in urine [1]. NSAID metabolism has also been investigated in vitro, particularly with human liver microsomes (HLM) as the enzyme source [2–4], and the liver is normally considered the major organ responsible for drug metabolic clearance [5]. However, significant glucuronidation activity towards endogenous compounds and xenobiotics has been reported for human kidney tissue. Studies using human kidney microsomes have demonstrated formation of the acyl glucuronides of diflunisal, flufenamic acid, mefenamic acid, naproxen, niflumic acid, ibuprofen, ketoprofen and flurbiprofen [3, 6–10].
Glucuronidation reactions are catalyzed by enzymes of the UDP-glucuronosyltransferase (UGT) superfamily, which have been classified into families and subfamilies based on sequence identity [11]. Eighteen catalytically active human UDP-glucuronosyltransferases (UGTs) are found in liver and kidney [12]. Of these, UGT 1A3, 1A6, 1A9 and 2B7 are the predominant enzymes reported to be involved in the glucuronidation of carboxylic acids, including many NSAIDs [2, 6, 13–15]. With respect to NSAID-glucuronidation, UGT2B7 is generally ranked highest in terms of activity with UGT2B7 > UGT1A9 > UGT1A3 ∼ UGT1A6.
Aldosterone (ALDO) is the major mineralocorticoid secreted by the adrenal cortex in response to physiological stimuli including angiotensin II, elevated plasma K+ concentration and Na+ deficiency. ALDO is metabolized to dihydro and tetrahydro derivatives that are subsequently glucuronidated in the liver [16]. Additionally, ALDO is glucuronidated directly to form aldosterone 18β-glucuronide (ALDO 18β-G) (Figure 1). Tetrahydroaldosterone glucuronide (THA-G) and ALDO 18β-G account for the majority of daily aldosterone elimination in urine. However, unlike THA-G, which is exclusively synthesized in the liver, the majority of ALDO 18β-G is thought to be formed in kidney (∼80%) with a minor hepatic component [16]. Girard et al.[17] have implicated UGT2B7 and UGT2B4 in the glucuronidation of ALDO and THA, respectively. The common involvement of UGT2B7 in the metabolism of ALDO and NSAIDs raises the possibility of a metabolic interaction between these compounds, especially in the kidney.
Figure 1.
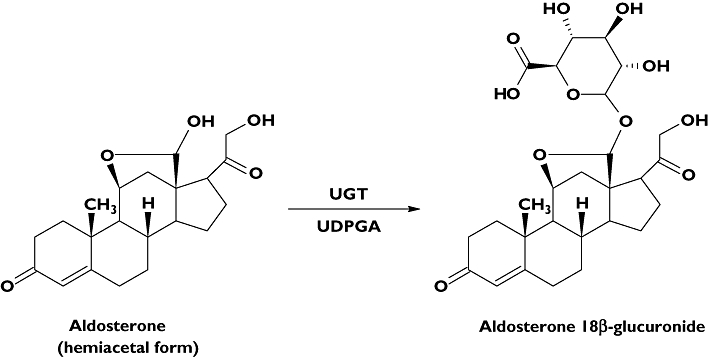
Structures of aldosterone (ALDO) and aldosterone 18β-glucuronide
NSAID use is associated with adverse renal effects. These include a reduction in renal perfusion, electrolyte disturbances (sodium and water retention) and, less commonly, acute renal failure and acute nephritis. It is generally considered that perturbations of renal function arise from reduced synthesis of prostaglandins such as PGE2, which causes vasodilation, influences sodium reabsorption in the renal tubule, and antagonizes the action of antidiuretic hormone in the collecting tubule. However, inhibition of prostaglandin synthesis as an exclusive explanation is not convincing as NSAIDs have been shown to exert adverse renal effects disproportionate to the level of inhibition of prostaglandin synthesis [18–20]. NSAIDs induce a state of hyperkalaemia in many individuals and this is believed to result from reduced potassium delivery to the nephron due to decreased renal perfusion subsequent to inhibition of PGI2 synthesis [21, 22]. Given ALDO promotes sodium and water retention and enhances potassium excretion, it is conceivable that the interindividual variability in serum potassium status with NSAID use may result from an imbalance between ALDO action and NSAID induced reduction in potassium secretion in the nephron. In turn, hyperkalaemia is a physiologic stimulus for ALDO secretion.
We hypothesize that the clinically observed increase in sodium and water retention that occurs with NSAID use is due, in part, to inhibition of ALDO glucuronidation, particularly locally at its sites of action in the kidney. Reduced ‘local’ clearance of ALDO would be expected to manifest as persistence of action at intra-renal sites. The aims of this study were: i) to define the kinetics of ALDO 18β-glucuronidation by HLM and human kidney cortical microsomes (HKCM), ii) identify using a ‘panel ‘of recombinant human UGTs, the enzyme(s) responsible for ALDO 18β-glucuronidation and iii) investigate whether non-selective NSAIDs inhibit hepatic and renal microsomal ALDO 18β-glucuronidation.
Methods
Alamethicin (from Trichoderma viride), β-glucuronidase (from Escherichia coli), UDP-glucuronic acid (UDPGA) trisodium salt, d-ALDO, S-naproxen and S-ibuprofen were purchased from Sigma-Aldrich (Sydney, Australia). Diclofenac and pirprofen were obtained from CIBA-GEIGY (Sydney, Australia), alclofenac from Continental Pharma (Brussels, Belgium), cicloprofen from E.R. Squibb (NJ, USA), diflunisal and indomethacin from Merck Sharp & Dohme (NJ, USA), fenoprofen from Eli Lilly (Sydney, Australia), R-ibuprofen from Boots (UK), ketoprofen from May & Baker (Melbourne, Australia), meclofenamic and mefenamic acid from Parke-Davis (Sydney, Australia) and tiaprofenic acid from Roussel Uclaf (Romainville, France). Solvents and other reagents were analytical reagent grade available from commercial sources.
Human liver microsomes and human kidney cortical microsomes
HLM were prepared by differential ultra-centrifugation from six livers from Caucasian donors (H7, 44 year-old female; H12, 66 year-old male; H24, 49 year-old female; H29, 45 year-old male; H40, 54 year-old female, H44, 42 year-old female), as described by Bowalgaha et al.[2]. Livers were obtained from the human liver bank of the Department of Clinical Pharmacology of Flinders University. HKCM were isolated from fresh renal tissue from five Caucasian donors (K5, 72 year-old male; K7, 43 year-old male; K9, 79 year-old male; K10, 64 year-old male; K11, 65 year-old male) immediately following surgery, according to Tsoutsikos et al.[23]. Approval for the use of human liver and kidney tissue in drug metabolism studies was obtained from the Flinders Clinical Research Ethics Committee, Flinders University and the Research and Ethics Committee of the Repatriation General Hospital, Adelaide, Australia. HLM and HKCM were activated by pre-incubation with alamethicin (50 µg mg−1 protein) on ice for 30 min prior to use in incubations [24].
Expression of UGT proteins
UGT 1A1, 1A3, 1A4, 1A5, 1A6, 1A7, 1A8, 1A9, 1A10, 2B4, 2B7, 2B10, 2B15, 2B17 and 2B28 cDNAs were stably expressed in a human embryonic kidney cell line (HEK293), as described previously [25, 26]. Cells were subsequently lysed by sonication using a Vibra Cell VCX130 Ultrasonic Processor (Sonics and Materials, Newtown, CT, USA) set to an amplitude of 40%. Cells expressing UGT1A proteins were sonicated with four 2 s ‘bursts’, each separated by 1 min cooling on ice. For UGT2B subfamily enzymes, sonication was limited to 1 s ‘bursts’. Lysed samples were centrifuged at 12 000 g for 1 min at 4°C, and the supernatant fraction was separated and stored at −80°C until use. Expression of each UGT protein was demonstrated by immunoblotting with a commercial UGT1A antibody and a non-selective UGT antibody [26]. Activities of recombinant UGTs (except UGT1A4 and UGT2B10) were confirmed using the non-selective substrate 4-methylumbelliferone, as described by Uchaipichat et al.[26] and Rowland et al.[27]. The activities of recombinant UGT1A4 and UGT2B10 were demonstrated using trifluoperazine [28] and cotinine (J.O. Miners, unpublished data) as substrate, respectively. Activities of all recombinant UGT enzyme preparations were within ±10% of values reported in references 26–28.
Aldosterone 18β–glucuronidation assay
Incubation mixtures (200 µl), in borosilicate glass tubes, contained either activated HLM (0.5 mg ml−1) or HKCM (0.2 mg ml−1), or HEK293 cell lysate expressing recombinant UGT (1 mg ml−1 in screening studies, or 0.5 mg ml−1 (UGT1A10) or 0.8 mg ml−1 (UGT2B7) in kinetic studies), UDPGA (5 mm) and MgCl2 (4 mm) in phosphate buffer (0.1 m, pH 7.4). ALDO was added to incubations as a solution in DMSO such that the final solvent concentration was 1% v/v; this concentration of DMSO has been shown to have a minor effect on most UGT activities [26]. ALDO was present at three concentrations (200, 800 and 2000 µm) in activity screening studies or 12 concentrations in the range 10–2000 µm in kinetic studies. After a 5 min pre-incubation at 37°C in a shaking water bath, reactions were initiated by the addition of UDPGA. Separate ‘blank’ incubations (minus UDPGA) were included in all experiments. Incubation duration was 45 min for HLM, 20 min for HKCM, 90 min for UGT1A10 and 50 min for UGT2B7. ALDO18β-G formation was linear with respect to both protein concentration (up to 1.25 mg for all enzyme sources) and incubation duration (up to 60 min with HLM and UGT2B7, and 90 min with HKCM and UGT1A10) for each enzyme source. Incubations at each ALDO concentration were performed in duplicate for HLM and recombinant UGTs. As duplicate estimations of ALDO18β-G differed by less than 4% with HLM and recombinant UGT proteins as the enzyme sources, incubations with HKCM were performed in singlicate due to the limited availability of human kidney tissue. Reactions were terminated by the addition of an equal volume (200 µl) of ice-cold 4% acetic acid/96% methanol and centrifugation at 5000 g for 10 min at 4°C. An aliquot (35 µl) of the supernatant fraction was injected onto an Agilent 1100 high performance liquid chromatography (HPLC) system (Agilent Technologies, Palo Alto, CA, USA).
Chromatographic separation of ALDO and ALDO 18β-G was achieved using a Waters Nova-Pak 18 column (150 × 3.9 mm, 4 µm; Milford, MA, USA) at a mobile phase flow rate of 1 ml min−1. The mobile phase comprised two components: A (95% water, 5% acetonitrile, 0.002% v/v acetic acid) and B (100% acetonitrile). Initial conditions were 95%A : 5%B held for 1 min then changed to 35%A : 65%B over 8 min. Analytes were monitored by ultraviolet absorbance at 241 nm. Under these conditions, retention times for ALDO18β-G and ALDO were 4.68 and 6.36 min, respectively. ALDO and ALDO 18β-G were quantified by comparison of peak areas to those of an ALDO external standard curve prepared over the concentration range 1–1000 µm. In the absence of commercially available ALDO 18β-G this approach was considered valid because the extinction coefficients of hydroxy-steroids and their glucuronide conjugates are generally similar (J.O. Miners & D.J. Elliot, unpublished observations). In addition, mass balance was demonstrated in incubations (i.e. the sum of the concentrations of ALDO and ALDO 18β-G present at the end of an incubation equated to the initial ALDO concentration added). Nevertheless, rates of ALDO 18β-glucuronidation and Vmax values presented in Results should be considered ‘apparent’ in the absence of a standard curve based on the authentic metabolite. ALDO standard curves were linear over the concentration range used with r2 values > 0.99. The coefficient of variation for the slopes of the ALDO standard curves (n= 10) was 2.1%, indicating minimal inter-day variability. Overall within-day assay reproducibility was determined by measuring ALDO 18β-G formation at two ALDO concentrations (100 and 1000 µm) in six replicate incubations of the same batch of HLM. Within-day coefficients of variation were <6% at 100 µm and <2% at 1000 µm ALDO. The lower limit of quantification, defined as five times baseline signal to noise ratio, was 0.15 µm and 0.2 µm for ALDO 18β-G and ALDO, respectively.
The identity of the ALDO 18β-G formed in incubations was confirmed by acid lability (hydrolysis at pH 2.5), resistance to hydrolysis by β-glucuronidase (1000 units, 2 h at 37°C) and liquid chromatography-tandem mass spectrometry (LC-MS/MS). For the latter experiment, ALDO (800 µm) was incubated with alamethicin-activated HLM (0.5 mg ml−1), MgCl2 (4 mm) and UDPGA (5 mm) in phosphate buffer (0.1 m, pH 7.4, 37°C) for 45 min and the reaction terminated as described above. An aliquot of the supernatant fraction from the incubation sample was analyzed using a Micromass Quattro micro tandem quadropole mass spectrometer (Waters Associates, Manchester, UK). Separation of incubation constituents was achieved using a Waters liquid chromatograph (Waters, Milford, USA) consisting of a Module 2695 Separation Module and Model 2487 dual wavelength detector. The column mobile phase gradient was as stated for HPLC analysis of ALDO 18β-G. Electrospray mass spectrometry was performed in the negative ion mode for the duration of the LC run, with scan duration 1 s, mass range 80–1000 m/z, capillary voltage −3.5 kV, cone voltage 25 V, source temperature 80°C, desolvation temperature 350°C and desolvation gas flow 410 l h−1.
Non-specific binding of ALDO to HLM and HEK293 cell lysate
Potential non-specific binding of ALDO to HLM and HEK293 cell lysate was investigated by equilibrium dialysis according to previously published procedures from this laboratory [27, 28]. Binding measurements were performed using a Dianorm equilibrium dialysis apparatus (Dianorm, Munich, Germany) that comprised Teflon dialysis cells (capacity of 1200 µl per side) separated into two compartments with dialysis membrane (molecular weight cut off 12 kDa; Sigma Aldrich). Membranes were conditioned overnight at 4°C in phosphate buffer (0.1 m, pH 7.4). One side of the dialysis cell was loaded with 1 ml of a solution of ALDO (100, 500 or 2000 µm) in phosphate buffer (0.1 m, pH 7.4). The other compartment was loaded with 1 ml of a suspension of HLM (0.5 mg) or HEK293 cell lysate (1 mg). The dialysis cell assembly was immersed in a water bath maintained at 37°C and rotated at 12 rev min−1 for 5 h. A 200 µl aliquot was collected from each compartment, treated with an equal volume of ice-cold methanol containing 4% glacial acetic acid, and cooled on ice. Samples were subsequently centrifuged at 5000 g for 10 min and an aliquot of the supernatant fraction was analyzed by HPLC, as described above. Experiments were performed at each ALDO concentration in duplicate. The mean (±SD) recovery of ALDO from dialysis cells was 102 ± 2%.
ALDO 18β-G inhibition studies
Inhibition of ALDO 18β-G formation, at a substrate concentration of 300 µm (the approximate apparent Km for ALDO; see Results section), by the UGT2B7 selective inhibitor fluconazole (2.5 mm) was characterized using pooled HLM, pooled HKCM and recombinant UGT2B7 employing the incubation conditions described previously. In addition, the effects of 15 NSAIDs on ALDO 18β-G formation by pooled HLM, pooled HKCM and recombinant UGT2B7 were determined at an ALDO concentration corresponding to the approximate Km for each enzyme source (HLM, 500 µm; HKCM 300 µm; and UGT2B7, 300 µm; see Results section). IC50 values were assessed using at least four NSAID concentrations in the following ranges: fenoprofen, ketoprofen, tiaprofenic acid and S-naproxen (20–800 µm); cicloprofen, 5–200 µm; diclofenac, 0.5–200 µm; mefenamic acid 0.05–5 µm; meclofenamic acid, 0.1–5 µm; alclofenac, 150–800 µm; diflunisal, 150–600 µm; ketorolac, 700–2000 µm; indomethacin, 50–350 µm; pirprofen, 25–250 µm; and R- and S-ibuprofen 200–1000 µm. Ki values and mechanisms of inhibition were subsequently determined for five representative NSAIDs. Inhibition by four concentrations of each NSAID (mefenamic acid, 0.05–0.5 µm; S-naproxen, 5–20 µm; diclofenac, 0.5–3 µm; indomethacin, 10–250 µm; and S-ibuprofen, 50–500 µm) was determined at each of three ALDO concentrations (150, 300 and 600 µm).
Data analysis
Data points represent the mean of duplicate determinations, except where indicated. The kinetic parameters apparent Km and Vmax for ALDO 18β- glucuronidation by HLM, HKCM and recombinant UGT1A10 and UGT2B7 were derived from fitting untransformed data to the Michaelis-Menten (MM) equation. Kinetic data are shown as Eadie-Hofstee plots for ease of visualization. Inhibition constants (Ki) were determined by fitting untransformed data to the equations for competitive, non-competitive and mixed inhibition using a non-linear least-squares modelling program (EnzFitter, version 2.0.18.0: Biosoft, Cambridge, UK). The goodness of fit was determined by comparison of statistical parameters (coefficient of determination (r2), parameter standard errors, 95% confidence intervals for the curve fit, and F-statistic). In vitro intrinsic clearance was calculated as Vmax/Km. IC50 values were similarly generated using EnzFitter by fitting untransformed data to the equation:
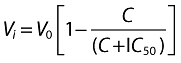 |
where V0 is the control activity and Vi is the activity in the presence of inhibitor concentration C.
Statistical analyses of kinetic constants for ALDO 18β-glucuronide formation by HLM and HKCM were performed using an unpaired Student's t-test. Values of P < 0.05 were considered statistically significant.
Results
HPLC analysis of incubation samples containing either HLM, HKCM or recombinant UGTs, ALDO and UDPGA revealed a peak eluting at 4.68 min, which was not observed in the absence of either ALDO or UDPGA. The identity of the peak was confirmed as ALDO 18β-G by susceptibility to acid hydrolysis, resistance to β-glucuronidase hydrolysis (data not shown) and LC-MS analysis. For the peak eluting at 4.68 min the MS analysis in the negative ion mode gave a molecular ion ([M-H]-) with m/z of 535 (c.f. known molecular mass of ALDO 18β-G, 536.5 g mol−1) and a fragment ion [M-GA-H]-m/z 359.0, which corresponds to the loss of the glucuronic acid moiety.
ALDO 18β-glucuronidation by microsomes from all six human livers and five human kidneys studied exhibited Michaelis-Menten kinetics (Figure 2). The derived kinetic parameters for ALDO 18β-G formation by HLM and HKCM are shown in Table 1. Mean (±SD) Km, Vmax and CLint values for HLM and HKCM were 509 ± 137 and 367 ± 170 µm, 1075 ± 429 and 1110 ± 522 pmol min−1 mg−1, and 2.36 ± 1.12 and 3.91 ± 2.35 µl min−1 mg−1, respectively. Differences in kinetic parameters between the enzyme sources were not statistically significant (P > 0.05). ALDO did not bind non-specifically to HLM or HEK293 cell lysate; mean (±SD) fraction of ALDO unbound at 100, 500 and 2000 µm was 99 ± 2%. Based on these data, it is assumed that ALDO does not bind to HKCM (which was not investigated in binding experiments due to the limited availability of tissue). Thus, no correction of in vitro kinetic parameters for non-specific binding was required. Moreover, it is known that the acidic drugs (used in the inhibition studies) bind negligibly to HLM [29].
Figure 2.
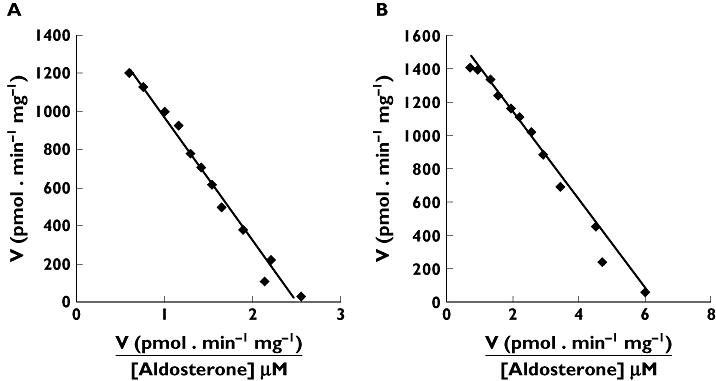
Representative Eadie-Hofstee plots for aldosterone 18β-glucuronidation by A) human liver microsomes (H12) and B) human kidney cortical microsomes (K10). Points are experimentally determined values and the solid lines are the model fitted curves of best fit
Table 1.
Derived kinetic parameters for aldosterone 18β-glucuronidation by human liver microsomes and human kidney cortical microsomes
| Kinetic parameters | |||
|---|---|---|---|
| Km | Vmax | CLint | |
| Enzyme source | (µm) | (pmol min−1 mg−1) | (µl min−1 mg−1) |
| HLM | |||
| H7 | 417 | 1471 | 3.53 |
| H12 | 646 | 1615 | 2.50 |
| H24 | 677 | 439 | 0.64 |
| H29 | 602 | 629 | 1.05 |
| H40 | 326 | 1004 | 3.08 |
| H44 | 386 | 1293 | 3.35 |
| HKCM | |||
| K5 | 437 | 523 | 1.20 |
| K7 | 664 | 707 | 1.06 |
| K9 | 183 | 850 | 4.64 |
| K10 | 264 | 1673 | 6.34 |
| K11 | 285 | 1796 | 6.30 |
UGT 1A1, 1A3, 1A4, 1A5, 1A6, 1A7, 1A8, 1A9, 1A10, 2B4, 2B7, 2B10, 2B15, 2B17, and 2B28 were screened for ALDO 18β-G formation at substrate concentrations of 200, 800 and 2000 µm. Of the recombinant UGT proteins, only UGT1A10 and UGT2B7 converted ALDO to its 18β-glucuronide. Activities at 200, 800 and 2000 µm with UGT1A10 were 20, 46 and 63 pmol min−1 mg−1, respectively. The corresponding respective activities with UGT2B7 as the enzyme source were 10, 36 and 42 pmol min−1 mg−1. Kinetic data for both UGT1A10 and UGT2B7 were well fitted to the Michaelis-Menten equation with Km values of 389 and 367 µm and Vmax values of 98 and 43 pmol min−1 mg−1, respectively. Eadie-Hofstee plots are shown in Figure 3.
Figure 3.
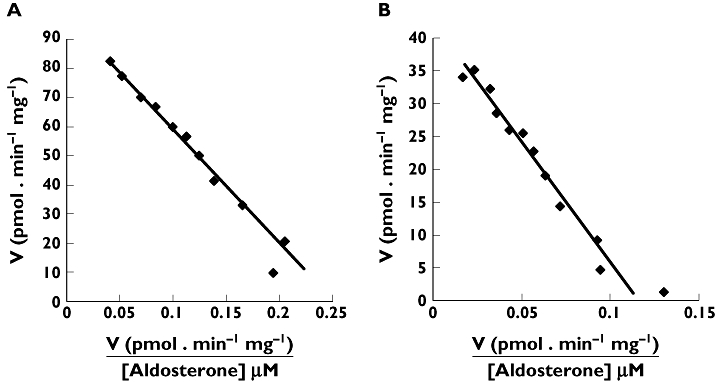
Eadie-Hofstee plots for aldosterone 18β-glucuronidation by recombinant A) UGT1A10 and B) UGT2B7. Points are experimentally determined values, while the solid lines are model fitted curves of best fit
Fluconazole (2.5 mm), a selective inhibitor of UGT2B7 [30], inhibited HKCM- and UGT2B7-catalyzed ALDO 18β-glucuronidation to a similar extent (70%–79%). However, somewhat lesser inhibition (53%) was observed using HLM as the enzyme source. IC50 values for the inhibition of ALDO 18β-G formation by 15 NSAIDs (alclofenac, cicloprofen, diclofenac, diflunisal, fenoprofen, R- and S-ibuprofen, indomethacin, ketoprofen, ketorolac, meclofenamic acid, mefenamic acid, S-naproxen, pirprofen and tiaprofenic acid) were determined for pooled HLM, pooled HKCM and recombinant UGT2B7 at an ALDO concentration corresponding to the approximate apparent Km for each enzyme source (HLM, 500 µm; HKCM 300 µm; and UGT2B7, 300 µm). All NSAIDs investigated inhibited ALDO 18β-G formation by the three enzyme sources (Table 2). Inhibition varied between each NSAID and each protein source and, with the exception of mefenamic acid and diflunisal, the IC50 of each NSAID was higher with HLM than with either HKCM or UGT2B7. Meclofenamic and mefenamic acid were more the most potent inhibitors of ALDO 18β-glucuronidation. Mechanisms of inhibition were characterized for diclofenac, indomethacin, S-ibuprofen, mefenamic acid and S-naproxen with HKCM as the enzyme source. Ki values ranged from 0.5 to 441 µm (Table 3, Figure 4). Similar to the IC50 data, mefenamic acid was the most potent inhibitor of renal aldosterone glucuronidation. Studies from our laboratory have demonstrated previously that mefenamic acid is a potent inhibitor of 4-methylumbelliferone glucuronidation by HKCM and UGT2B7 [9].
Table 2.
IC50 values for NSAID inhibition of aldosterone 18β-glucuronidation by HLM, HKCM and recombinant UGT2B7
| IC50 (µm) | |||
|---|---|---|---|
| NSAID | HLM | HKCM | UGT2B7 |
| Arylpropionates | |||
| Cicloprofen | 20 | 7 | 7 |
| Fenoprofen | 125 | 46 | 90 |
| R-ibuprofen | 588 | 364 | 350 |
| S-ibuprofen | 754 | 425 | 532 |
| Ketoprofen | 278 | 97 | 197 |
| S-naproxen | 132 | 67 | 97 |
| Pirprofen | 121 | 31 | 63 |
| Tiaprofenic acid | 262 | 73 | 151 |
| N-phenylanthranilic acids | |||
| Meclofenamic acid | 0.5 | 0.07 | 0.08 |
| Mefenamic acid | 0.3 | 1.0 | 0.1 |
| Heteroaryl acetic acids | |||
| Alclofenac | 660 | 183 | 296 |
| Diclofenac | 44 | 16 | 13 |
| Ketorolac | >1000 | >1000 | >1000 |
| Others | |||
| Diflunisal | 369 | 343 | 419 |
| Indomethacin | 234 | 115 | 133 |
Table 3.
Inhibition constant (Ki) for NSAID inhibition of aldosterone 18β-glucuronidation by HKCM
| NSAID | Ki (µm) |
|---|---|
| Diclofenac | 8.4 ± 0.5 |
| S-ibuprofen | 441 ± 2.4 |
| Indomethacin | 113 ± 7.1 |
| Mefenamic acid | 0.5 ± 0.02 |
| S-naproxen | 48.7 ± 4.5 |
Ki (µm) is expressed as the mean ± SE of the parameter estimate derived from model fitting.
Figure 4.
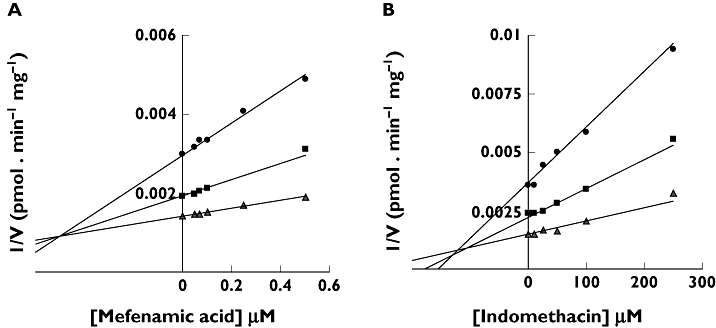
Dixon plots (1/V vs[I]) for inhibition of HKCM catalyzed aldosterone 18β-glucuronidation by A) mefenamic acid and B) indomethacin. Points are experimentally determined values, while the solid lines are model fitted curves of best fit
Discussion
Inhibition of prostaglandin synthesis does not fully explain the renal toxicity associated with NSAIDs. This study was undertaken based on the premise that the clinically observed increase in sodium and water retention that occurs with NSAID use is due, in part, to inhibition of ALDO glucuronidation, particularly at intra-renal sites of ALDO action. In order to evaluate this hypothesis a sensitive, reliable and reproducible method for the quantification of ALDO 18β-glucuronidation in vitro was developed. Authentication of ALDO 18β-G formation was established initially by dependency of metabolite formation on the presence of active protein and cofactor (UDPGA), and acid lability of the metabolite but resistance to β-glucuronidase hydrolysis. (In contrast, glucuronides formed from 21 to hydroxy-steroids are susceptible to β-glucuronidase but not to acid hydrolysis (D.J. Elliot & J.O. Miners, unpublished results)). Negative ion mass spectrometric analysis confirmed a parent ion at m/z 535 and a fragment ion at m/z 359, as expected for ALDO 18β-G. These experimental results are consistent with the report that humans excrete the 18β-glucuronide of ALDO [16].
HLM and HKCM both efficiently catalyzed ALDO 18β-glucuronidation. The mean Km for the reaction catalyzed by HLM (509 ± 137 µm) was approximately 40% higher than that for HKCM (367 ± 170 µm), although this difference was not statistically significant (P > 0.05) due to the relatively high inter-liver/kidney variability in kinetics. Mean Vmax values were similar (1075 ± 429 vs 1110 ± 522 pmol min−1 mg−1). Consequently, CLint (calculated as Vmax/Km) was higher for HKCM than HLM (3.91 ± 2.35 vs 2.36 ± 1.12 µl min−1 mg−1), but again the difference was not statistically significant (P > 0.05). These data accord with studies published previously by this group that demonstrate comparable CLint for liver and kidney glucuronidation of S-naproxen [2, 10], 4-methylumbelliferone [23, 31] and furosemide [32]. It should be noted that, given the major involvement of UGT2B7 in ALDO 18β-glucuronidation by HKCM and HLM, derived Km values are almost certainly over-estimated (and hence CLint is under-estimated) due to the inhibitory effects of long-chain unsaturated fatty acids released during the course of microsomal incubations [33–35]. Indeed, Km values obtained with HLM are normally an order of magnitude higher than the ‘true’Km values of UGT2B7 substrates [33]. BSA may be added to incubations to sequester the inhibitory fatty acids, but this approach was not adopted in the present study as our major aim was to characterize inhibition of ALDO 18β-glucuronidation by NSAIDs, all of which bind extensively (>98%) to albumin.
Although a comparison of microsomal CLint values suggests that the kidney might be more important than the liver in terms of ALDO clearance via 18β-glucuronidation, scaling to a whole organ value takes into account microsomal content (or yield) and organ weight [36]. Average microsome yield from liver (38 mg g−1 tissue) is higher than from kidney (6 mg g−1 tissue) [33, 36], and liver weight is also higher than that of kidney (on average 1500 g vs 300 g). Thus, the scaled whole liver and whole kidney mean CLint values for ALDO 18β-glucuronidation are 135 and 7 ml min−1, respectively. These values indicate that the liver is quantitatively more important for systemic ALDO clearance, while ALDO 18β-glucuronidation is more likely to be of significance for regulating the intra-renal concentration of this hormone.
Of the recombinant human UGT enzymes screened for ALDO 18β-glucuronidation activity only UGT1A10 and UGT2B7 metabolized ALDO. Consistent with the observations for HLM and HKCM, ALDO 18β-glucuronidation by UGT1A10 and UGT2B7 exhibited Michaelis-Menten kinetics, with similar Km values 389 µm and 367 µm, respectively. Importantly, however, UGT1A10 is expressed only in the gastrointestinal tract [37]. Glucuronidation of ALDO in the gastrointestinal tract is not surprising given that ALDO acts in this tissue to increase dietary sodium and water absorption. Hence glucuronidation may serve to limit the ‘local’ biological activity of ALDO with either subsequent excretion of the glucuronide in faeces or reabsorption from the gastrointestinal tract following hydrolysis by β-glucuronidase. In contrast, UGT2B7 is expressed in liver, kidney and gastrointestinal tract [10, 37] suggesting that UGT2B7 is the enzyme largely responsible for ALDO 18β-glucuronidation in human kidney and liver.
A comparison of mean Km values for HLM-, HKCM-, and UGT2B7-catalyzed ALDO 18β-glucuronidation indicates that all are similar; 509 ± 137 µm, 367 ± 170 µm, and 367 µm, respectively. Although not significantly different to HKCM, the somewhat higher Km for the HLM-catalysed reaction may indicate that another, as yet unidentified, UGT also contributes to hepatic ALDO 18β-glucuronidation. Evidence in support of this is provided by inhibition studies with fluconazole, a known selective inhibitor of UGT2B7 [30]. Fluconazole (2.5 mm) inhibited HKCM- and UGT2B7-catalyzed ALDO 18β-glucuronidation to the same extent (70–79%), but lesser inhibition (53%) was observed for HLM-catalyzed ALDO 18β-glucuronidation.
ALDO glucuronidation by UGT2B7, but not other UGT2B enzymes (2B4, 2B15, 2B17 and 2B28), has been reported by Girard et al.[17]. These authors further demonstrated ALDO glucuronidation by human kidney microsomes and HLM. The Km reported for the reaction catalyzed by recombinant UGT2B7 was 0.4 µm. This value is three orders of magnitude lower than the Km determined here. Unlike the present study a non-specific radiometric thin-layer chromatographic method, which utilises 14C-UDPGA as cofactor, was used to identify and quantify product formation [17]. The ‘ALDO glucuronide’ metabolite separated by thin-layer chromatography was not authenticated, whereas in the present study several approaches were adopted to confirm metabolite identity. Additionally, Girard et al.[17] did not publish any kinetic plots. To exclude the possibility of a very high affinity component of ALDO 18β-glucuronidation, kinetic studies were performed with HLM from one liver using substrate concentrations to 1 µm. There was no evidence of deviation from Michaelis-Menten kinetics, precluding a very high affinity reaction similar to that reported by Girard et al.[17]. However, we observed during method validation studies an impurity in some batches of the commercial ALDO that increased in abundance with storage time. The impurity was efficiently glucuronidated by HLM and UGT2B7 and may account for the ‘glucuronide’ measured in the previous work.
Of the non-selective NSAIDs studied here, the rank order of inhibition (IC50) of renal and hepatic ALDO 18β-glucuronidation in vitro followed the general trend: fenamates > diclofenac (heteroaryl acetic acid) > arylpropionates. Cicloprofen was an exception being the only arylpropionate to have a lower IC50 value than diclofenac with all protein sources. Unlike the in vivo situation this in vitro study has the advantage that the metabolism of ALDO was not influenced by the sodium and potassium status, posture and diurnal variation. Consistent with the observation of inhibition of renal and hepatic glucuronidation, NSAIDs also inhibited ALDO 18β-glucuronidation by recombinant UGT2B7. The mechanism of the inhibition of HKCM catalyzed ALDO 18β-glucuronidation by diclofenac, S-naproxen and mefenamic acid was competitive. As expected for this mechanism, Ki values were approximately 50% of the estimated IC50 values (since the substrate concentration employed in the IC50 studies corresponded to the Km). In contrast the mechanism of inhibition of ALDO 18β-glucuronidation by indomethacin and S-ibuprofen was ‘mixed’. The rank order of inhibition, as determined from the Ki values, was mefenamic acid > diclofenac >> indomethacin >> S-ibuprofen.
Like the Km values generated for ALDO 18β-glucuronidation the Ki values reported here are most certainly over-estimates due to ‘additive’ inhibition by membrane long-chain unsaturated acids released during the course of the incubations. Indeed, based on previous studies in this laboratory [27, 30, 33] the Ki values generated for NSAID inhibition of renal ALDO 18β-G formation may be over-estimated by as much as an order of magnitude. Thus, the extent of inhibition in vivo is under estimated. As noted previously, however, the very high binding of NSAIDs to albumin precludes the inclusion of BSA in incubations to sequester the released fatty acids. Despite, the over-estimation of Ki values, the Ki for mefenamic acid (0.5 µm) is similar to the ‘peak’ unbound concentration reported in plasma (0.28 µm, [38]). If a 10-fold over-estimation of Ki is assumed [27, 30, 33], ratios of the peak unbound plasma concentration to Ki would also suggest potential inhibition of ALDO glucuronidation by S-naproxen and possibly diclofenac [39, 40], especially since renal tubular cell concentrations of NSAIDs may be elevated due to reabsorption from urine. However, interactions with indomethacin and S-ibuprofen, which have uncorrected Ki values > 100 µm, are unlikely.
The results obtained here support our hypothesis that NSAID-ALDO interactions may occur in vivo. Inhibition of ALDO 18β-glucuronidation in the kidney would be expected to result in elevated intra-renal concentrations of this hormone. In turn, this could result in an enhanced and prolonged decrement in the natriuretic excretory function of the kidney. The net retention of sodium and water would contribute to volume expansion and ultimately to a rise in blood pressure, hallmarks of NSAID use in ‘at-risk’ individuals [41]. Prostaglandins are known to cause vasodilation of renal vasculature down-regulating the pressure-natriuresis mechanism while ALDO increases the pressure-natriuresis set point [42]. Additionally, NSAIDs are known to blunt the antihypertensive effects of diuretics and/or antihypertensive drugs. Differences in the extent of this effect are not consistent across all NSAID chemical classes, which has been attributed solely to differences in the extent of COX inhibition at a given dose [43]. In our view the duality of non-selective NSAIDs as inhibitors of both the intra-renal synthesis of vasodilatory prostaglandins and of the glucuronidation of ALDO adds further to explaining the adverse renal effects of NSAIDs and their effects on antihypertensive drug response.
In conclusion, ALDO was glucuronidated by HLM, HKCM, UGT1A10 and UGT2B7. UGT2B7 appears to be the major UGT enzyme involved in ALDO 18β-glucuronidation in both liver and kidney. These data clearly highlight the importance of intra-renal glucuronidation of ALDO as a mechanism to terminate the action of this mineralocorticoid. The common involvement of UGT2B7 in both the glucuronidation of carboxylic acid NSAIDs and aldosterone predicates a significant NSAID-ALDO interaction in vivo that may explain in part the clinical observations of variable degrees of fluid retention and blood pressure effects with NSAID use.
Competing interests
None declared.
The research was supported by grants from the National Health and Medical Research Council of Australia.
REFERENCES
- 1.Murray MD, Brater DC. Renal toxicity of the nonsteroidal anti-inflammatory drugs. Annu Rev Pharmacol Toxicol. 1993;32:435–65. doi: 10.1146/annurev.pa.33.040193.002251. [DOI] [PubMed] [Google Scholar]
- 2.Bowalgaha K, Elliot DJ, Mackenzie PI, Knights KM, Swedmark S, Miners JO. Naproxen and desmethylnaproxen glucuronidation by human liver microsomes and recombinant human UDP-glucuronosyltransferases (UGT): role of UGT2B7 in the elimination of naproxen. Br J Clin Pharmacol. 2005;60:423–33. doi: 10.1111/j.1365-2125.2005.02446.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brunelle FM, Verbeeck RK. Glucuronidation of diflunisal in liver and kidney microsomes of rat and man. Xenobiotica. 1996;26:123–31. doi: 10.3109/00498259609046694. [DOI] [PubMed] [Google Scholar]
- 4.King C, Tang W, Nqui J, Tephly T, Braun M. Characterization of rat and human UDP-glucuronosyltransferases responsible for the in vitro glucuronidation of diclofenac. Toxicol Sci. 2001;61:49–53. doi: 10.1093/toxsci/61.1.49. [DOI] [PubMed] [Google Scholar]
- 5.Miners JO, Knights KM, Houston JB, Mackenzie PI. In vitro-in vivo correlation for drugs and other compounds eliminated by glucuronidation in humans; Pitfalls and promises. Biochem Pharmacol. 2006;71:1531–9. doi: 10.1016/j.bcp.2005.12.019. [DOI] [PubMed] [Google Scholar]
- 6.McGurk KA, Brierley CH, Burchell B. Drug glucuronidation by human renal UDP-glucuronosyltransferases. Biochem Pharmacol. 1998;55:1005–12. doi: 10.1016/s0006-2952(97)00534-0. [DOI] [PubMed] [Google Scholar]
- 7.Soars MG, Riley RJ, Findlay KA, Coffey MJ, Burchell B. Evidence for significant differences in microsomal drug glucuronidation by canine and human liver and kidney. Drug Metab Dispos. 2001;29:121–6. [PubMed] [Google Scholar]
- 8.Soars MG, Burchell B, Riley RJ. In vitro analysis of human drug glucuronidation and prediction of in vivo metabolic clearance. J Pharmacol Exp Ther. 2002;301:382–90. doi: 10.1124/jpet.301.1.382. [DOI] [PubMed] [Google Scholar]
- 9.Gaganis P, Miners JO, Knights KM. Glucuronidation of fenamates: kinetic studies using human kidney cortical microsomes and recombinant UDP-glucuronosyltransferase (UGT) 1A9 and 2B7. Biochem Pharmacol. 2007;73:1683–91. doi: 10.1016/j.bcp.2007.01.030. [DOI] [PubMed] [Google Scholar]
- 10.Gaganis P, Miners JO, Brennan JS, Thomas A, Knights KM. Human renal cortical and medullary UDP-glucuronosyltransferases (UGTs): immunohistochemical localization of UGT2B7 and UGT1A enzymes and kinetic characterization of S-naproxen glucuronidation. J Pharmacol Exp Ther. 2007;323:1–9. doi: 10.1124/jpet.107.128603. [DOI] [PubMed] [Google Scholar]
- 11.Miners JO, Smith PA, Sorich MJ, McKinnon RA, Mackenzie PI. Predicting human drug glucuronidation parameters: application of in vitro and in silico modelling approaches. Annu Rev Pharmacol Toxicol. 2004;44:1–25. doi: 10.1146/annurev.pharmtox.44.101802.121546. [DOI] [PubMed] [Google Scholar]
- 12.Mackenzie PI, Bock KW, Burchell B, Guillemette C, Ikushiro S, Iyanagi T, Miners JO, Owens IS, Nebert DW. Nomenclature update for the mammalian UDP glycosyltransferase (UGT) gene superfamily. Pharmacogenet Genomics. 2005;15:677–85. doi: 10.1097/01.fpc.0000173483.13689.56. [DOI] [PubMed] [Google Scholar]
- 13.Sorich MJ, McKinnon RA, Miners JO, Smith PA. The importance of local chemical structure for chemical metabolism by human uridine 5′-diphosphate glucuronosyltransferase. J Chem Inf Model. 2006;46:2692–7. doi: 10.1021/ci600248e. [DOI] [PubMed] [Google Scholar]
- 14.Jin C, Miners JO, Lillywhite KJ, Mackenzie PI. Complementary deoxyribonucleic acid cloning and expression of human liver uridine diphosphate-glucuronosyltransferase glucuronidating carboxylic acid-containing drugs. J Phamacol Exp Ther. 1993;264:475–9. [PubMed] [Google Scholar]
- 15.Sakaguchi K, Green M, Stock N, Reger TS, Zunic J, King C. Glucuronidation of carboxylic acid containing compounds by UDP-glucuronosyltransferase isoforms. Arch Biochem Biophys. 2004;424:219–25. doi: 10.1016/j.abb.2004.02.004. [DOI] [PubMed] [Google Scholar]
- 16.Bledsoe T, Liddle GW, Riondel A, Island DP, Bloomfield D, Sinclair-Smith B. Comparative fates of intravenously and orally administered aldosterone: evidence for extrahepatic formation of acid-hydrolyzable conjugate in man. J Clin Invest. 1966;45:264–9. doi: 10.1172/JCI105339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Girard C, Barbier O, Veilleux G, El-Alfy M, Belanger A. Human uridine diphosphate-glucuronosyltransferase UGT2B7 conjugates mineralocorticoid and glucocorticoid metabolites. Endocrinology. 2003;144:2659–68. doi: 10.1210/en.2002-0052. [DOI] [PubMed] [Google Scholar]
- 18.Brater DC, Harris C, Redfern JS, Gertz BJ. Renal effects of COX-2 selective inhibitors. Am J Nephrol. 2001;21:1–15. doi: 10.1159/000046212. [DOI] [PubMed] [Google Scholar]
- 19.Kokko JP. Effect of prostaglandins on renal epithelial electrolyte transport. Kidney Int. 1981;19:791–6. doi: 10.1038/ki.1981.82. [DOI] [PubMed] [Google Scholar]
- 20.Guan Y, Zhang Y, Breyer RM, Fowler B, Davis L, Hebert RL, Breyer MD. Prostaglandin E2 inhibits renal collecting duct Na+ absorption by activating the EP1 receptor. J Clin Invest. 1998;102:194–201. doi: 10.1172/JCI2872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.de Broe ME, Porter GA, Bennett WM, Verpooten GA. Clinical Nephrotoxins: Renal Injury From Drugs and Chemicals. 2nd edn. London: Kluwer Academic Publishers; 2003. pp. 280–306. [Google Scholar]
- 22.Whelton A, Hamilton CW. Nonsteroidal anti-inflammatory drugs: effects on kidney function. J Clin Pharmacol. 1991;31:588–98. doi: 10.1002/j.1552-4604.1991.tb03743.x. [DOI] [PubMed] [Google Scholar]
- 23.Tsoutsikos P, Miners JO, Stapleton A, Thomas A, Sallustio BC, Knights KM. Evidence that unsaturated fatty acids are potent inhibitors of renal UDP-glucuronosyltransferases (UGT): kinetic studies using human kidney cortical microsomes and recombinant UGT1A9 and UGT2B7. Biochem Pharmacol. 2004;67:191–9. doi: 10.1016/j.bcp.2003.08.025. [DOI] [PubMed] [Google Scholar]
- 24.Boase S, Miners JO. In vitro-in vivo correlations for drugs eliminated by glucuronidation: investigations with the model substrate zidovudine. Br J Clin Pharmacol. 2002;54:493–503. doi: 10.1046/j.1365-2125.2002.01669.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Stone AN, Mackenzie PI, Galetin A, Houston JB, Miners JO. Isoform selectivity and kinetics of morphine 3- and 6-glucuronidation by human UDP-glucuronosyltransferases: evidence for atypical glucuronidation kinetics by UGT2B7. Drug Metab Dispos. 2003;31:1086–9. doi: 10.1124/dmd.31.9.1086. [DOI] [PubMed] [Google Scholar]
- 26.Uchaipichat V, Mackenzie PI, Guo XH, Gardner-Stephen D, Galetin A, Houston JB, Miners JO. Human UDP-glucuronosyltransferases: isoform selectivity and kinetics of 4-methylumbelliferone and 1-naphthol glucuronidation, effects of organic solvents, and inhibition by diclofenac and probenecid. Drug Metab Dispos. 2004;32:413–23. doi: 10.1124/dmd.32.4.413. [DOI] [PubMed] [Google Scholar]
- 27.Rowland A, Elliot DJ, Williams JA, Mackenzie PI, Dickinson RG, Miners JO. In vitro characterization of lamotrigine N2-glucuronidation and the lamotrigine-valproic acid interaction. Drug Metab Dispos. 2006;34:1055–62. doi: 10.1124/dmd.106.009340. [DOI] [PubMed] [Google Scholar]
- 28.Uchaipichat V, Mackenzie PI, Elliot DJ, Miners JO. Selectivity of substrate (trifluoperazine) and inhibitor (amitriptyline, androsterone, canrenoic acid, hecogenin, phenylbutazone, quinidine, quinine, and sulfinpyrazone) ‘probes’ for human UDP-glucuronosyltransferases. Drug Metab Dispos. 2006;34:449–56. doi: 10.1124/dmd.105.007369. [DOI] [PubMed] [Google Scholar]
- 29.Sykes M, Sorich M, Miners JO. Molecular approaches for the prediction of the non-specific binding of drugs to hepatic microsomes. J Chem Inf Model. 2006;46:2661–73. doi: 10.1021/ci600221h. [DOI] [PubMed] [Google Scholar]
- 30.Uchaipichat V, Winner LK, Mackenzie PI, Elliot DJ, Williams A, Miners JO. Quantitative prediction of in vivo inhibitory interactions involving glucuronidated drugs from in vitro data: the effect of fluconazole on zidovudine glucuronidation. Br J Clin Pharmacol. 2006;61:427–39. doi: 10.1111/j.1365-2125.2006.02588.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Miners JO, Lillywhite KJ, Matthews AP, Jones ME, Birkett DJ. Kinetic and inhibitor studies of 4-methylumbelliferone and 1-naphthol glucuronidation in human liver microsomes. Biochem Pharmacol. 1988;37:665–71. doi: 10.1016/0006-2952(88)90140-2. [DOI] [PubMed] [Google Scholar]
- 32.Kerdpin O, Knights KM, Elliot DJ, Miners JO. In vitro characterisation of human renal and hepatic frusemide glucuronidation and identification of the UDP-glucuronosyltransferase enzymes involved in this pathway. Biochem Pharmacol. 2008;76:249–57. doi: 10.1016/j.bcp.2008.04.014. [DOI] [PubMed] [Google Scholar]
- 33.Rowland A, Gaganis P, Elliot DJ, Mackenzie PI, Knights KM, Miners JO. Binding of inhibitory fatty acids is responsible for the enhancement of UDP-glucuronosyltransferase 2B7 activity by albumin: implications for in vitro-in vivo extrapolation. J. Pharmacol Ther. 2007;321:137–47. doi: 10.1124/jpet.106.118216. [DOI] [PubMed] [Google Scholar]
- 34.Rowland A, Knights KM, Mackenzie PI, Miners JO. The ‘albumin effect’ and in vitro-in vivo extrapolation: sequestration of long-chain unsaturated fatty acids enhances phenytoin hydroxylation by human liver microsomal and recombinant cytochrome P450 2C9. Drug Metab Dispos. 2008;36:870–7. doi: 10.1124/dmd.107.019885. [DOI] [PubMed] [Google Scholar]
- 35.Rowland A, Knights KM, Mackenzie PI, Miners JO. The ‘albumin effect’ and drug glucuronidation: bovine serum albumin and fatty acid free albumin enhance the glucuronidation of UGT1A9 substrates but not UGT1A1 and UGT1A6 activities. Drug Metab Dispos. 2008;36:1056–62. doi: 10.1124/dmd.108.021105. [DOI] [PubMed] [Google Scholar]
- 36.Bowalgaha K, Miners JO. The glucuronidation of mycophenolic acid by human liver, kidney and jejunum microsomes. Br J Clin Pharmacol. 2001;52:605–9. doi: 10.1046/j.0306-5251.2001.01487.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tukey RH, Strassburg CP. Human UDP-glucuronosyltransferases: metabolism, expression, and disease. Annu Rev Pharmacol Toxicol. 2000;40:581–616. doi: 10.1146/annurev.pharmtox.40.1.581. [DOI] [PubMed] [Google Scholar]
- 38.Neuvonen PJ, Kivisto KT. Effect of magnesium hydroxide on the absorption of tolfenamic and mefenamic acids. Eur J Clin Pharmacol. 1988;35:495–501. doi: 10.1007/BF00558244. [DOI] [PubMed] [Google Scholar]
- 39.Hinz B, Chevts J, Renner B, Wuttke H, Rau T, Schmidt A, Szelenyi I, Brune K, Werner U. Bioavailability of diclofenac potassium at low doses. Br J Clin Pharmacol. 2005;59:80–4. doi: 10.1111/j.1365-2125.2005.02226.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Charles BG, Mogg GA. Comparative in vitro and in vivo bioavailability of naproxen from tablet and caplet formulations. Biopharm Drug Dispos. 1994;15:121–8. doi: 10.1002/bdd.2510150204. [DOI] [PubMed] [Google Scholar]
- 41.Gambro G, Perazella MA. Adverse renal effects of anti-inflammatory agents: evaluation of selective and nonselective cyclooxygenase inhibitors. J Intern Med. 2003;253:643–52. doi: 10.1046/j.1365-2796.2003.01146.x. [DOI] [PubMed] [Google Scholar]
- 42.Lopez-Hernandez F, Lopez-Novoa JM. The lord of the ring: mandatory role of the kidney in drug therapy of hypertension. Pharmacol Ther. 2006;111:53–80. doi: 10.1016/j.pharmthera.2005.08.002. [DOI] [PubMed] [Google Scholar]
- 43.Morgan T, Anderson A. The effect of nonsteroidal anti-inflammatory drugs on blood pressure in patients treated with different antihypertensive drugs. J Clin Hypertens. 2003;5:53–7. doi: 10.1111/j.1524-6175.2003.00514.x. [DOI] [PMC free article] [PubMed] [Google Scholar]