

Abstract
We introduce “library based Monte Carlo” (LBMC) simulation, which performs Boltzmann sampling of molecular systems based on pre-calculated statistical libraries of molecular-fragment configurations, energies, and interactions. The library for each fragment can be Boltzmann distributed and thus account for all correlations internal to the fragment. LBMC can be applied to both atomistic and coarse-grained models, as we demonstrate in this “proof of principle” report. We first verify the approach in a toy model and in implicitly solvated poly-alanine systems. We next study five proteins, up to 309 residues in size. Based on atomistic equilibrium libraries of peptide-plane configurations, the proteins are modeled with fully atomistic backbones and simplified Gō-like interactions among residues. We show that full equilibrium sampling can be obtained in days to weeks on a single processor, suggesting that more accurate models are well within reach. For the future, LBMC provides a convenient platform for constructing adjustable or mixed-resolution models: the configurations of all atoms can be stored at no run-time cost, while an arbitrary subset of interactions is “turned on.”
Keywords: library-based Monte Carlo, Metropolized Independence Sampling, Metropolis-Hastings Monte Carlo, Gō potential, CDC25B, GGBP
I. Introduction
Proteins are tiny machines that perform their functions accompanied by motion in many cases. It has been recently suggested that proteins evolved not only to have a specific structure but also dynamic properties to perform certain biological functions. Conformational fluctuations are believed to be important for many protein functions such as signaling 1, catalysis 2,3, and intracellular transport 4. Indeed, experimental techniques now allow observation of protein motions at a wide range of different time scales 5-7. However, the simultaneous determination of structure and dynamics remains a challenging task 8 and has motivated the development of simulation methods.
Among computational methods for studying protein dynamics, molecular dynamics (MD) simulation is one of the most popular, due to its all-atom realism and the reasonable accuracy of current forcefields. However, a considerable gap remains between the timescales of many biologically important motions and timescales accessible to MD simulations – see, e.g., Ref. 9 about the attempted folding of a WW domain. To bridge this timescale gap, numerous atomistic algorithms have been suggested based on generalized ensembles 10-14. Another approach is to bias simulations based on high temperature sampling 15 or structural knowledge (e.g., pdb structures). Subsequently, these simulations must be unbiased to preserve the correct equilibrium distribution.
Coarse-grained (CG) and multi-resolution approaches for modeling proteins are gaining in popularity for the study of long-timescale phenomena 16-20. Recently our group developed the “resolution exchange” (ResEx) algorithm that utilizes a ladder of several different coarse-grained models with simulations simultaneously running at different levels and occasional exchange attempts between different levels 21,22. In the more efficient top-down implementation 22 ResEx relies on converged sampling at the most coarse-grained level and gradual “de-coarsening” to higher resolution levels. Beyond equilibrium sampling, it can be anticipated that CG models will be needed for path sampling of dynamic processes 23.
The success of multi-resolution simulation approaches hinges, in part, on model design: the most coarse-grained model in a multi-resolution protocol should be amenable to full equilibrium sampling. Therefore, a key question is: How simplified must a protein model be to permit good sampling?24 To put it more positively, what is the most detailed model that can be fully sampled? Answering these questions is essential for progress in multi-resolution sampling. We have already developed tools for assessing sampling quality 25,26. The main motivation for the present work is to develop a flexible algorithmic strategy for sampling increasingly more realistic “coarse-grained” models – in fact, “semi-atomistic” models with some all-atom features. We previously developed a semi-atomistic coarse-grained model based on rigid peptide-plane geometry 24, but technical considerations (rather than sampling difficulties) limited that approach to relatively small proteins.
This report presents an apparently novel Monte Carlo scheme for flexibly incorporating atomic detail into a model using pre-sampled molecular fragments. A molecule is divided into fragments (e.g., residues) for which large libraries of configurations are generated in advance. It is important that the approach can be applied to “semi-atomistic” coarse-grained models as well as to implicitly solvated, standard all-atom forcefields. For the case of semi-atomistic models, the sampling speed of a model can be tuned by turning on or off interactions among pre-stored fully atomic fragments. This tuning is independent of the atomic detail stored in the fragments, permitting tremendous flexibility in model construction within a single computer program. For instance, one can store and update atomic coordinates of every atom of a molecule (at negligible run-time cost) while enabling rapid sampling by turning off an arbitrary subset of interactions. We term the method “library based Monte Carlo” (LBMC) because libraries are central to storing configurations and generating trial moves.
This study is a “proof of principle” demonstrating the potential future utility of the LBMC approach. The models employed are purposefully simplistic in this initial study. In a sense, the goal of this study is to provide a “lower bound” on the degree of chemical accuracy which permits full sampling of proteins. We will employ significantly improved models in future studies aimed at addressing specific biophysical and biochemical questions.
Our report first presents derivations of two LBMC Metropolis acceptance criteria, in addition to alternative derivations in the Appendix. Separate criteria are derived for trial moves which are simple swaps with random library configurations and for “neighbor-list” swap moves with library configurations similar to the current conformation. The procedure can be applied to arbitrary non-overlapping molecular fragments. The LBMC method is first carefully verified by comparison to Brownian dynamics simulation of a simple toy system, as well as to implicitly solvated all-atom peptides sampled with Langevin dynamics. The peptide simulations are considerably more efficient than expected based on the modest savings in energy calculation. The technique is then applied to several proteins described by a simple semi-atomistic model. The largest protein contains more than 300 residues. The convergence analysis indicates that full sampling is easily obtained in about a month of single processor time.
II. Library-based Monte Carlo (LBMC)
The library-based Monte Carlo approach is very general and can be used with arbitrary molecular fragments – e.g., peptide-planes, full residues, or multi-residue fragments. LBMC is also applicable to arbitrary forcefields, whether all-atom or simplified.
Library-based techniques should be of increasing interest in the future, with the fast growth and declining cost of computer memory. Intuitively, it seems clear that vast computer resources are devoted to redundant calculations (e.g. of bond energy) which could more parsimoniously be retrieved from memory. Libraries which are themselves Boltzmann-factor distributed, furthermore, store correlations internal to a fragment, and this appears to be quite important in the effectiveness of LBMC.
In LBMC a molecule is split into fragments, with a library of configurations for each fragment generated in advance. Such a strategy is natural for proteins, which consists of only 20 building blocks. During a simulation, fragment configurations in the present state of the system are swapped with configurations in the library and the new state is accepted according to the corresponding Metropolis criterion derived below. The interactions within a fragment do not have to be calculated again during a simulation: libraries should be generated only once and then can be used in repeated simulations. The idea of using libraries of pre-calculated configurations has been around for some time in the polymer growth field and several library-based algorithms have previously been suggested 27-29.
Our technique is partly inspired by the success of the fragment assembly software Rosetta, in which short fragments of known protein structures are assembled into native-like conformation using ad hoc library-based Monte Carlo 30. Rosetta is one of the most successful methods for de novo protein structure prediction 31. There are several differences between our approach and Rosetta. Algorithmically, LBMC generates canonical sampling whereas Rosetta's Monte Carlo does not yield a defined distribution. Rosetta's libraries are not distributed according to a mathematically defined distribution. On the other hand Rosetta employs significantly larger fragments than used in this initial LBMC study. Rosetta's forcefield is quite different than those used here – and much more chemically accurate than the simple protein model employed here. For peptides, however, we employ the OPLS-AA forcefield 32 with a simple implicit solvent model.
As in all Monte Carlo (MC) simulations, different types of trial moves can be used alternately or in combination. For example, one can make hybrid trial moves in which fragment configurations are swapped along with making random changes to degrees of freedom which are not included with the libraries. If desired, all degrees of freedom can be pre-sampled in libraries.
The acceptance criterion for a library-based swap move depends on the distribution of the library. If the degrees of freedom in the fragment are already distributed according to the Boltzmann factor (for those coordinates alone), the acceptance criterion becomes particularly simple and independent of fragment energy as shown below. However, as we also discuss, in some cases it may be useful to employ other library distributions.
It is important to emphasize that LBMC simulation has several strengths: (i) the storage of energy terms reduces run-time calculation cost; (ii) distinct from the run-time savings there is addition benefit from the sampling and correlations stored in the libraries, and (iii) there is flexibility to adjust the types and quantity of interactions while storing as many atomic positions as desired at negligible run-time cost. The first characteristic is the most obvious: the run-time savings in energy calculation compared with ordinary MC due to the storage of terms internal to fragments – and possibly of interaction energies between covalently bound fragments 33. This storage guarantees that LBMC can compute the energy faster, but the fractional savings may only be modest, depending on the interactions in the forcefield, the size of the fragments, and the size of the molecule. Consider a molecule consisting of M fragments, each with f atoms: the number of stored energy terms will scale as Mf2 , whereas the minimum number of distance calculations varies as M2 (e.g., with a single long-ranged interaction site per fragment in a simplified model). Whether LBMC leads to a significant advantage clearly depends on fragment size and the number of inter-fragment interactions present in the forcefield.
The fact that canonically distributed libraries store correlations is a strength distinct from the energy savings. Indeed, our initial all-atom peptide results indicate it is far more important than saving on energy calculation! To see why, imagine libraries are used to generate trial moves but the energy is calculated without using stored terms. This type of LBMC seems at first like a minor variation from internal-coordinate Monte Carlo (ICMC). In fact, the difference is substantial. Every library configuration is guaranteed to account for all steric and other non-bonded correlations internal to the fragment, in sharp contrast to ICMC. Sufficiently large fragments will store a substantial amount of correlation information. In casual terms, the significant sampling time invested (a single time) in generating the libraries pays off every time the libraries are used.
The final strength lies in the unique ability of LBMC to keep track of locations of practically limitless number of atoms at negligible run-time cost. For instance, every atom in a protein can be tracked based on appropriate fragments. Then (as for any code) different interactions can be specified as desired. Yet the ability of LBMC to store all atoms appears to make it particularly suitable for multi-resolution methods which can employ a single LBMC program with a range of interaction choices.
II.A. Defining fragments and the energy decomposition
To be fully general, we first consider an arbitrary division of a molecule into M non-overlapping fragments. On the individual fragment level, a configuration of fragment i containing Ni degrees of freedom is described by r⃗I = {xi1,…,xiNi}. Such fragments need not correspond to groups of atoms. For instance, in the protein model used bellow, we have excluded the ψ backbone dihedral angle from our peptide-plane fragments – and all of the ψ dihedrals together can be considered to constitute a single fragment. Regardless of the division into fragments, the usual total potential energy of the system (e.g., from a standard forcefield) can then be decomposed as
| (1) |
where contains all energy terms which depend solely on the coordinates of fragment i but with no interactions between fragments. Urest (r⃗1,…,r⃗M) simply includes the balance of the energy terms, and thus encompasses all interactions between fragments and any energy terms dependent on the coupling between fragments. In a typical molecular system, will contain all van der Waals, electrostatics and bonded terms internal to the fragment.
II.B. Monte Carlo simulation with libraries
The acceptance criteria necessary for library-based MC can be derived in more than one way. In the Appendix we present derivations based on a continuum description, while the present section uses statistical ideas more directly relevant to the discrete nature of the libraries.
Our present derivation is based on the fact that LBMC is nothing more than the use of Metropolis MC to perform re-weighting – or more precisely “resampling” 34. This can be understood based on standard simulation ideas.
First, why do we need to re-weight? Quite simply, the configuration space spanned by our libraries has a built-in bias. The library for fragment i consists of a set of configurations distributed according to some function which will be specified below. If we draw a random configuration from each library, we will generate full molecular configurations distributed according to the simple product of the single-fragment distributions,
| (2) |
In any realistic system, this simple-product distribution will differ from the desired equilibrium distribution peq, which in our case is proportional to the usual Boltzmann factor of the full potential, Eq. 1, namely,
| (3) |
At a minimum, the full Boltzmann factor will contain terms which couple the fragments and which are absent from the fragment libraries by construction. Regardless of the precise differences, the simple-product distribution of Eq. 2 will need to be reweighted in order to recover the correct distribution.
The ideas of reweighting and resampling are textbook subjects 34 and can readily be understood. The basic idea is that one has an incorrect distribution, plib (r), instead of the desired target distribution peq r, with in our case. This means that the calculation of a canonical average in the peq ensemble requires that every configuration from the plib distribution be assigned a weight 35
| (4) |
The denominator exactly cancels the incorrect frequency from the plib distribution. We note that overall normalization is irrelevant in a weighted average.
Another way to understand the weights of Eq. 4 is by observing that a partition function can always be re-written using a “sampling function” g. Specifically, the identity:
| (5) |
where the bracketed function denotes either uniform or g-distributed sampling, implies the weights of configurations must be e−βUtot (r) or e−βUtot (r)/g (r), respectively. In our case g = plib.
“Resampling” describes the procedure for generating a representative ensemble of unweighted configurations in the peq ensemble from a sample of plib 34. Quite simply, configurations from an ensemble distributed according to plib should be included in the peq ensemble with probability proportional to the weight w(r). Operationally, resampling can be performed in a number of different ways. As an example, the simplest method examines each configuration r in the original plib sample and accepts or rejects it with probability w(r)/wmax, where wmax is the maximum weight among all sampled configurations.
Direct resampling as just described is effectively impossible in our case, because of the astronomical number of full molecular configurations under consideration. In a typical case, we will employ over 105 configurations per fragment library, with fragments representing individual amino acids. Thus, for a protein of 100 residues, there are over 10500 possible configurations, and we could never consider every one.
Metropolis Monte Carlo is designed precisely for such a situation, where all possible configurations cannot be considered directly. Further, due to the finite precision of digital computing, ordinary “continuum” MC is, in fact, discrete. Thus, standard MC can be seen as a method to sample a set of Boltzmann-factor-distributed configurations based on a much larger “library” of finely and uniformly discretized Cartesian configurations 36. One can call this “sampling” or even “resampling,” but the net effect is to transform one distribution into another. Our goal is the same. The key difference in our case is that we wish to sample our discrete space of biased library-based configurations with probability proportional to w of Eq. 4. The word “biased” here really only means that our initial distribution is neither uniform nor equilibrium.
Based on this logic, it is straightforward to adapt Metropolis MC to the necessary resampling. Ordinary MC samples a distribution p based on the condition of detailed balance, namely,
| (6) |
where pgen and pacc are the usual generating and acceptance conditional probabilities, while “o” and “n” are shorthand for the old and new (trial) configurations, respectively. The usual case of setting p = peq corresponds to (re)sampling the “library” of finite precision configurations into the Boltzmann distribution, as already noted.
In our case, we want to set p = w in Eq. 6, in order to re-weight our library distribution, which has been customized for molecular fragments. In the resampling case, then, the correct acceptance criterion becomes:
| (7) |
where overall normalization of w is irrelevant as in standard Metropolis MC. Our Results section carefully demonstrates the validity of this acceptance criterion in simple, verifiable systems.
It is important to note that, as in standard Metropolis Monte Carlo, once the distribution or weighting function is selected for Eqs. 6 and 7, arbitrary trial moves among the library configurations can be employed, so long as any asymmetries in generating probabilities are accounted for. Further, whenever the generating probability is symmetric, i.e., when pgen(i→j)=pgen(j→i) for all i and j, we obtain the simpler form
| (8) |
The acceptance criterion of Eq. 8 is related to “Metropolized independence sampling,” originally derived by Hastings 36.
II.C. Library Monte Carlo with molecular fragments
In order to employ library-based Monte Carlo with our molecular fragment libraries, we need to derive the appropriate weight function of Eq. 4 for use in the acceptance criterion of Eq. 8. As just described, this weight function is the ratio of the targeted equilibrium distribution of Eq. 3 to the distribution based on the pre-calculated fragment configurations. The fragment-based distribution of Eq. 2 results from separate libraries for each fragment in the present case. In our study, library configurations r⃗i for each fragment i will be distributed according to the “internal” Boltzmann factors, implying:
| (9) |
where the fragment energy has been defined in Eq. 1. In essence, the “global” library of full configurations we are considering is constructed from independent configurations for each fragment, and is therefore distributed according to the simple product of fragment distributions of Eq. 9.
We can now directly calculate the necessary weight function 4 based on Eqs. 2, 3 and 9 , which yields:
| (10) |
This is the weight function which should be employed in the acceptance criteria of Eqs. 7 and 8 when using libraries distributed as in Eq. 9. Thus, the acceptance probability for trial moves with symmetric generating probability reduces to the simple expression
| (11) |
where ΔUrest=Urest (n)−Urest (o). The appendix provides a more traditional derivation of this acceptance criterion based on detailed-balance arguments, which is equivalent to “Metropolized independence sampling”34 originally derived by Hastings 36.
By design, all of the fragment energies have cancelled out from Eq. 11, which can serve two purposes. First, the cancellation reduces the number of interactions which need to be calculated in testing for acceptance via Eq. 8 although the reduction may be very modest depending on the system. Second, because all interactions internal to the fragments have been pre-sampled, one can hope that the discretized configuration space embodied in the libraries has more overlap with the target distribution than would a uniform sampling implicit in the uniform trial moves of a typical Monte Carlo simulation. That is, trial moves based on library swaps will automatically account for interactions internal to the swapped fragment.
For completeness, we note that the ψ dihedral angles are treated as standard continuum variables in the simple protein model studied. This is a minor technical point, and is equivalent to placing the associated energy terms in Urest rather than as a separate fragment term. No bias results from this choice, and indeed it underscores the freedom to employ libraries for some variables but not others. We anticipate that future protein models will include all degrees of freedom in libraries.
II.D. Neighbor lists in library-based Monte Carlo
The MC scheme described above has poor control of the acceptance rate for trial moves where fragment configurations are selected at random from one of the fragment libraries. We found that with peptide-plane fragment libraries the acceptance rate is very small (0.6-3%) for all proteins studied. The reason is the “small angle disaster” – i.e., even a difference of a few degrees in angle accumulated over one fragment may result into large conformational change 20-30 Å away from the swapped fragment. To improve the acceptance rate, library configurations can be classified into neighbor lists so that each configuration can be assigned a number of similar neighbor configurations. Instead of selecting configurations randomly from the library they can be selected from the neighbor list, thereby increasing the acceptance rate.
We thus need to determine the two generating probabilities in Eq. 7 which correspond to using neighbor lists. For simplicity, we are solely interested in trial moves which change a single fragment configuration – i.e., the overall configurations “o” and “n” differ only in a single fragment. That is, we wish to consider “neighbors” of a given configuration within a single library (generalizations to multi-fragment trial moves are straightforward, but not presented here).
Fortunately, once a list of neighbors has been determined for a given fragment configuration (as described below), the generating probabilities take a trivial form. Indeed, pgen (i → j) answers the question, “What is the probability the computer program will choose configuration j as a trial move, given that i is the present configuration?” If configuration i has ki neighbors, of which one is selected at random, then:
| (12) |
Regardless of how the library was generated, once the neighbor list is determined, the (conditional) generating probability takes this simple form.
The acceptance criterion appropriate for neighbor lists is therefore given by
| (13) |
where ko must be understood as the number of neighbors for the particular (single) fragment configuration selected for a trial move, and kn is the number of neighbors of the trial fragment configuration. The criterion in Eq. 13 is also derived from a continuous picture in the Appendix.
If neighbor lists are constructed to have the same number of neighbors for all configurations in a given library, then the acceptance criterion in Eq. 13 reduces to the simpler symmetric form of Eq. 11. In the present study, all neighbor lists were constructed to be the same size ( ki=10 for all i), as we now describe. Neighbor lists were not used in the peptide simulations.
In practice, fragment configurations can be classified into (arbitrary) “neighbor lists” using some metric describing the similarity of configurations to each other. This metric can be, for example, RMSD or the sum of absolute differences over all backbone bond and dihedral angles. This metric can be calculated between all pairs of configurations in the library and sorted based on similarity to each other. When building the neighbor list, configurations most similar to the chosen one should be selected to be in its neighbor list. Further, to satisfy microscopic reversibility, if configuration i has in its neighbor list configuration j then j must have i. In the present study, neighbor lists were generated using an approximate procedure, which cannot guarantee the best possible neighbors.
One potential problem with the neighbor lists is that they may form isolated clusters. We checked our neighbor lists, however, and found that all configurations are connected to each other and there are no disconnected clusters. We emphasize that the algorithm remains correct for arbitrary neighbor lists, so long as trial moves to disconnected configurations are occasionally attempted.
We found that implementation of the neighbor lists increased the acceptance rate of LBMC swap moves by 8-20 times depending on the system. The acceptance rate increased from just 2-3% to 15-35% for the small proteins containing less than 100 residues. Even for the large 309 residue protein the acceptance rate increased from 0.6 % to 5 %.
II.E. Using libraries with non-Boltzmann distributions
We found that, for some systems, libraries distributed according to the Boltzmann factor are not effective because fragment configurations may have very low population densities in high-energy transition regions of configuration space that may be important in a full system. For example, high free energy regions connecting the left (φ<0) and right (φ>0) sides of the Ramachandran plot are very rarely populated in equilibrium libraries of amino acids. However, these regions are functionally important for some proteins and ignoring them would make it difficult to sample potentially important configurations. Inefficiency also arises when a fragment library has more configurations than necessary in the low free energy regions.
With these points in mind, the fragment libraries can be improved by over-representing some regions of configuration space and under-representing others. In principle, the fragment configurations can be biased toward known protein structures similar to the Rosetta method 30. When biased, configurations must be assigned weights based on the ratio of the desired library size to the true (unbiased) equilibrium populations.
One way to bias libraries employs discretization of configuration space into distinct regions. If every library configuration j is classified into some state s(j) – i.e., some region of configuration space – then its weight is
| (14) |
where Pbias (s) is the biased library size (fractional population) for state s and Peq (s) is the true equilibrium population in the same state. Note that Peq (s) is simply proportional to the unbiased library counts for the fragment i under consideration, as well as to the local partition function . We assume that within each state, a library is unbiased – i.e. configurations are distributed according to the Boltzmann factor. The states can represent, for example, different regions of the Ramachandran plot.
Many other biasing schemes are possible. For example, individual configurations could be distributed according to an alternative forcefield or energy terms accounting for neighboring fragments.
When the biased library is used, the library distribution of Eq. 2 should be modified to account for the introduced bias
| (15) |
This library distribution can be used along with the target distribution of Eq. 3 to find the necessary weighting function:
| (16) |
Using this weighting function, the acceptance criterion for a swap move with biased libraries can be obtained
| (17) |
Note that the product of biasing weights needs to be calculated only for fragments which were swapped in the current MC move. For the rest of the fragments (which were not swapped) the biasing weights are the same and cancel out.
II.F. Practical library generation and the use of dummy atoms
Libraries of fragment configurations can be generated using any standard canonical sampling method – for example, Langevin dynamics or Metropolis MC. The only statistical requirement for a library is that it should represent the true equilibrium distribution – that is, it must be consistent with Eq. 9 – for all the degrees of freedom in the fragment and the chosen energy function .
Dummy atoms
In practice, the efficiency of LBMC depends on how the system is split into fragments and what degrees of freedom are included in each fragment. Because fragments are sampled separately and independently from each other it is usually convenient to include and sample the extra six degrees of freedom that specify the orientation of fragments relative to each other. These are rigid-body translation and rotation degrees of freedom, for any pair of configurations in neighboring fragments. Because the dummy atoms are not considered part of the fragment, their interactions with the real fragment atoms should not contribute to Utot. This can be achieved either by making the dummy atoms non-interacting or, if they are interacting, their interaction energy must be accounted for in the acceptance criterion as described bellow. We found that the interacting dummy atoms improve the overlap with the full molecular distribution by mimicking the neighboring fragment atoms. The improved overlap can increase the acceptance rate and improve the efficiency of LBMC.
When interacting dummy atoms are employed, care must be taken because they may introduce addition degrees of freedom which are not part of the system. Here, we only consider the case where these additional degrees of freedom are fixed, leading to a fairly simple acceptance criterion.
The acceptance criterion of Eq. 11 must be modified to account for interactions of real and dummy atoms within a fragment. To derive the criterion, we can start with the overall fragment potential energy, which now includes the additional interactions:
| (18) |
where the full set of fragment coordinates is denoted by , with describing the additional degrees of freedom which are introduced by the dummy atoms. In Eq. 18 is the potential energy of the real fragment atoms and is the interaction energy between real and dummy atoms.
Importantly, because we choose the additional dummy internal coordinates to be fixed for every fragment configuration, the relevant energy differences depend only on the coordinates of real atoms r⃗i.
To derive the final acceptance criterion for fragments with interacting dummy atoms, the generating probability of selecting configurations randomly from the libraries must account for the additional interactions of Eq. 18. The resulting acceptance criterion is
| (19) |
where .
Two library types were used in our LBMC simulations: one type is based on the peptide planes and the other is based on the full residues. In this study the peptide-plane libraries were used for all protein LBMC simulations and the residue libraries were used only for simulations of all-atom peptides. The main differences between these two library types are the degrees of freedom included with the fragments and the forcefield used. Both library types include interacting dummy atoms. All the library details are provided in the Supplemental Material. All libraries are available on our website: www.epdb.pitt.edu.
In fact, for future protein models, we prefer the residue libraries, largely because they explicitly embody Ramachandran correlations – but the residue libraries were actually constructed after the plane libraries.
III. Protein Model
In our previous work we showed that complete sampling of equilibrium ensemble is possible with a simple rigid peptide plane protein model in several weeks of single CPU time 24. Building on our previous work we further improve the model by including all-atom based backbone flexibility at a small computational cost.
In our protein backbone model, a fragment is represented by a peptide plane configuration containing all of the atomic backbone degrees of freedom except ψ which is sampled as a standard continuous variable. The planes are coupled through interactions sited, for simplicity, at alpha-carbons, as described below. Our protein backbone model is schematically shown in Figure 1.
Figure 1.

The protein backbone model used in this study is represented by a set of peptide plane configurations. A library of atomistic peptide-plane configurations is generated in advance and used for trial moves in library-based Monte Carlo, allowing full flexibility of the atomic backbone.
Three different types of peptide planes are used, corresponding to Ala, Gly and Pro residues needed for the correct atomic model of the backbone. In our protein model all non-Gly and non-Pro residues are reduced to a “pseudo-Ala” plane containing a beta carbon without hydrogen atoms; this is a natural choice for a backbone model since all non-Gly and non-Pro residues have similar Ramachandran propensities 37. For the Pro residues all of the ring atoms are included with the peptide plane because they affect backbone configurations.
To stabilize the native state and also allow fluctuations, this initial study employs “structure-based” or Gō interactions among alpha-carbons 38,39. Our previous studies showed that the Gō potential can reproduce reasonable protein fluctuations compared to experimental data 40,24. Neighboring peptide planes are excluded from Gō interactions so that residue i can interact only starting from residue i+3. The Cα interaction centers for the Gō potential consist of native and non-native interactions. Thus the total potential energy not internal to the fragments (peptide-planes) is
| (20) |
where is the total energy for native contacts and is the total energy for non-native contacts. All residues that are separated by a distance less than a cutoff, Rcut, in the experimental structure are given native interaction energies defined by a square well
| (21) |
where rij is the distance between Cα atoms of residues i and j, is the distance between residues in the experimental structure, ε determines the energy scale of Gō interactions, and δ sets the width of the square well. All residues that are separated by more than Rcut in the experimental structure are given non-native interaction energies defined by
| (22) |
where ρi is the hard-core radius of residue i, defined at half the Cα distance to the nearest non-covalently bonded residue in the experimental structure, and h determines the strength of the repulsive interactions.
For this study, parameters were chosen to be similar to those in Ref. 24, i.e., ε=1.0, h=0.3, δ=0.2, and Rcut=8.0 Å.
IV. Results and Discussion
IV.A. Toy system
We performed a “reality check” on the LBMC technique by applying it to a simple one-dimensional toy system represented by two particles connected to harmonic springs. Two cases were considered: in the first case, the particles are non-interacting, whereas in the second case particles interact via a repulsive Coulombic (r−1) potential. For each case, the probability density function (pdf) along the one-dimensional coordinate for each particle was calculated using LBMC and checked using standard Brownian dynamics (BD). For LBMC a library of 106 configurations was generated in advance for a system composed of one particle connected to a harmonic spring. During LBMC simulation both particles were sampled using the same library. For BD simulations a standard over-damped Langevin Dynamics procedure without velocities was used 41. The results for LBMC and BD are compared in Figure 2B for non-interacting particles and in Figure 2C for interacting particles. The agreement between LBMC and BD for both cases is excellent. Note that in Figure 2C the equilibrium distance between particles increased due to Coulomb repulsion that was correctly reproduced by LBMC.
Figure 2.
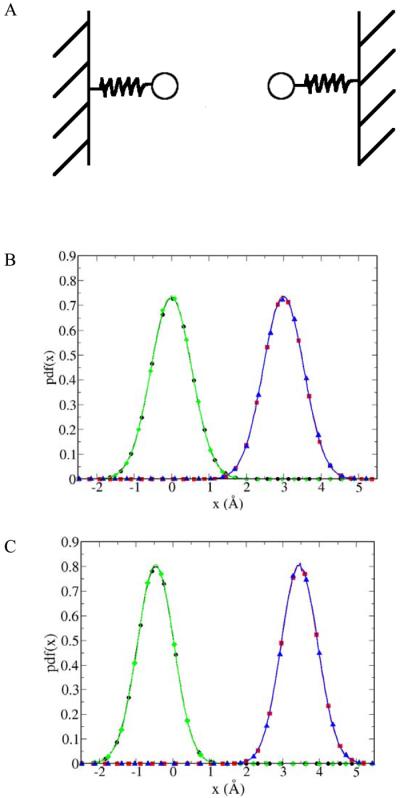
The one-dimensional toy system used as a “reality check” of library-based Monte Carlo (LBMC) is shown schematically in (A) and consists of two particles connected to harmonic springs and restrained in one-dimension. The equilibrium position of the first particle was set at the origin and the second 3 Å away from the origin. The probability density function (pdf) along the one-dimensional coordinate calculated LBMC and checked with BD for two cases: (B) particles do not interact with each other, and (C) particles interact with each other via Coulombic repulsion. BD results of the first particle are denoted by black line with circles and the second particle by red line with squares. LBMC results are denoted by green line with diamonds for the first particles and by blue line with triangles for the second particle. Note that the equilibrium distance between particles increased when the Coulombic repulsion was switched on (C) which was correctly reproduced by LBMC. Both particles were restrained with a spring constant of 0.5 kcal/(mol*Å2) at the temperature of 300 K. In (C) the partial charges of particles were set to 0.2 e and the dielectric constant was set to 1.
IV.B All-atom peptides
We generated equilibrium ensembles for two all-atom peptides: Ace-(Ala)4-Nme and Ace-(Ala)8-Nme, both to verify the algorithm and to assess efficiency in a molecular contest. As discussed previously, there may not be a significant savings in the calculation of the energy, so we did not expect significant efficiency improvement. In fact, our initial tests reported here used a far-from-optimal implementation – in that many energy terms which could have been stored were not (i.e., interactions among fragments 33 were not stored). Nevertheless, there were very significant efficiency gains as described below.
Our peptide simulations employed the OPLS-AA forcefield 32 and a highly simplified continuum solvent model (constant dielectric, with dielectric contains of 60). The solvent model was chosen only to perform the proof-of-principle calculations reported here, and the value of 60 was selected based on trial-and-error comparisons with GBSA simulations of poly-alanine systems. Details of the libraries are given in the Supplementary Material, but we note that these libraries differ from the peptide-plane libraries used for proteins.
The production run for Ace-(Ala)4-Nme consisted of 105 MC steps with frames saved every 10 MC steps. The production run for Ace-(Ala)8-Nme was 107 MC steps with configurations saved every 102 MC steps. No neighbor lists were required for these flexible systems. It took 10 seconds to complete the production run for Ace-(Ala)4-Nme and 20 minutes for Ace-(Ala)8-Nme. For both systems the LBMC swap move consisted of swapping only one fragment (i.e., residue) configuration per MC step.
We first verified that the algorithm reproduced the correct equilibrium ensembles. To do so, we prepared the “structural histograms” 25,26 in Figure 3, which show the fractional populations in a set of configuration-space regions or bins. The data for Ace-(Ala)4-Nme and Ace-(Ala)8-Nme show that the LBMC results agree well with independent Langevin simulations performed using Tinker 42 at T=298 K and friction constant 5 ps−1. The bins were constructed based on a Voronoi classification of configuration space as described earlier 33. Langevin simulations of 100 ns in length required 16 hours and 43.5 hours respectively, for Ace-(Ala)4-Nme and Ace-(Ala)8-Nme.
Figure 3.

Fractional populations of Voronoi bins for LBMC (red line) and Langevin (black line) simulations for two all-atom peptides: (A) Ace-(Ala)4-Nme, and (B) Ace-(Ala)8-Nme. The bins were constructed based on a Voronoi classification of configuration space as described in Ref. 33. The Langevin simulations for both peptides were run for 100 ns, whereas LBMC runs employed 105 and 107 MC steps, respectively in (A) and (B). Error bars represent one standard deviation for each bin, estimated from 10 independent simulations for both LBMC and Langevin.
We also performed an efficiency analysis. For each simulation we calculated the CPU time required to generate one statistically independent configuration – based on the overall “structural decorrelation time”.25,26 This is roughly equivalent to the reduction in computer time necessary to achieve the same degree of statistical precision in the bin populations depicted in Figure 3. Although our implementation did not significantly decrease the computation time per energy call, substantial efficiency gains were obtained compared to Langevin simulation. The CPU time was reduced by factors of ~2000 and ~140, for Ace-(Ala)4-Nme and Ace-(Ala)8-Nme, respectively, compared to Langevin.
A detailed analysis of the effectiveness of LBMC for implicitly solvated peptides will be given in a separate publication, but we can briefly state the main points: (i) the efficiency does not result from what might be described as mimicking internal-coordinate MC (ICMC), as tests with ICMC indicate that approach is much less efficient than LBMC (data not shown); (ii) we believe the efficiency stems from the correlations stored in the libraries After all, it is extremely unlikely to randomly perturb a molecule (or fragment) and yield a physically reasonable configuration, but our extensive one-time investment in generating large libraries has already done this for the fragments.
In summary, tests with all-atom peptides in a simple continuum solvent clearly verified the correctness of the LBMC algorithm, and also suggested that LBMC can be efficient even when the energy calculation is not significantly accelerated.
IV.C. Proteins
We studied five proteins using the simplified protein model described in Sec. III – briefly, an all-atom backbone based on peptide-plane fragments and Gō interactions sited at the alpha-carbons. For the protein test systems, there are two goals: (i) to determine whether the model can indeed be fully sampled, based on the “structural decorrelation time” described previously,25,26 and (ii) to gain an initial perspective on the types of fluctuations that arise from full sampling of a semi-atomistic model. The present simple model is not designed to exhibit residue-specific chemical realism, and thus must be viewed as merely a first step in assessing the potential of LBMC and semi-atomistic models.
We studied two groups of proteins. The first group consisted of three small “test proteins” (<100 residues) studied in our previous work 24. The second group consisted of two much larger proteins (177 and 309 residues) that we used to explore size limitations of our approach.
For all studied proteins trial moves consisted of swapping three consecutive peptide planes and/or changing the corresponding ψ dihedrals. The acceptance rate was tuned to approximately 25% by adjusting two parameters. The first parameter controls the ratio of local moves from the neighbor lists to the “global” ones in which configurations are randomly selected from the neighbor lists of neighbor configurations. The second parameter is the ratio of ψ-only moves to the full moves consisting of swapping peptide planes plus changing the corresponding ψ dihedrals.
Test proteins
To compare our LBMC technique with our previously developed rigid peptide plane model 24 we performed LBMC calculations for the same three proteins previously studied with rigid planes. These were the binding domain of protein G (PDB code 1PGB, residues 1-56), the N-terminal domain of calmodulin (PDB code 1CLL, residues 5-75), and barstar (PDB code 1A19, residues 1-89).
As in our previous study we chose the dimensionless simulation temperature kBT/ε to be slightly below the unfolding temperature of the protein. The unfolding temperatures for each protein were determined via short simulations of 5*107 MC steps for 13 different temperatures. The temperatures determined for the production runs turned out to be the same as used in the previous study i.e. kBT/ε=0.5 for protein G, kBT/ε=0.4 for calmodulin and kBT/ε=0.6 for barstar.
Each protein system was first equilibrated for 108 MC steps followed by the production runs of 2*109 MC steps. For the production runs frames were saved at the interval of 1000 MC steps, generating equilibrium ensembles of 2*106 frames. The LBMC simulations were performed on a single Xeon 3.6 GHz CPU and it took 33 hours to complete the production run for protein G, 48 hours for calmodulin, and 52 hours for barstar.
The root mean square deviations (RMSD) along the trajectory relative to the experimental structure for three different proteins are shown in Figure 4. All backbone heavy atoms were used for RMSD calculations. These plots demonstrate the ability of our model to sample large conformational fluctuations of proteins along with the apparent convergence of the trajectories characterized by the stable behavior, in contrast to typical MD plots.
Figure 4.

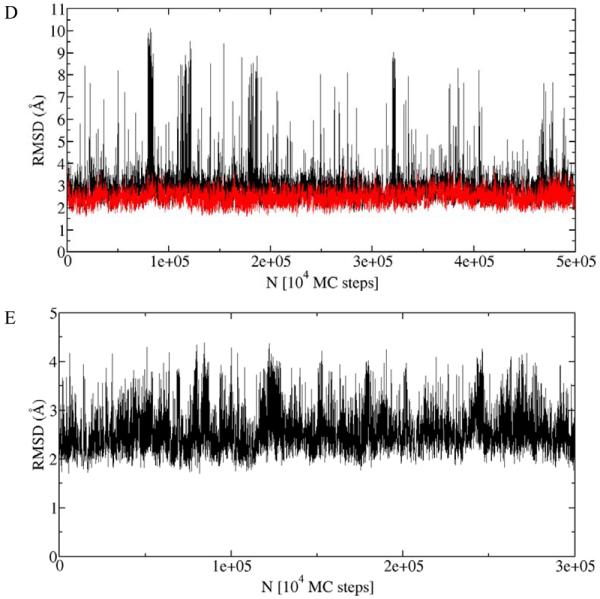
Root mean square deviation (RMSD) along the library-based Monte Carlo (LBMC) trajectory relative to the experimental structure for five different proteins: (A) protein G, (B) calmodulin, (C) barstar, (D) CDC25B, and (E) GGBP. RMSD was calculated based on all backbone heavy atoms. For CDC25B (D) RMSD was calculated based on the whole protein (black) and only on the stable part of the protein (residues 374-524) (red). These RMSD plots show that the LBMC technique is capable of sampling large conformational fluctuations along with apparent convergence characterized by stable behavior. The simulations were performed in less than three weeks on a single commercial processor.
A quantitative convergence analysis was performed using the method reported in Ref. 26, i.e. convergence was analyzed by studying the variance of the structural-histogram bin populations. The analysis determines the necessary time between trajectory frames so that statistically decorrelated behavior occurs – i.e. “the decorrelation time”. Figure 5 shows the convergence properties of LBMC simulations based on the ratio σ2 of the average population variance to that expected for independent sampling. A normalized variance (σ2) of one indicates statistical independence. The decorrelation time was therefore estimated from the graphs as the point where the normalized variance reaches the value of one within error bars for the first time (indicated by vertical arrows in Figure 5). The resulting decorrelation times are reported in Table 1 and indicate that the LBMC technique allows high quality sampling (ca. 100 decorrelation times) in several weeks of single CPU wall-clock time.
Figure 5.
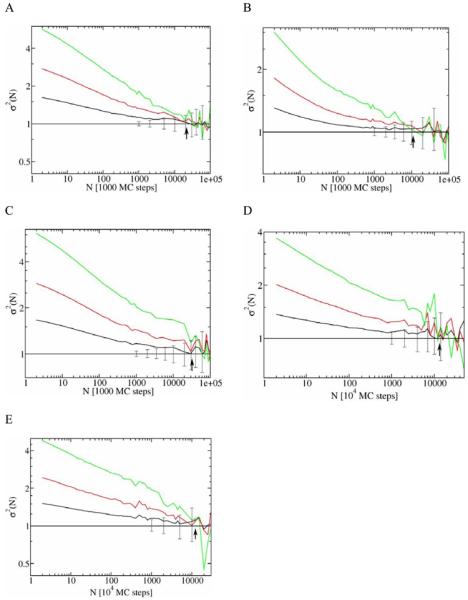
Convergence analysis of LBMC simulations for five different proteins: (A) protein G, (B) calmodulin, (C) barstar, (D) CDC25B, and (E) GGBP. Each plot shows the convergence properties analyzed using the procedure described in Ref. 26. N denotes the interval between frames along the trajectory at which the convergence properties are calculated. The number of frames required for the normalized variance (σ2) to reach the value of one (horizontal line) is an approximation of the structural decorrelation time marked by vertical arrows. The three curves on each plot are results for different subsample size and demonstrate the robustness of the value for the decorrelation time; see Ref. 26. The error bars correspond to estimates of an 80 % confidence interval intrinsic to the number of subsamples studied for the green line.
Table 1.
Decorrelation time for five different proteins in units of wallclock time of a single CPU using a method described in Ref. 26.
| Protein | Number of residues | Time |
|---|---|---|
| Protein G | 56 | 20 min |
| Calmodulin | 71 | 14.4 min |
| Barstar | 89 | 47 min |
| CDC25B | 177 | 9 h |
| GGBP | 309 | 24 h |
Although such a simple model cannot be expected to yield residue-specific properties pertinent to most experimental studies, it is interesting to examine the overall fluctuations resulting from the model. These fluctuations are implicitly shown in Figure 4, and can be compared to the range of fluctuations exhibited by NMR models. Although NMR “ensembles” are based on solution-state data, there has been considerable debate as to whether they suitably model the true fluctuations 40,24. Intriguingly, RMSD values for NMR structures (from PDB files 1GB1, 1AK8, and 1BTB), relative to the respective crystal structures (PDB codes 1PGB, 1CLL, and 1A19), exhibit significantly smaller fluctuations than our LBMC simulations. The average NMR values are 1.1 Å, 1.3 Å, and 1.0 Å respectively for protein G, calmodulin and barstar, which can be compared to Figure 4A, B, and C. While the simplicity of our model would seem to suggest the disparity is purely artifactual, it is noteworthy that the NMR fluctuations correspond to the variation seen in LBMC ensembles based on much lower temperatures – less than 20% of the simulated temperature in all cases (data not shown). These low temperatures, we note, are less than 20% of the melting temperatures (for our model) in all three proteins. Although it is premature to draw any reliable conclusions from the present comparison, the relatively small NMR fluctuations (equivalently, large simple-model fluctuations) merit further investigation in more realistic models which can be fully sampled.
CDC25B
As discussed above, one of the main advantages of the flexible peptide-plane model compared to a rigid plane model is that it can be used for proteins containing more than 100 residues. Thus we applied LBMC to human CDC25B catalytic domain (PDB code 1QB0, residues 374-550) containing 177 residues. This is a dual-specificity phosphatase with established links to cancer 43.
Simulation parameters were similar to those for test proteins. The simulation temperature, like for other proteins, was chosen slightly below the unfolding temperature based on 5 different temperature simulations and was determined to be kBT/ε=0.5. The system was equilibrated for 2*109 MC steps followed by the production run of 5*109 MC steps with frames saved every 104 frames generating the equilibrium ensemble of 5*105 frames. It took ca. 13 days of a single Xeon 3.6 GHz CPU wall-clock time to complete the production run.
The trajectory of RMSD values relative to the experimental structure for CDC25B is shown in Figure 4D. The RMSD based on the whole protein (black line in Figure 4D) shows very large conformational fluctuations. Inspection of trajectory configurations reveals that the C-terminus helix (residues 525-550) is flexible and partially unfolds during the simulation. The RMSD calculated based on the stable part of the protein (residues 374-524) is shown as a red line in Figure 4D and indicates that the rest of the protein is stable. Twenty different configurations along the trajectory are superposed in Figure 7 and show that the C-terminus helix is unstable (in our model) and partially unfolds during the simulation along with the stable behavior of the rest of the protein. The convergence analysis of the LBMC trajectory is shown in Figure 5D and the corresponding structural decorrelation time is reported in Table 1, indicating that converged sampling is possible in several weeks of a single CPU wallclock time.
Figure 7.
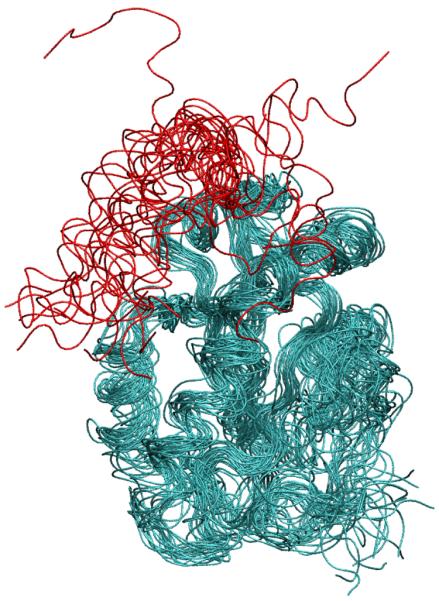
Superposition of 20 different backbone configurations of CDC25B along the LBMC trajectory. The stable part of the protein (residues 374-524) is shown in cyan and the flexible C-terminus helix in red. Interestingly, isoform A of CDC25 has a similar structure except for the C-terminus helix that is unfolded 44. In CDC25B this helix forms a cleft running along the protein body with an anion binding site at the end of the helix occupied by Cl−. Since the protein molecules in the crystal are in contact by C-termini these helices may be a crystal structure artifact caused by crystal packing forces and favorable electrostatic interactions. [Figure was produced using the program VMD 56].
It is worth noting that the crystal structures of CDC25 isoforms A and B are very similar except for the C-terminus region (corresponding to residue 529 and beyond in CDC25B) that is unfolded in isoform A 44. In isoform B this region forms an alpha-helix lying along the protein body with an anion binding site at the end occupied by one Cl− to stabilize the electrostatic charge in this region. Since the protein molecules in the crystal are in contact by C-termini, the stability of this region may be a crystal structure artifact caused by crystal packing forces and favorable electrostatic interactions 45.
GGBP
To test the limits of our LBMC technique we applied it to an even larger protein, the D-Galactose/D-glucose binding protein (GGBP) (PDB code 2GBP, residues 1-309) containing 309 residues. This is one of the first proteins used experimentally to study equilibrium fluctuations directly 46, and full details of these fluctuations will be examined in a future study.
As with other proteins, the simulation temperature was chosen slightly below the unfolding temperature based on 13 short simulations of 3*108 MC steps. The simulation temperature was determined to be kBT/ε =0.7. For the production run the system was run for 3*109 MC steps with frames saved every 104 MC steps, generating an equilibrium ensemble of 3*105 frames. It took ca. 22 days of single Xeon 3.6 GHz single CPU wallclock time to complete the production run.
The RMSD plot relative to the experimental structure is shown in Figure 4E. This plot demonstrates the ability of our technique to sample large conformational fluctuations for a large protein. The convergence analysis of the LBMC trajectory is shown in Figure 5E and the decorrelation time is reported in Table 1, indicating that a converged sampling is possible in about a month of single CPU wallclock time.
Are the large fluctuations in GGBP realistic? Comparison with disulfide-trapping data of Careaga and Falke 46 suggests large fluctuations akin to those in the LBMC simulation occur spontaneously in solution. For instance, Figure 6 shows the distribution of distances between the beta carbons of residues Gln26 and Asp267 produced by LBMC, with the red arrow indicating the minimum distance observed. Many of the LBMC distances are close enough (4-5 Å) for the observed disulfide formation exhibited by pairwise Cys mutants. The black arrow shows a typical experimental beta-carbon distance from disulfide groups in crystal structures. By contrast, four months of explicit solvent Langevin simulation (23 ns) yielded a much narrower distribution, with no disulfide-capable conformations (blue arrow indicates the minimum distance). A detailed analysis of the conformational fluctuations will be reported elsewhere.
Figure 6.

Distribution of distances between a pair of residues shown to form disulfide linkages experimentally in GGBP. The plot examines the distance between beta-carbons of Gln26 and Asp267, for which a pairwise Cys mutant exhibits disulfide bond formation on a sub-ms timescale in experiments 46. The LBMC simulation data (red line) were obtained in less than one month of single CPU wallclock time in contrast to the much narrower distribution from four months of explicit solvent Langevin simulation (blue line), which yielded 23 ns. The red and the blue arrows indicate the minimum distances observed in LBMC and Langevin simulations respectively. The black arrow shows the average distance required for disulfide bond formation, based on crystal structures. The vertical black line indicates the beta carbon distance between Gln26 and Asp267 in the crystal structure of GGBP (PDB code 2GBP).
For Langevin simulations, the crystal structure of GGBP (PDB code 2GBP) was used as the starting structure, which was solvated by 11,636 TIP3P water molecules.47 The total system size was 39,604 atoms. This system was simulated using Langevin dynamics as implemented in the NAMD software program 48 with CHARMM 27 all atom forcefield 49 at 298 K.
Approximate all-atom ensembles
For some applications, our statistical backbone ensembles can be converted into ad hoc all-atom ensembles. We explored this possibility by converting 500 configurations of CDC25B and 300 configurations of GGBP generated with LBMC to all-atom configurations by adding side chains using the program SCWRL 3.0 50. To remove steric clashes the side chain addition was followed by energy minimization using the OPLS-AA forcefield 32. These structures are available at www.epdb.pitt.edu. In CDC25B, inspection of the generated configurations revealed that ca. 40% of configurations have the disulfide bond formed between residues Cys426 and Cys473 which is not present in the original crystal structure (PDB code 1QB0). A literature search revealed that these Cys residues are conserved in CDC25 phosphatase family and easily oxidize to form a disulfide bond 44,45. It was speculated that formation of this disulfide bond may be important for self-inhibition during oxidative stress. The fact that our model can sample backbone conformations optimal for formation of the disulfide bond observed experimentally can be considered as a partial validation of our model.
V. Conclusions
We developed and tested a novel library-based Monte Carlo (LBMC) technique that allows the use of pre-sampled, flexible, all-atom components in arbitrary forcefields. A protein (or any molecule) is divided into atomistic molecular fragments which are pre-sampled, canonically and independent of other fragments. Configurations and energy terms are stored in libraries that are repeatedly re-used. In addition to avoiding computation of pre-calculated energy terms, the library ensembles embody non-trivial correlations internal to fragments: in this way, trial swap moves to other library configurations avoid clashes and incompatibilities which would arise from changing individual coordinates. LBMC is a rigorous Monte Carlo procedure which yields a canonical ensemble of the chosen model.
To underscore its flexibility, we applied LBMC to all-atom peptides using a standard forcefield, as well as to several proteins up to 309 residues in length modeled with an atomistic backbone and long-range Gō interactions sited at alpha-carbons. The peptide studies demonstrated the correctness and somewhat surprising efficiency of LBMC in an all-atom context. Studies of simplified proteins illustrated the capacity for full sampling of semi-atomistic models and the generation of fairly large but reasonable conformational fluctuations. A quantitative analysis demonstrates that fully sampled equilibrium ensembles can be obtained in about a month of single CPU wallclock time. Intriguing comparisons with several types of experimental data were also described. The LBMC approach with flexible atomistic peptide planes as described here greatly exceeds the practical limitation of our earlier semi-atomistic model employing rigid peptide planes 24.
The idea to use molecular fragments in computations is well established in the literature 51,29, and has been most successfully employed in the protein structure prediction software Rosetta 30. The main difference between the present LBMC technique compared to Rosetta's simulation method is that LBMC employs true statistical libraries and is statistically rigorous, allowing the generation of statistical ensembles and calculation of thermodynamic properties. We emphasize, however, that this initial study employed a much less chemically realistic forcefield than is employed in Rosetta.
Although all-atom based flexibility is present in our initial test model, it lacks side-chains and hence accurate physico-chemical interactions. Because our model can achieve converged sampling in such a short time, it readily can be enhanced by more accurate interactions. Additional potential energy terms such as Ramachandran propensities, hydrogen-bonding and hydrophobic interactions are currently being studied. Importantly, the LBMC approach permits the tracking of the locations of all atoms at negligible runtime cost, enabling additional interactions to be added readily as permitted by computational resources and the simulation goals. For instance, a set of residue libraries containing all atoms could interact very simply in most instances, except in a region of interest – e.g., a binding site – where full interactions could be used.
Potential applications of backbone ensembles generated with LBMC include docking and homology modeling. To make accurate predictions, these methods critically rely on ensembles of configurations, especially when substrate binding induces large conformational changes. For this purpose fully atomistic MD and MC simulations have been used before 52,53 but have practical limitations due to their computational cost and time scale limitations. Since our model can rapidly sample large conformational fluctuations and transitions, it may be useful for generating ensembles of configurations suitable for methods like docking and homology modeling. In fact, our CDC25B ensemble is already being employed for inhibitor-design studies (G. M. Arantes, personal communications 2008).
In principle LBMC can be applied to study protein folding although it should employ more accurate potential energy functions than used in this report. An example of such functions can be found in Ref. 54,55 where a new folding mechanism has been recently reported.
Further technical improvements certainly are possible. For instance, additional computation time may be saved by storage of interactions among neighboring residues. Larger fragments should facilitate more localized (crankshaft-like) trial moves and libraries biased to account for neighboring fragments should increase the acceptance rate. Also, the use of a fine grid 40 can permit storage of interactions among non-bonded residues.
Supplementary Material
Acknowledgements
We would like to thank Marty Ytreberg, Ed Lyman, Bin Zhang, Xin Zhang, Andrew Petersen, and Eran Eyal for helpful discussions. Funding was provided by the NIH through grants GM070987 and GM076569, as well as by the NSF through grant MCB-0643456.
Appendix
The acceptance criteria for trial moves which are library swaps can also be derived in a continuum picture by incorporating the library distribution into pgen. Let us start from the standard equation of detailed balance
| (23) |
where the trial configuration is fully described by the set of new fragment configurations, , and the old configuration is similarly given by . As in Eq. 3, peq is the Boltzmann factor of the full potential, while pgen and pacc are the conditional probabilities for generating and accepting trial moves, as above. From Eq. 23 the usual general expression for the acceptance criterion can be obtained, namely
| (24) |
Swap with a full fragment library
Let us first consider the case when a library-based trial move is the replacement (“swap”) of a single fragment configuration with a random choice from the full library of the same fragment i. That is, we are choosing the trial configuration , which differs from the configuration o by only a single fragment . But further, because the trial fragment configuration is chosen independently of the old configuration, the generating probability will depend only on the probability of choosing the new fragment from the library. In our case, each library is distributed according to the fragment Boltzmann factor, Eq. 9, so that the generating probability for a swap with the full library of fragment i is therefore
| (25) |
which is indeed independent of o. Here, is the normalizing partition function for fragment i, with limits of integration over the full hypervolume Vi of configuration space available to the fragment. We note that the strategy of choosing a trial configuration independent of the old configuration was originally suggested by Hastings 36, and is called “Metropolized independence sampling” 34.
The ratio of generating probabilities is obtained by a similar analysis of the reverse move, leading to the result
| (26) |
where the normalizing partition function has cancelled because the same fragment library is considered in both cases.
The full acceptance criterion can be derived by employing the ratio of Eq. 26 along with the equilibrium distribution of Eq. 3. Recalling the decomposition of the total potential energy of Eq. 1, all fragment energy terms cancel in the final acceptance criterion. The fragment terms aside from that of the swapped fragment cancel trivially because their configurations are unchanged, but the ratio of Eq. 26 leads – by design – to the cancellation of even the fragment term i with the equilibrium Boltzmann factor. The net result is that
| (27) |
for a swap generated using Eq. 25, where ΔUrest =Urest(n) −Urest(o). Eq. 27 is the same as Eq. 11 derived using the re-weighting approach.
Swap based on a neighbor list
Now let us consider a more general case, namely, when a trial fragment configuration is selected from a pre-generated neighbor list instead from the whole library. In a continuum description, this amounts to selecting a configuration from a pre-defined region of configuration space “neighboring” the old fragment configuration . (Note that the choice of the region is arbitrary, and it could be disconnected.) In our case, a trial fragment configuration will be selected according to the fragment Boltzmann factor solely within , which corresponds to the generating probability
| (28) |
which can be contrasted with Eq. 25.
To construct the ratio of generating probabilities, we must recognize that the normalizing local partition functions Zi,o and Zi,n (for pgen(o → n) and pgen(n → o), respectively) will not be the same now because generally the neighborhoods will differ, i.e., . The ratio of generating probabilities thus becomes, instead of Eq. 26,
| (29) |
We note that to satisfy microscopic reversibility, the neighborhoods should be defined so that if contains , then should contain .
The full acceptance criterion for neighbor lists can be derived by substituting Eqs. 3 and 29 into Eq. 24. Initially, one finds
| (30) |
but we want to eliminate the partition functions and convert the criterion for the discrete library case of interest. This can be done using the key fact that, because each fragment library is Boltzmann-distributed over its entire configuration space as in Eq. 9, the number of library configurations for fragment i which occur in the volume is simply proportional to the local partition function Zi,j. Further, the number of configurations is precisely the number of neighbors of configuration j for fragment i. Therefore, the ratio of partition functions occurring in Eq. 30 is equal to the ratio of the numbers of configurations in the corresponding neighbor lists, and the acceptance criterion simplifies to
| (31) |
Eq. 31 is indeed the same as Eq. 13 for the neighbor list swap moves derived using the re-weighing approach, completing our alternate derivation.
Footnotes
Supporting Information Available
The fragment libraries used in the LBMC simulations are described in detail. This information is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Volkman BF, Lipson D, Wemmer DE, Kern D. Science. 2001;291:2429–2433. doi: 10.1126/science.291.5512.2429. [DOI] [PubMed] [Google Scholar]
- 2.McCallum SA, Hitchens TK, Torborg C, Rule GS. Biochemistry. 2000;39:7343–7356. doi: 10.1021/bi992767d. [DOI] [PubMed] [Google Scholar]
- 3.Eisenmesser EZ, Bosco DA, Akke M, Kern D. Science. 2002;295:1520–1523. doi: 10.1126/science.1066176. [DOI] [PubMed] [Google Scholar]
- 4.Svoboda K, Mitra PP, Block SM. Proc. Natl. Acad. Sci. U. S. A. 1994;91:11782–11786. doi: 10.1073/pnas.91.25.11782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schotte F, Lim M, Jackson TA, Smirnov AV, Soman J, Olson JS, Phillips GN, Jr., Wulff M, Anfinrud PA. Science. 2003;300:1944–1947. doi: 10.1126/science.1078797. [DOI] [PubMed] [Google Scholar]
- 6.Vendruscolo M, Paci E, Dobson CM, Karplus M. J. Am. Chem. Soc. 2003;125:15686–15687. doi: 10.1021/ja036523z. [DOI] [PubMed] [Google Scholar]
- 7.Kitahara R, Yokoyama S, Akasaka K. J. Mol. Biol. 2005;347:277–285. doi: 10.1016/j.jmb.2005.01.052. [DOI] [PubMed] [Google Scholar]
- 8.Lindorff-Larsen K, Best RB, DePristo MA, Dobson CM, Vendruscolo M. Nature. 2005;433:128–132. doi: 10.1038/nature03199. [DOI] [PubMed] [Google Scholar]
- 9.Freddolino PL, Liu F, Gruebele M, Schulten K. Biophys. J. 2008;94:L75–77. doi: 10.1529/biophysj.108.131565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Swendsen RH, Wang J-S. Phys. Rev. Lett. 1986;57:2607–2609. doi: 10.1103/PhysRevLett.57.2607. [DOI] [PubMed] [Google Scholar]
- 11.Berg BA, Neuhaus T. Phys. Lett. B. 1991;267:249–253. [Google Scholar]
- 12.Hukushima K, Nemoto K. J. Phys. Soc. Jpn. 1996;65:1604–1608. [Google Scholar]
- 13.Hansmann UHE. Chem. Phys. Lett. 1997;281:140–150. [Google Scholar]
- 14.Nakajima N, Nakamura H, Kidera A. J. Phys. Chem. B. 1997;101:817–824. [Google Scholar]
- 15.Berg BA. Phys Rev Lett. 2003;90:180601. doi: 10.1103/PhysRevLett.90.180601. [DOI] [PubMed] [Google Scholar]
- 16.Okamoto Y. J. Mol. Graphics. 2004;22:425–439. doi: 10.1016/j.jmgm.2003.12.009. [DOI] [PubMed] [Google Scholar]
- 17.Tozzini V. Curr. Opin. Struct. Biol. 2005;15:144–150. doi: 10.1016/j.sbi.2005.02.005. [DOI] [PubMed] [Google Scholar]
- 18.Ayton GS, Noid WG, Voth GA. Curr. Opin. Struct. Biol. 2007;17:192–198. doi: 10.1016/j.sbi.2007.03.004. [DOI] [PubMed] [Google Scholar]
- 19.Clementi C. Curr. Opin. Struct. Biol. 2008;18:10–15. doi: 10.1016/j.sbi.2007.10.005. [DOI] [PubMed] [Google Scholar]
- 20.Kim YC, Hummer G. J. Mol. Biol. 2008;375:1416–1433. doi: 10.1016/j.jmb.2007.11.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lyman E, Ytreberg FM, Zuckerman DM. Phys. Rev. Lett. 2006;96:028105. doi: 10.1103/PhysRevLett.96.028105. [DOI] [PubMed] [Google Scholar]
- 22.Lyman E, Zuckerman DM. J. Chem. Theory Comput. 2006;2:656–666. doi: 10.1021/ct050337x. [DOI] [PubMed] [Google Scholar]
- 23.Zhang BW, Jasnow D, Zuckerman DM. Proc. Natl. Acad. Sci. U. S. A. 2007;104:18043–18048. doi: 10.1073/pnas.0706349104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ytreberg FM, Aroutiounian SK, Zuckerman DM. J. Chem. Theory Comput. 2007;3:1860–1866. doi: 10.1021/ct700076t. [DOI] [PubMed] [Google Scholar]
- 25.Lyman E, Zuckerman DM. Biophys. J. 2006;91:164–172. doi: 10.1529/biophysj.106.082941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lyman E, Zuckerman DM. J. Phys. Chem. B. 2007;111:12876–12882. doi: 10.1021/jp073061t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wall FT, Rubin RJ, Isaacson LM. J. Chem. Phys. 1957;27:186–188. [Google Scholar]
- 28.Alexandrowicz Z. J. Chem. Phys. 1969;51:561–565. [Google Scholar]
- 29.Macedonia MD, Maginn EJ. Mol. Phys. 1999;96:1375–1390. [Google Scholar]
- 30.Rohl CA, Strauss CEM, Misura KMS, Baker D. Methods Enzymol. 2004;383:66–93. doi: 10.1016/S0076-6879(04)83004-0. [DOI] [PubMed] [Google Scholar]
- 31.Das R, Qian B, Raman S, Vernon R, Thompson J, Bradley P, Khare S, Tyka MD, Bhat D, Chivian D, Kim DE, Sheffler WH, Malmstrom L, Wollacott AM, Wang C, Andre I, Baker D. Proteins. 2007;69:118–128. doi: 10.1002/prot.21636. [DOI] [PubMed] [Google Scholar]
- 32.Jorgensen WL, Maxwell DS, Tirado-Rives J. J. Am. Chem. Soc. 1996;118:11225–11236. [Google Scholar]
- 33.Zhang X, Mamonov AB, Zuckerman DM. J. Comput. Chem. 2009 doi: 10.1002/jcc.21337. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liu JS. Monte Carlo strategies in scientific computing. New York; Springer: 2001. [Google Scholar]
- 35.Ferrenberg AM, Swendsen RH. Phys. Rev. Lett. 1988;61:2635–2638. doi: 10.1103/PhysRevLett.61.2635. [DOI] [PubMed] [Google Scholar]
- 36.Hastings WK. Biometrika. 1970;57:97–109. [Google Scholar]
- 37.Lovell SC, Davis IW, Adrendall WB, de Bakker PIW, Word JM, Prisant MG, Richardson JS, Richardson DC. Proteins. 2003;50:437–450. doi: 10.1002/prot.10286. [DOI] [PubMed] [Google Scholar]
- 38.Ueda Y, Taketomi H, Go N. Int. J. Pept. Protein Res. 1975;7:445–459. [PubMed] [Google Scholar]
- 39.Ueda Y, Taketomi H, Go N. Biopolymers. 1978;17:1531–1548. [Google Scholar]
- 40.Zuckerman DM. J. Phys. Chem. B. 2004;108:5127–5137. [Google Scholar]
- 41.Allen MP, Tildesley DJ. Computer simulation of liquids. Oxford University Press; New York: 2001. [Google Scholar]
- 42.Ponder JW, Richard FM. J. Comput. Chem. 1987;8:1016–1024. [Google Scholar]
- 43.Galaktionov K, Lee A, Eckstein J, Draetta G, Meckler J, Loda M, Beach D. Science. 1995;269:1575–1577. doi: 10.1126/science.7667636. [DOI] [PubMed] [Google Scholar]
- 44.Fauman EB, Cogswell JP, Lovejoy B, Rocque WJ, Holmes W, Montana VG, Piwnica-Worms H, Rink MJ, Saper MA. Cell. 1998;93:617–625. doi: 10.1016/s0092-8674(00)81190-3. [DOI] [PubMed] [Google Scholar]
- 45.Reynolds RA, Yem AW, Wolfe CL, Deibel MR, Chidester CG, Watenpaugh KD. J. Mol. Biol. 1999;293:559–568. doi: 10.1006/jmbi.1999.3168. [DOI] [PubMed] [Google Scholar]
- 46.Careaga CL, Falke JJ. Biophys. J. 1992;62:209–219. doi: 10.1016/S0006-3495(92)81806-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML. J. Chem. Phys. 1983;79:926–935. [Google Scholar]
- 48.Phillips JC, Braun R, Wang W, Gumbart J, Tajkhorshid E, Villa E, Chipot C, Skeel RD, Kale L, Schulten K. J Comput Chem. 2005;26:1781–1802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.MacKerell AD, Bashford D, Bellott M, Dunbrack RL, Evanseck JD, Field MJ, Fischer S, Gao J, Guo H, Ha S, Joseph-McCarthy D, Kuchnir L, Kuczera K, Lau FTK, Mattos C, Michnick S, Ngo T, Nguyen DT, Prodhom B, Reiher WE, Roux B, Schlenkrich M, Smith JC, Stote R, Straub J, Watanabe M, Wiorkiewicz-Kuczera J, Yin D, Karplus M. The Journal of Physical Chemistry B. 1998;102:3586–3616. doi: 10.1021/jp973084f. [DOI] [PubMed] [Google Scholar]
- 50.Canutescu AA, Shelenkov AA, Dunbrack RL. Protein Sci. 2003;12:2001–2014. doi: 10.1110/ps.03154503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Miranker A, Karplus M. Proteins: Structure, Function, and Genetics. 1991;11:29–34. doi: 10.1002/prot.340110104. [DOI] [PubMed] [Google Scholar]
- 52.Lin JH, Perryman AL, Schames JR, McCammon JA. J. Am. Chem. Soc. 2002;124:5632–5633. doi: 10.1021/ja0260162. [DOI] [PubMed] [Google Scholar]
- 53.Lin JH, Perryman AL, Schames JR, McCammon JA. Biopolymers. 2003;68:47–62. doi: 10.1002/bip.10218. [DOI] [PubMed] [Google Scholar]
- 54.Mohanty S, Hansmann UH. J. Phys. Chem. B. 2008;112:15134–15139. doi: 10.1021/jp804661t. [DOI] [PubMed] [Google Scholar]
- 55.Mohanty S, Meinke JH, Zimmermann O, Hansmann UH. Proc. Natl. Acad. Sci. U. S. A. 2008;105:8004–8007. doi: 10.1073/pnas.0708411105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Humphrey W, Dalke A, Schulten K. J. Mol. Graphics. 1996;14:33–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.